
Abstract
Our recent findings indicate an induced upregulation of 14-3-3γ mRNA and protein in ischemic cortical astrocytes. Despite being brain-specific, the functional role of 14-3-3γ in the brain still remains largely unknown. In this study, we show that among all the 14-3-3 isoforms, only the γ isoform is inducible under ischemia in astrocytes. Furthermore, this upregulation of 14-3-3γ may play a specific protective role in astrocytes under ischemia. Overexpression experiments and antisense treatment show that an elevation of 14-3-3γ protein in astrocytes promotes survival, while a decrease in 14-3-3γ enhances apoptosis in astrocytes under ischemia. Under ischemia, endogenous 14-3-3γ binds p-Bad, thus preventing Bad from entering mitochondria to initiate apoptosis. Therefore, 14-3-3γ is selectively induced during ischemia to protect astrocytes from apoptosis through p-Bad-related signaling.
Introduction
Complex organs, such as the brain, are composed of a wide variety of regulatory proteins to maintain their proper biologic functions. The acidic and soluble 14-3-3 protein family members are abundant in brain tissues (Moore and Perez, 1967; Boston et al, 1982), but their functions remain elusive. Some reports indicate, however, that 14-3-3 may play a critical role in signaling for cell survival (Zha et al, 1996; Masters et al, 2001; Tzivion et al, 2001; van Hemert et al, 2001). The 14-3-3 family has seven known subtypes, β, ɛ, γ, η, σ, τ, and ζ, with 14-3-3γ identified in mammalian cells as brain- and neuron-specific (Moore and Perez, 1967; Aitken et al, 1992; Watanabe et al, 1993). Our previous findings show that 14-3-3γ is also expressed in cultured astrocytes (Chen and Yu, 2002; Chen et al, 2003), indicating that 14-3-3γ may be more than just brain-specific. A few studies have shown elevated 14-3-3γ protein levels in several brain regions in patients with Alzheimer's disease and Down's syndrome (Burkhard et al, 2001), and in the cerebrospinal fluid in patients with Creutzfeldt–Jakob disease (Wiltfang et al, 1999), indicating an involvement of 14-3-3γ in neurodegenerative diseases. Upregulation of 14-3-3γ has been previously shown in cultured astrocytes subjected to ischemic injury (Chen et al, 2003). Furthermore, 14-3-3γ is associated with actin (Chen and Yu, 2002) and Raf (Chen et al, 2003) in cultured astrocytes during ischemia, suggesting a possible role for 14-3-3γ in ischemic signaling.
In this study, we show that upregulation of 14-3-3γ is crucial to protect astrocytes from ischemia-induced apoptosis. The protective mechanism may involve the binding of 14-3-3γ to phosphorylated Bad, which thus prevents the entry of Bad into mitochondria to trigger proapoptotic events.
Materials and methods
Primary Cultures of Cerebral Cortical Astrocytes
Astrocyte cultures were prepared from cerebral cortices of newborn ICR mice, as previously reported (Jiang et al, 2002, 2003). Typically, each dissociated cerebrum would be distributed into 13 culture dishes (35 mm; Becton Dickinson & Company, San Jose, CA, USA) containing 2 mL culture medium. All cultures were incubated in a Napco CO2 incubator (Precision Scientific Inc., Chicago, IL, USA) at 37°C with 95% air/5% CO2 (v/v) and 95% humidity. Culture medium was changed 2 days after initial seeding, and subsequently, twice per week with Dulbecco's modified Eagle medium (DMEM) (Invitrogen Corp., Burlington, Ontario, Canada) containing 10% (v/v) fetal calf serum (FCS) (HyClone, Logan, UT, USA) for the first 2 weeks, and 7% (v/v) FCS thereafter. Confluent cultures (approximately 106 cells/mL) used for experiments were at least 4 weeks old.
Anaerobic Chamber-Induced Ischemia and Inhibitor Treatment
The anaerobic chamber-induced ischemia model has been used in various studies (Chen and Yu, 2002; Chen et al, 2003). Briefly, ischemia media (free of glucose and serum) were degassed for 30 mins with N2 and re-gassed with N2:CO2:H2 (85:5:10) for 20 mins before use. The ischemia media and cultures were then transferred into an anaerobic chamber (Forma Scientific, Thermo Electron Corp., Massachusetts, USA) saturated with N2:CO2:H2 (85:5:10). The oxygen concentration in the ischemia media was monitored by a dissolved oxygen meter (HI 9142 model, Hanna Instruments Inc., Italy) to ensure ischemic conditions. The cultures were washed three times with ischemia media, after which the cells were covered with 6.4 mL (or 0.78 mL) ischemia media for 100-mm (or 35-mm) dishes. All culture dishes were wrapped with a parafilm to prevent evaporation during ischemic incubation of various durations (1, 2, 4, and 6 hours). Normoxic cultures (0-hour ischemia) were used as controls. Cells were harvested immediately after ischemic treatment without reperfusion.
Quantitative Reverse Transcription-PCR
Total RNA from astrocyte cultures was isolated with TRIZOL®Reagent (Invitrogen), according to the manufacturer's protocol. Reverse transcription was performed using 1 μg total RNA, 20 μmol/L dNTP (Invitrogen), 1 μmol/L random hexamers (Invitrogen), and 100 U reverse transcriptase (Invitrogen) in a total volume of 10 μL. PCR (with 28 cycles of 94°C, 45 secs; 68°C, 45 secs; 72°C, 45 secs, and a final extension at 72°C for 5 mins) was performed using 1 μL cDNA in a total volume of 10 μL. The forward and reverse primers for amplifying the 14-3-3 isoforms were based on GenBank sequences of murine 14-3-3 isoforms: for γ, 5′-gttggtctggctcttcatcat-3′ and 5′-aggtgcagagtagacttgggtg-3′; for β, 5′-ctcttcctggcgtgtcatct-3′ and 5′-actttgctttctgcctgggt-3′; for ɛ, 5′-ccccattcgtttaggtcttg-3′ and 5′-ggtccacagcgtcaggttat-3′; for η, 5′-atgggcatttgctggactg-3 and 5′-aaggaatgagttgtcgctgtg-3′; for ζ, 5′-tgctggtgatgacaagaaagg-3′ and 5′-gaggcagacaaaggttggaag-3′. Murine glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers (5′-tgatgacatcaagaaggtggtgaag-3′ and 5′-tccttggaggccatgtaggccat-3′) were used as internal controls. DNA sequencing was performed to confirm that the PCR amplification products were the expected 14-3-3 isoforms and GAPDH. The expression levels of 14-3-3 isoforms during ischemia were computed as relative to those of GAPDH and were compared with 0-hour ischemia.
Western Blot and Coimmunoprecipitation Analyses
Western blot analysis was performed, as previously described (Chen and Yu, 2002), using diluted rabbit polyclonal antibody (all with 1/1000 dilution) against 14-3-3γ, green fluorescent protein (GFP) (both from Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), Bad, p-Bad 112, p-Bad 136 or p-Bad 155 (Cell Signaling Technology Inc., Beverly, MA, USA). Specificity of the 14-3-3γ antibody has been shown previously (Chen et al, 2003). Coimmunoprecipitation (Co-IP) was performed according to the methods described previously (Chen and Yu, 2002), using antibody (1/1000 dilution) against 14-3-3γ or Bad.
Immunostaining and Terminal Deoxynucleotidyl Transferase End-labeling Staining
Cultured astrocytes were washed twice with phosphate buffered saline (PBS) before staining. All staining procedures were performed at room temperature. Cells were fixed with 4% paraformaldehyde for 15 mins. After washing with PBS twice, cells were permeabilized with 0.2% Triton X-100 for 15 mins and blocked with 3% BSA for 2 hours. After incubation with primary antibody for 2 hours, cells were washed three times and incubated with FITC or rhodamine-conjugated secondary antibody for 1 hour. Hoechst 33342 (2 μg/mL) was used to stain the nucleus for 5 mins. Terminal deoxynucleotidyl transferase end-labeling (TUNEL) (Roche Diagnostics Corp., Indianapolis, IN, USA) staining was performed as reported previously (Yu et al, 2001) before Hoechst 33342 staining. After extensive washing, the coverslip was mounted before observation using regular fluorescent microscopy.
Lipofection of Primary Culture of Astrocytes
The coding region of the 14-3-3γ gene was PCR-amplified using total RNA extracted from ischemic astrocytes and a high fidelity Taq ELONGASE™ Enzyme (Invitrogen). To optimize fidelity, 25 cycles of PCR were performed. Primer sequences were derived from the mouse 14-3-3γ mRNA (GenBank Accession No. AF058799; nucleotides 183 to 223 and 906 to 926). The PCR-amplified 14-3-3γ fragment was cloned into pDsRed1-N1 (Clontech, Becton Dickinson & Company) to construct the 14-3-3γ sense and antisense vectors. Expression of sense or antisense 14-3-3γ was driven by the CMV promoter present in the pDsRed1-N1 vector. The cloned plasmids were sequenced to confirm correct DNA identity. The mouse Bad DNA fragment, derived from pEBG-mBad (Cell Signaling Technology Inc.), was subcloned into pDsRed-N1. Plasmid DNA for transfection was extracted with a Wizard® Plus Midipreps (or Maxipreps) DNA purification kit (Promega Corp., Madison, WI, USA). For transient transfection of confluent cultures of astrocytes, 3 μg of plasmid DNA (in a maximum volume of 10 μL) and 6 μL of LipofectAMINE™ 2000 (LF2000) (Invitrogen) were diluted separately in 0.15 mL of Opti-MEM for 5 mins at room temperature, then combined and incubated for 30 mins. Afterwards, the DNA/LF2000 mixture was added directly to cultures grown in 35-mm dishes containing 2 mL medium, and incubated for 6 hours under normoxic conditions. Transfected cultures were subsequently washed twice with fresh DMEM medium and maintained in fresh DMEM medium for 48 hours before experiments.
For cotransfection, p-EGFP-N1 vector expressing GFP was used as a marker for transfected cells and apoptosis was scored by nuclear condensation, as previously described (Gillardon et al, 1996; Liu et al, 2003). The plasmid ratio was optimized by cotransfecting p-EGFP-N1 and p-DsRed1-N1 in astrocytes. In a ratio of 1:3, all p-EGFP-N1-transfected astrocytes (green) also expressed pDsRed1-N1 (red), thus subsequent cotransfection followed a 1:3 ratio for p-EGFP-N1, and p-DsRed1-N1 respectively.
Statistical Analysis
All data were presented as mean±s.e.m. from at least three independent experiments. For statistical analysis of cell numbers, averages of cells counted in nine randomly selected fields were used. To quantify the results of Western blot analysis, the intensity of the bands was measured by a densitometer. Statistical analysis was performed by Student's unpaired t-test at a confidence interval of at least 95% (i.e. P⩽0.05).
Results
Ischemia-Induced 14-3-3γ Protects Astrocytes from Apoptosis
Ischemia stimulated 14-3-3γ protein expression in astrocytes and this elevation of 14-3-3γ (1 hour) occurred earlier than the death of astrocytes (4 hours) under ischemia. We examined the expression levels of the 14-3-3γ protein in surviving and apoptotic astrocytes under ischemia. After 6- and 8-hour ischemia, many astrocytes shrank and their cell boundaries became more prominent, allowing the stained 14-3-3γ in individual astrocytes to be clearly distinguished. The nuclei of apoptotic astrocytes under ischemia, when stained with Hoechst 3342, appeared to be highly condensed (Chen and Yu, 2002; Jiang et al, 2002, 2003). We found that the intensity of 14-3-3γ protein staining in these apoptotic astrocytes (Figure 1A, indicated by arrows) was lower than in the surrounding surviving astrocytes, which exhibited contracted but not heavily condensed nuclei (Figure 1A, indicated by concave arrowheads). 14-3-3γ protein levels were elevated in all surviving astrocytes (Figure 1A, concave arrowheads). These results indicate that the protein levels of 14-3-3γ in the surviving astrocytes under ischemia are much higher than in apoptotic astrocytes.
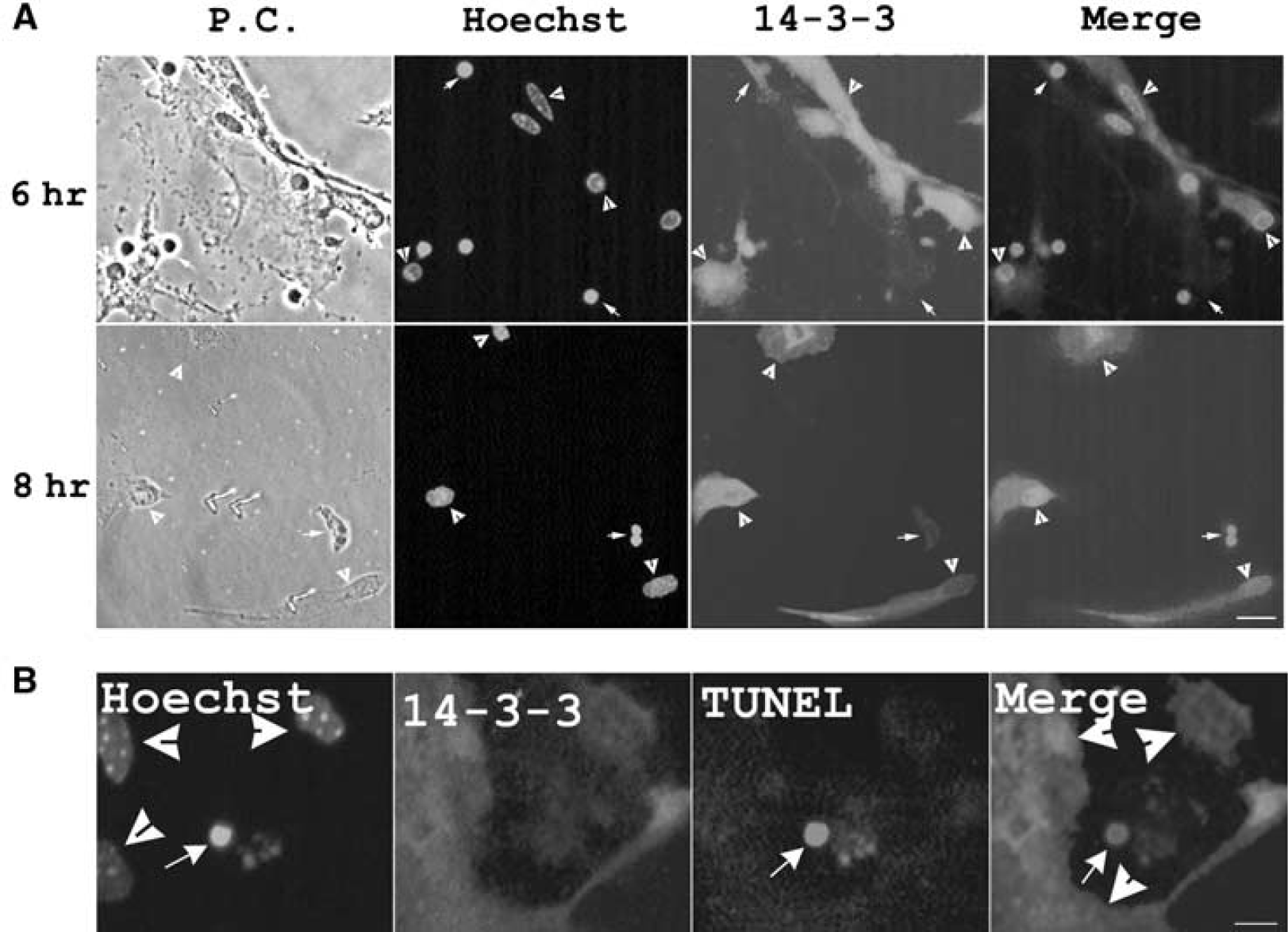
14-3-3γ protein level is elevated in surviving astrocytes but decreased in apoptotic astrocytes under ischemia. (
We further investigated whether an early elevation of 14-3-3γ protein is critical to protect astrocytes from ischemia insults. Under 4 hours of ischemia, early apoptotic signs appear but apoptotic death is rare (Yu and Lau, 2000). The level of 14-3-3γ protein in the astrocyte committed to apoptosis (TUNEL-positive nuclei) was found to be significantly lower than the surrounding TUNEL-negative astrocytes (Figure 1B, indicated by concave arrowheads). This again indicates that an upregulation of 14-3-3γ protein is critical to protect astrocytes from apoptosis.
Transient Transfection of 14-3-3γ Enhances Survival of Ischemic Astrocytes
We verified the protective effect of 14-3-3γ protein by transfecting a wild-type 14-3-3γ gene to astrocytes in primary cultures. To visualize transfected astrocytes, 14-3-3γ and GFP genes (in a 3:1 ratio) were cotransfected into astrocytes. The effect of overexpressing 14-3-3γ in cultured astrocyte was determined by subjecting transfected cultures to 6 hours in vitro ischemia treatment (Figure 2A). While some dead or dying cells remained attached to the culture (Figure 2A, arrowheads), the empty space was because of dead cells being washed off during the staining process. The successfully transfected astrocytes survived (Figure 2A, green, arrows) with normal nuclear morphology. Surviving and apoptotic cells could be distinguished by Hoechst staining (Chen and Yu, 2002; Jiang et al, 2002, 2003), which thus allowed an estimation of the percentage of apoptotic cells in cultures (Gillardon et al, 1996; Liu et al, 2003). Considering astrocytes transfected with GFP alone, the percentage of apoptotic cells (GFP-positive) was approximately 50% (Figure 2B, leftmost bar), similar to that observed in untransfected cultures (Figure 2B, rightmost bar). For astrocytes cotransfected with 14-3-3γ and GFP genes, the percentage of apoptotic cells (GFP-positive) was significantly reduced to approximately 25% (Figure 2B, center bar).
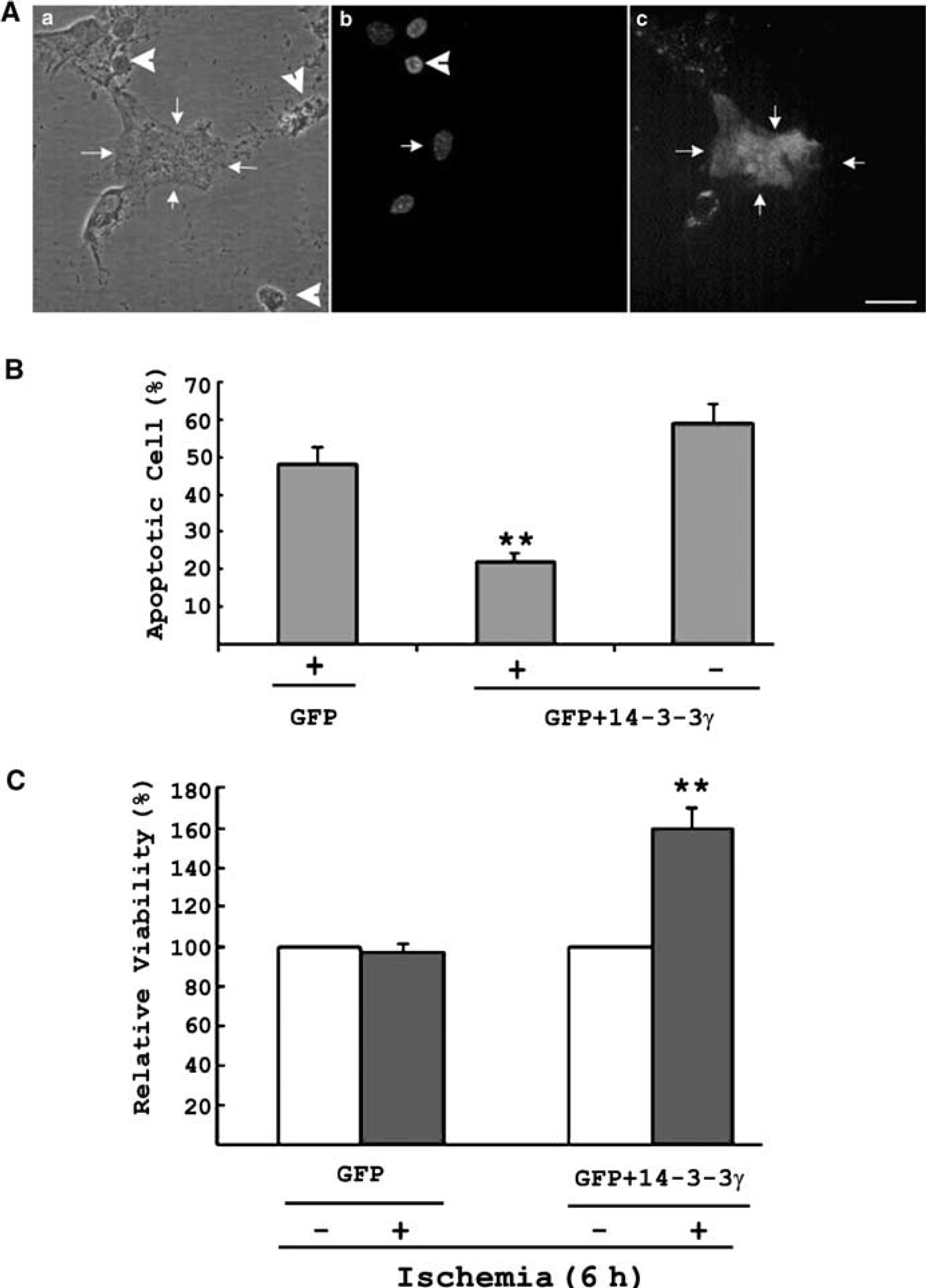
Transient transfection of 14-3-3γ enhances cell survival and prevents apoptosis in astrocytes under ischemia. (
The relative viability of transfected cells was determined by measuring the relative levels of GFP (indicating the amount of transfected cells that survived) to actin (indicating the amount of total cells that survived) through Western blot analysis. In cultures transfected with GFP alone, the percentage of transfected astrocytes was approximately 12%. The GFP/actin levels did not change under ischemia (Figure 2C, left panel), supporting the assumption that GFP transfection alone did not affect the viability of astrocytes under ischemia. In cultures cotransfected with 14-3-3γ and GFP, GFP/actin level significantly increased (Figure 2C) after ischemia, consistent with our hypothesis of the protective effect of 14-3-3γ transfection. This was verified by counting the number of apoptotic cells (Figure 2B). The apoptotic cell count together with the measurement of relative GFP/actin levels in ischemic cultures confirmed the protective role of 14-3-3γ in astrocyte under ischemia.
Antisense 14-3-3γ Promotes Apoptosis in Ischemic Astrocytes
To further elucidate the protective role of 14-3-3γ, the effect of antisense 14-3-3γ gene treatment was examined by cotransfecting the antisense construct (75%) and p-EGFP-N1 vector (25%) into astrocytes. Cultures transfected with antisense 14-3-3γ produced apparently less 14-3-3γ protein when compared with the cells transfected with sense 14-3-3γ (Figure 3A). The antisense-transfected cells did not exhibit apoptosis before ischemic incubation as compared with untransfected and sense 14-3-3γ transfected cultures. We examined cultured astrocytes undergoing 4-hour ischemia and observed that astrocytes transfected with antisense 14-3-3γ displayed apoptotic characteristics (Figure 3B). These astrocytes presented highly condensed nuclei (Figure 3B, arrows), in contrast to neighboring untransfected cells (Figure 3B, concave arrowheads). The GFP/actin ratio was then measured to quantify the apoptotic effect of antisense 14-3-3γ in astrocytes under ischemia for 6 hours (Figure 3C). Again, GFP transfection alone did not affect astrocyte viability under ischemia (Figure 3C, left panel). Green fluorescent protein/actin ratios in astrocytes cotransfected with antisense 14-3-3γ and the GFP gene also remained unchanged under normal conditions (i.e., 0-hour ischemia). However, a reduction of 50% in relative viability was observed in cotransfected cultures under ischemia for 6 hours, indicating that antisense treatment can abolish the upregulation of 14-3-3γ and thus enhancing cell death under ischemia.
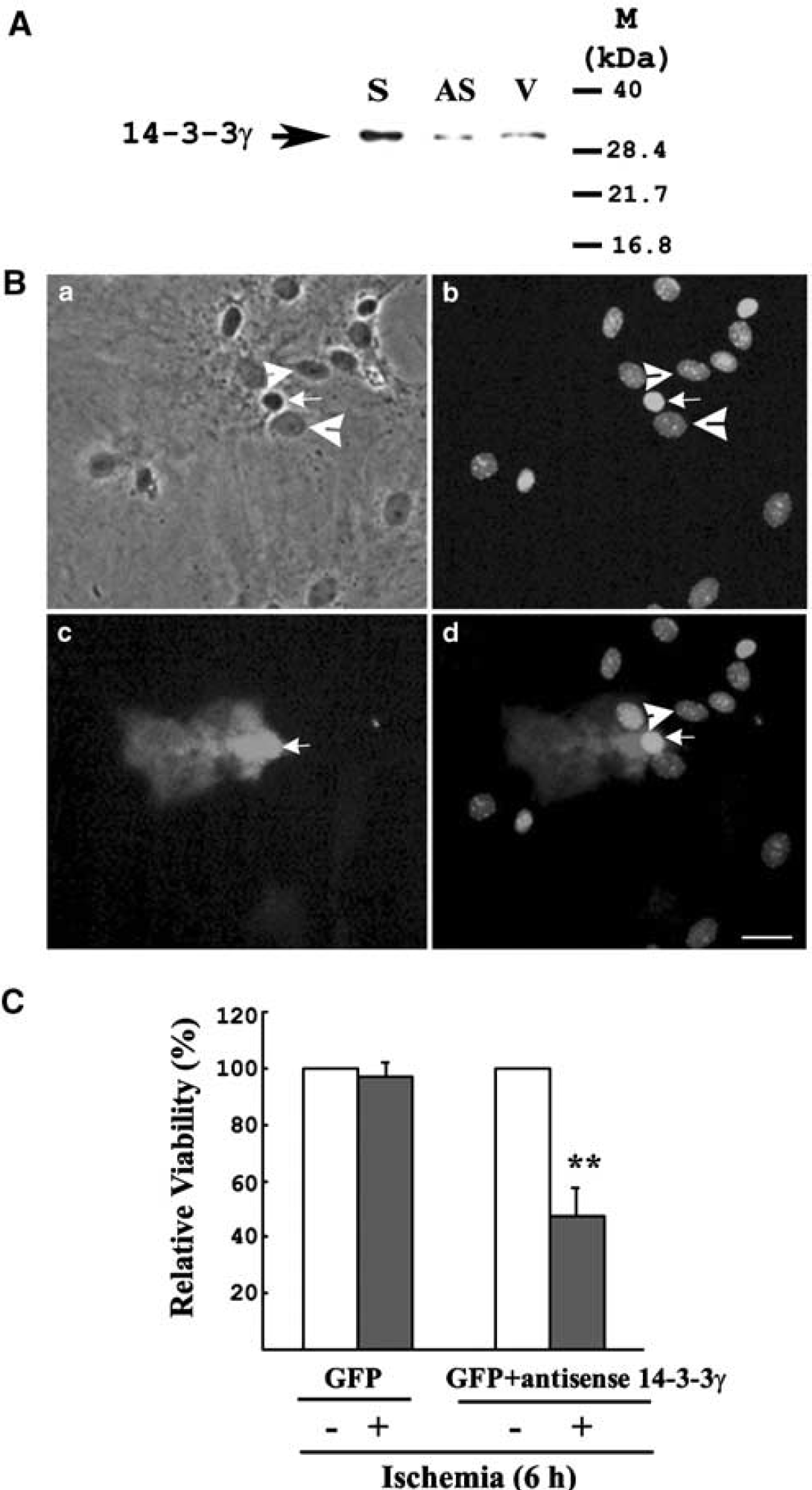
Transient transfection of antisense 14-3-3γ promotes apoptosis in ischemic astrocytes. (
Specific Role of the γ Isoform During Ischemia
Five major members (β, ɛ, γ, η and ζ) of the 14-3-3 family have been reported in the brain. The role of each of these isoforms, however, remains elusive. It is therefore important to determine if the ischemia-induced 14-3-3γ upregulation observed in cultured astrocytes is isoform-specific. In cultured astrocytes, the expression of τ and σ isoforms of 14-3-3 was not detected by RT-PCR, while the five 14-3-3 isoforms (β, ɛ, γ, η and ζ) were expressed in astrocytes (Figure 4A). Interestingly, only the γ isoform was induced by ischemic insult in astrocytes. The expression levels of the 14-3-3γ gene steadily increased during the ischemic treatment, and after peaking 2-hour ischemia, the levels remained above the basal level at 4- and 6-hour ischemia (Figure 4A). In contrast, gene expression levels of 14-3-3 β, ɛ, η and ζ did not significantly change after various durations of ischemic incubation. The expression levels of all the isoforms of 14-3-3 were normalized to GAPDH and compared with levels at 0 hour of ischemia. Statistical analysis showed that the upregulation of 14-3-3γ under ischemia, especially at 2 hours, was significant (Figure 4B). The strong induction of 14-3-3γ on ischemic insult indicates a distinct functional role of this 14-3-3 isoform in ischemic astrocytes.
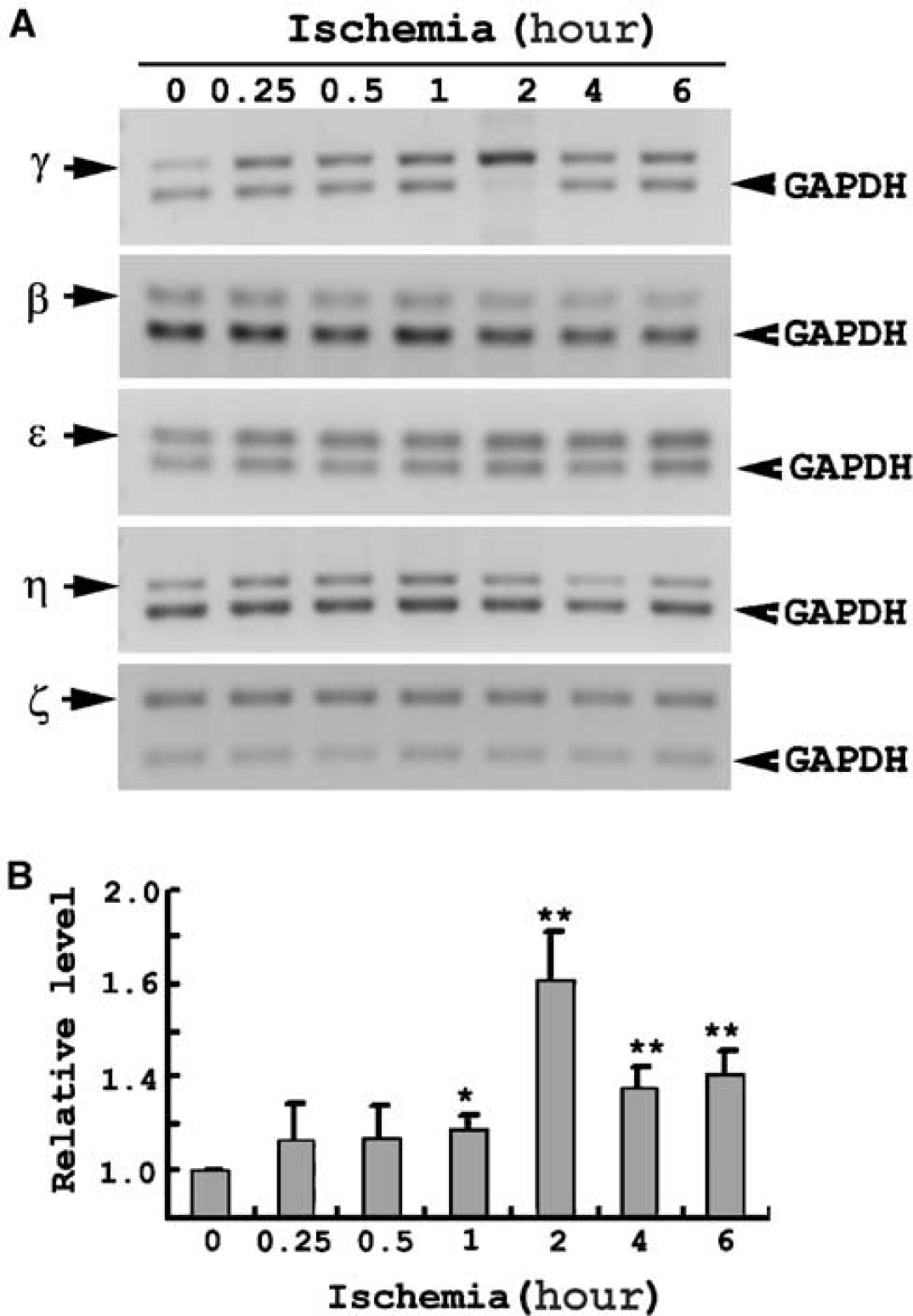
Selective upregulation of 14-3-3γ gene expression by ischemia. (
14-3-3γ Reduces Ischemia-Induced Apoptosis Through Specific Bad Signaling Pathway
To explore how 14-3-3γ attenuates astrocytes from apoptosis under ischemia, we studied the binding of endogenous 14-3-3γ protein to Bad, a critical protein involved in apoptotic pathways. In a co-IP study with 14-3-3γ antibody, a basal level of phosphorylated Bad (p-Bad) was detected in the precipitates (Figure 5A). The amount of 14-3-3γ-bound p-Bad apparently increased during ischemia of various time. Western blot results are consistent with the co-IP finding, demonstrating increases of 14-3-3γ and Bad protein levels (peaking at 4 hours) in astrocytes under ischemia (Figure 5B). Association of Bad with 14-3-3γ could be mediated by binding of 14-3-3γ to the p-Bad 112, 136, and/or 155 forms (Figure 5C). Further quantification of the levels of p-Bad 112, 136 and 155 during ischemia would identify which p-Bad form is more likely to mediate such binding to alleviate ischemia-induced apoptosis in astrocytes.
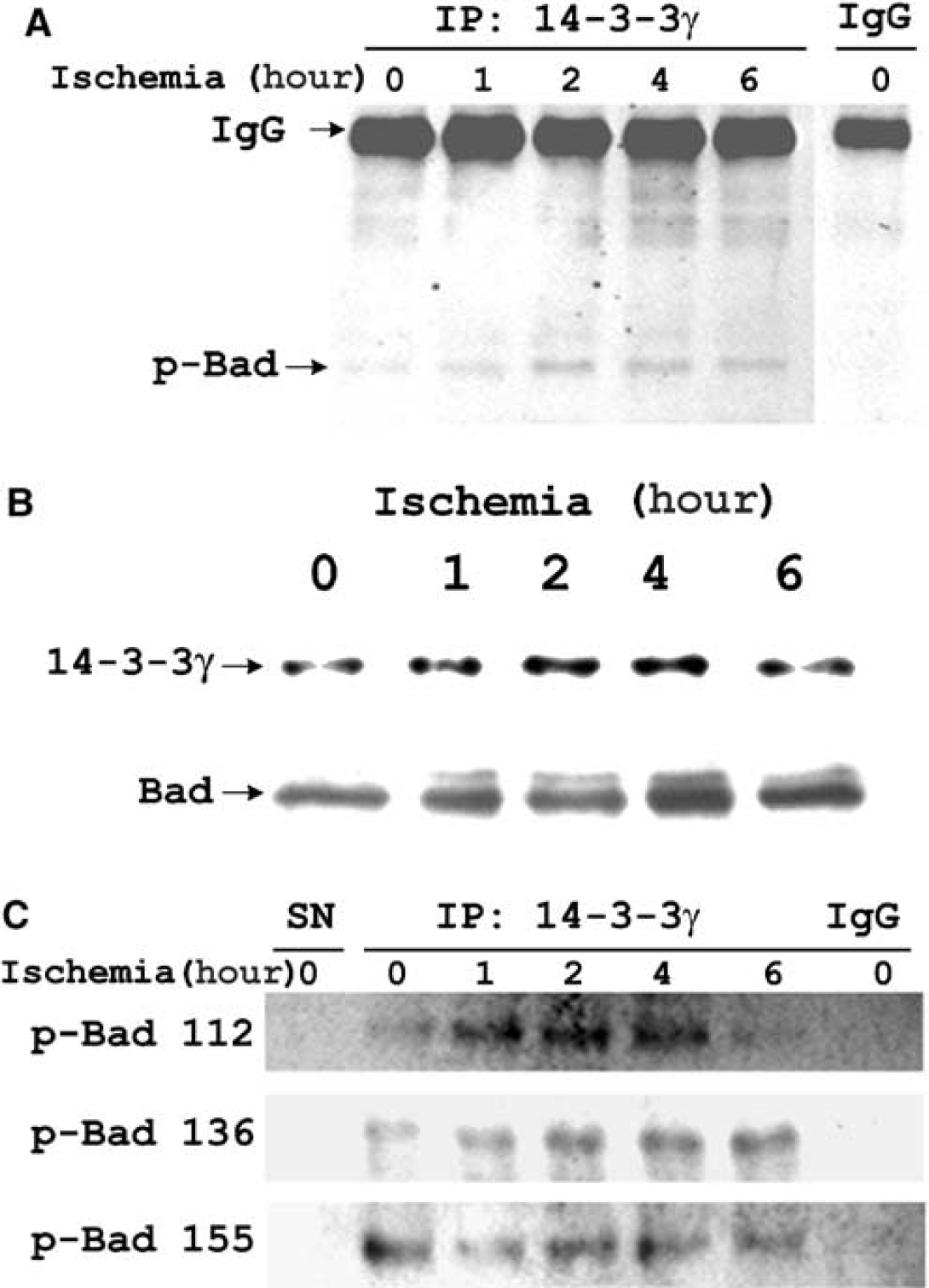
Endogenous 14-3-3γ binds to p-Bad 112, p-Bad 136, and p-Bad 155 in ischemic astrocytes. (
14-3-3γ is Essential for Preventing Bad-Induced Apoptosis under Ischemia
The increase in the levels of 14-3-3γ and Bad proteins, and their bindings, in astrocytes under ischemia led to the question of whether elevated 14-3-3γ protein levels were essential for preventing Bad-induced apoptosis under ischemia. Bad (43%), and either 14-3-3γ or antisense 14-3-3γ gene (43%) together with a GFP gene (14%) were cotransfected into astrocytes. In this plasmid ratio, astrocytes were successfully transfected with GFP/Bad/14-3-3γ or GFP/Bad/antisense 14-3-3γ genes (Figure 6A). Before ischemic incubation, the amount of GFP was similar in astrocytes cotransfected with either GFP/Bad/14-3-3γ or GFP/Bad/antisense 14-3-3γ (Figure 6A). After 4-hour ischemia, the amount of GFP was not significantly affected in astrocytes cotransfected with 14-3-3γ constructs. However, GFP levels decreased markedly (Figure 6A) in astrocytes cotransfected with antisense 14-3-3γ construct. Statistical analysis of transfected cell viability based on the relative GFP/actin ratios showed that cotransfection with GFP/Bad/14-3-3γ did not significantly alter cell viability, while cotransfection with GFP/Bad/antisense 14-3-3γ resulted in less than 20% relative viability in astrocytes at 4-hour ischemia (Figure 6B). Thus, the 14-3-3γ protein was largely responsible for suppressing Bad-induced apoptosis under ischemia.
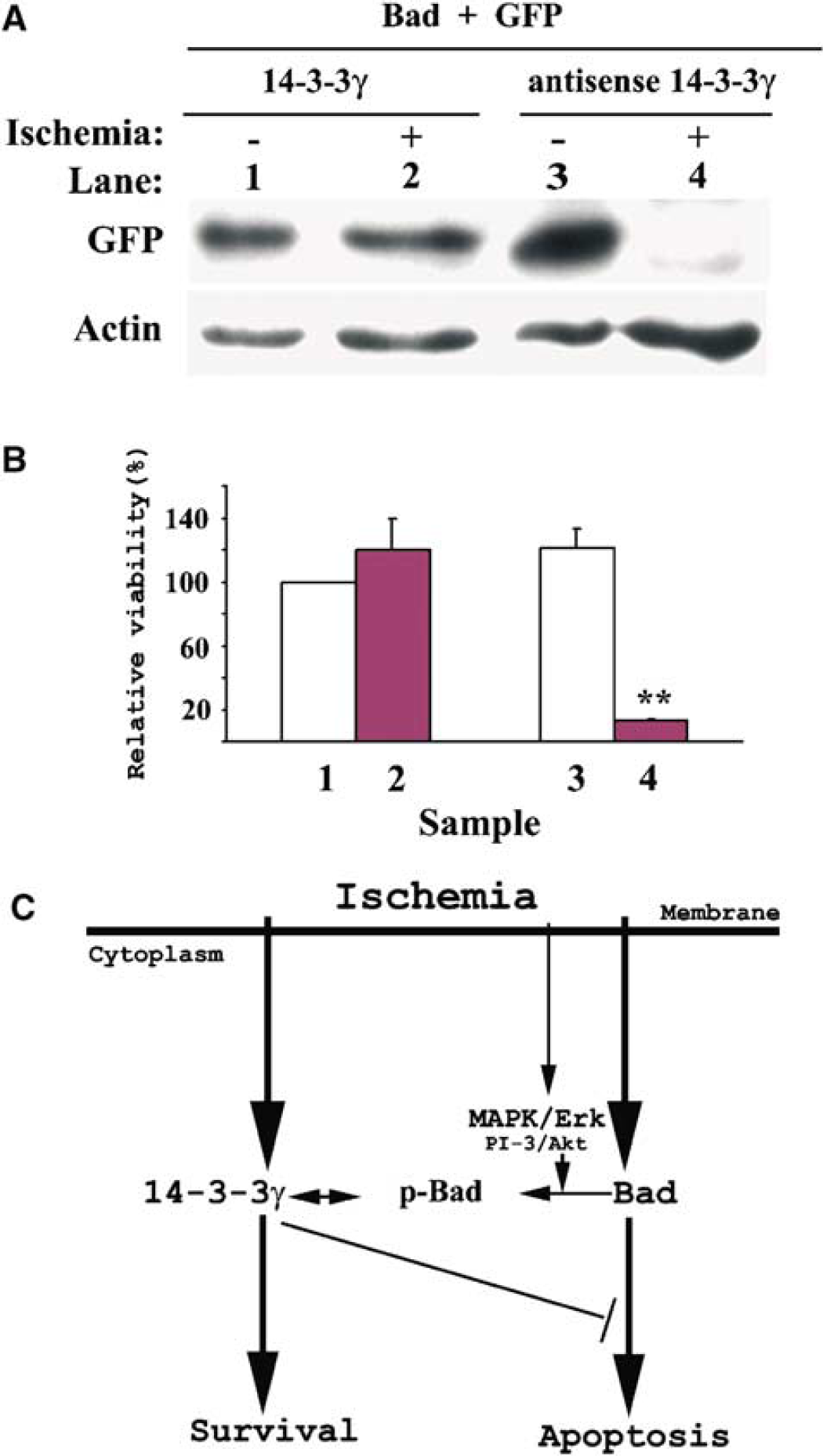
14-3-3γ is required to prevent Bad-induced apoptosis in astrocytes under ischemia. (
Discussion
It is known that ischemia causes apoptotic cell death in the brain (Gillardon et al, 1996; Nicotera et al, 1999; Zhu et al, 2002) and cultured astrocytes (Ho et al, 2001; Yu et al, 2001; Jiang et al, 2002, 2003). It has also been hypothesized that certain endogenous cellular protective mechanisms might be induced during injury to protect cells (Jiang et al, 2003). Presently, the most convincing evidence is that ischemia upregulates Bcl-2 (Gillardon et al, 1996; Chen et al, 1997; Schwarz et al, 2002) to protect cells from injury. However, in astrocytes, the most abundant glial cells in the brain, Bcl-2 is not upregulated by ischemia (Jiang et al, 2002), suggesting that Bcl-2 does not play a role in protecting astrocytes under ischemia insult. In this study, we show that ischemia-induced upregulation of 14-3-3γ protein in astrocytes promotes survival (or prevents apoptosis) through binding to p-Bad (Figure 6C).
The evidence to support the hypothesis that 14-3-3γ protein plays a protective role in astrocytes under ischemia can be summarized as follows: (1) 14-3-3γ gene is selectively upregulated by ischemia and the induction is sustained during the whole process of ischemic insult; (2) elevation of 14-3-3γ protein levels after prolonged ischemia is observed in all surviving astrocytes; (3) decreases in 14-3-3γ protein are observed in apoptotic astrocytes during ischemia; (4) overexpression of 14-3-3γ promotes cell survival under ischemia; (5) expression of a 14-3-3γ antisense construct enhances apoptotic death under ischemia; (6) 14-3-3γ is essential for preventing Bad-induced apoptosis under ischemia by binding to Bad.
Our hypothesis that ischemia-induced upregulation of 14-3-3γ protects astrocytes is supported by two other studies on 14-3-3. In Cos-7 cells, overexpression of a peptide blocking the interaction of 14-3-3 with other proteins caused 50% of the cells to die (Masters and Fu, 2001). Although this study did not focus on any particular isoform, it suggested that the binding of 14-3-3 to injury-related proteins could promote cell survival or prevent cell death. In a separate study with fibroblasts, blocking 14-3-3ζ expression enhanced cell death 48 hours after UV irradiation (Xing et al, 2000), suggesting a protective role for the ζ isoform specific to UV irradiation. In addition, several studies have reported that various growth factors (e.g. nerve growth factor) and cytokines (e.g. interleukin-3) promote the binding of 14-3-3 to Bad, FKHRL1, ASK1, Raf-1 or Cdc25, which are known to correlate with cell survival (Tzivion et al, 2001; van Hemert et al, 2001). Although most studies have not addressed the functions of specific isoforms, it is likely that all isoforms of 14-3-3 share some basic functional similarities (Aitken et al, 1992).
The specific protective role for the 14-3-3γ isoform in ischemic astrocytes is supported by our previous finding that the γ isoform is induced in astrocytes only under ischemia, but not under heat shock or scratch-wound injury (Chen et al, 2003). Others have shown that the level of β, but not the γ isoform, is elevated in the spinal cord after crush injury (Springer et al, 2000). In this study, our results shows that only the γ isoform, among all the 14-3-3 isoforms, is upregulated in astrocytes by ischemia. Blocking 14-3-3γ expression in astrocytes by antisense 14-3-3γ treatment enhances apoptosis in astrocytes under ischemia, but not in normoxia, further clarifying the protective role of upregulated γ isoform during ischemia. A study showing that the inhibition of 14-3-3ζ expression in fibroblasts enhances cell death after exposure to UV irradiation, but not under normal conditions, again lends support to our hypothesis.
14-3-3γ upregulation by ischemia might be a crucial protective mechanism in the brain under ischemic insult. 14-3-3 proteins are abundant in the brain and the γ isoform is brain-specific (Boston et al, 1982; Watanabe et al, 1993). Early induction of 14-3-3γ during ischemia suggests that such induction might be a primary protective response of the brain against ischemic insult. This is supported by our findings of activated PI-3/Akt and MAPK/Erk pathways in ischemic astrocytes (Jiang et al, 2002). With the use of LY294002 and U0126, specific inhibitors of PI-3/Akt and MAPK/Erk, respectively, 14-3-3γ expression remained unaffected (Chen et al, 2003), indicating the involvement of additional signaling pathways other than the PI-3/Akt and MAPK/Erk ones for 14-3-3γ to be activated.
In this study, we showed that 14-3-3γ binds to phosphorylated forms of Bad, namely p-Bad 112, 136 and 155, consistent with previous findings of 14-3-3 binding to p-Bad 112, 136, or 155 (Zha et al, 1996; Harada et al, 1999; Datta et al, 2000; Tan et al, 2000). A recent report demonstrating the inhibition of Bad-induced cell death by interaction between 14-3-3ζ and p-Bad 136 may indicate a more predominant role of the p-Bad 136 (Masters et al, 2001). The binding of 14-3-3γ to p-Bad may sequester ischemia-induced Bad in the cytoplasm, thus blocking Bad from disturbing the balance of pro- and antiapoptotic Bcl-2 members in the mitochondria (Gross, 2001). 14-3-3γ protein plays a crucial role in maintaining this balance because our experiments have shown that overexpressing 14-3-3γ alone completely blocks the apoptotic effect of Bad, while blocking the expression of 14-3-3γ allows Bad to induce apoptosis under ischemia (Figure 6).
In conclusion, selectively elevated 14-3-3γ plays a critical role in preventing apoptosis of astrocytes under ischemia. This protective effect is most likely mediated by specific binding to p-Bad. This finding highlights the importance of the up-regulation of 14-3-3γ in astrocytes under ischemia. We also discuss a possible mechanism by which 14-3-3γ exerts its protective effects against ischemic injury: binding of 14-3-3γ to p-Bad blocks the translocation of Bad into the mitochondria, thus protecting the ischemic cells from apoptosis. The findings presented here have not only given us a better understanding of the self-defense mechanisms in astrocytes subjected to ischemia, but also opened a new window in developing protective therapy to counter the effects of ischemic insult on the central nervous system.
Footnotes
Acknowledgements
The work was supported by grants from the Hong Kong Research Grants Council (HKUST6177/97M), the North American Medical Association Foundation (Hong Kong) (NAMA 94/95.SC01), the National Natural Science Foundation of China (30270426) and the Beijing Natural Science Foundation (7032026) to ACHY.