
Abstract
Neurons express a variety of plasma-membrane potassium channels that play important roles in regulating neuronal excitability and synaptic transmission, but also contain mitochondrial ATP-sensitive potassium channels, the functions of which are unknown. Studies of cardiac cells suggest that similar mitochondrial ATP-sensitive potassium channels are involved in the process of ischemic preconditioning, suggesting a role in regulating cell survival. The authors report that mice given diazoxide, an activator of mitochondrial ATP-sensitive potassium channels, exhibited a large (60% to 70%) decrease in cortical infarct size after permanent occlusion of the middle cerebral artery. Diazoxide decreases neuronal apoptosis and increases astrocyte survival and activation in the penumbral region of the ischemic cortex. The neuroprotective effect of diazoxide is abolished by 5-hydroxydecanoate, a selective antagonist of mitochondrial ATP-sensitive potassium channels. Studies of cultured hippocampal neurons reveal that diazoxide depolarizes mitochondria, prevents cytochrome c release, and protects cells against death induced by staurosporine and chemical hypoxia. Diazoxide increased the levels of Bcl2 and inhibited the association of Bax with mitochondria in neurons exposed to an apoptotic insult, suggesting that activation of mitochondrial ATP-sensitive potassium channels may stabilize mitochondrial function by differentially modulating proapoptotic and anti-apoptotic proteins. Collectively, the data suggest that mitochondrial ATP-sensitive potassium channels play a key role in modulating neuronal survival under ischemic conditions, and identify agents that activate mitochondrial ATP-sensitive potassium channels as potential therapeutics for stroke and related neurodegenerative conditions.
Keywords
Eukaryotic cells contain one or more members of a family of evolutionarily conserved potassium channels that modulate plasma-membrane potential and thereby regulate cell excitability (Robertson, 1997). Abnormalities in potassium channels have been linked to a variety of diseases, with the nervous and cardiovascular systems being the most commonly affected (Jentsch, 2000). Although most research of potassium channels has focused on those in the plasma membrane, recent studies have shown that mitochondria also contain potassium channels (Inoue et al., 1991; Grover and Garlid, 2000). Whereas membrane potassium channels consist of a pore-forming subunit (Kir6.1 or Kir6.2) and a sulfonylurea receptor (SUR1, SUR2A, or SUR2B), the protein composition of mitochondrial ATP-sensitive potassium channels (Mito-KATP) channels has not been established. However, a distinct pharmacology of Mito-KATP channels has been established such that the channels are selectively activated by low concentrations of diazoxide and blocked by 5-hydroxydecanoate (5HD) (Ghosh et al., 2000; Sato et al., 2000; Liu et al., 2001). Recent studies have identified candidate Mito-KATP channel proteins. A 28-kd protein was isolated from heart muscle mitochondria based on its selective binding to the sulfonylurea compound glibenclamide (Szewczyk et al., 1997), though this protein may not be a component of diazoxide-sensitive Mito-KATP channels. Bajgar et al. (2001) recently isolated and purified a novel Mito-KATP channel protein from rat brain mitochondria that exhibits ligand-binding properties similar to those of heart Mito-KATP channels. Data in the latter study indicate that the amount of Mito-KATP channels in brain cells is at least sixfold higher than in heart cells, suggesting an important role for these channels in neuronal function.
When heart or brain cells are subjected to brief mild ischemia, they become resistant to a more severe ischemic insult such as that which occurs during a myocardial infarction or stroke (Yellon and Dana, 2000; Matsushima and Hakim, 1995; Barone et al., 1998; Stagliano et al., 1999; Shimizu et al., 2001). Data suggest that such “ischemic preconditioning” activates transcription factors that induce increased production of cytoprotective proteins such as antioxidant enzymes and antiapoptotic proteins (Das et al., 1999). Studies of cardiac myocytes have provided convincing evidence that Mito-KATP channels play a central role in ischemic preconditioning. Thus, agents such as diazoxide that selectively open Mito-KATP channels can mimic the cytoprotective effect of preconditioning, whereas drugs such as 5HD that selectively block Mito-KATP channels can abolish ischemic preconditioning (Takashi et al., 1999; Eells et al., 2000; O'Rourke, 2000). The mechanism of ischemic preconditioning may involve a cascade of events including the liberation of adenosine, stimulation of adenosine A1 receptors, and the opening of Mito-KATP via these receptors (Heurteaux et al., 1995). It has been proposed that mild ischemia activates Mito-KATP by a mechanism involving oxyradical production and activation of protein kinase C (Takashi et al., 1999; Pain et al., 2000).
Stroke, a major cause of disability and death worldwide, occurs when a cerebral blood vessel becomes occluded or ruptures, resulting in ischemic damage and death of cells in the brain tissue supplied by the affected vessel (Elkind and Sacco, 1998). There are currently no treatments that can reduce the extent of brain damage after a stroke. Studies of the pathogenesis of stroke suggest that overactivation of glutamate receptors (excitotoxicity), oxidative stress, and cellular calcium overload contribute to the death of neurons (Lee et al., 2001). As in the heart, ischemic preconditioning can increase the resistance of neurons in the brain to a stroke (Reis et al., 1997; Yu and Mattson, 1999). Recent findings suggest that much of the brain damage in stroke involves a form of programmed cell death called apoptosis (Mattson et al., 2000; Graham and Chen, 2001). Pivotal events in neuronal apoptosis include oxyradical production, dysregulation of cellular ion homeostasis, mitochondrial dysfunction, release of cytochrome c, and activation of proteases of the caspase family (Bratton and Cohen, 2000). Because mitochondrial changes including membrane permeability transition and release of cytochrome c are thought to be pivotal events in apoptosis (Fiskum, 2000; Gorman et al., 2000), there is considerable interest in establishing the regulatory systems in mitochondria that determine whether apoptosis occurs. Although the role of Mito-KATP channels in ischemic stroke is unknown, it has been reported that the Mito-KATP channel opener diazoxide can protect neurons against insults relevant to stroke, including glutamate toxicity and oxidative stress (Abele and Miller, 1990; Goodman and Mattson, 1996). Data obtained in the present study suggest that Mito-KATP channels play a pivotal role in modulating neuronal vulnerability in ischemic stroke.
MATERIALS AND METHODS
Mice, drug administration, and focal cerebral ischemia model
Adult male 3-month-old C57BL/6 mice weighing 25 to 28 g were obtained from the National Cancer Institute and maintained on a 12-hour light/12-hour dark cycle with continuous access to food and water. Food was withdrawn from all mice 12 to 16 hours before surgery. Diazoxide and 5HD were infused intravenously in the femoral vein in doses of 5 and 20 mg/kg, respectively, 20 to 30 minutes before middle cerebral artery occlusion (MCAO). Glibenclamide was infused into the left lateral ventricle (0.1 mg/kg) under stereotaxic guidance using methods similar to those described previously (Duan et al., 2001).
Permanent focal cerebral ischemia was induced by cauterizing the left middle cerebral artery using methods described previously (Nawashiro et al., 2000). Briefly, mice were anesthetized using isoflurane administered as a vapor; body temperature was maintained at 37°C throughout the surgical procedure and recovery period. The left middle carotid artery was exposed through a 1-cm vertical incision between the left eye and ear. The temporal muscle was split and a portion of the skull at the junction of the zygomatic arch and squamous bone was removed. Cerebral blood flow was measured continuously by laser-Doppler flowmetry using a flexible 0.5-mm fiberoptic probe (Perimed, Stockholm, Sweden) beginning 5 to 10 minutes before drug administration and ending 20 to 30 minutes after MCAO. A probe used for brain temperature measurement was placed directly on the brain surface. Focal ischemia was produced by permanent occlusion of the left middle cerebral artery by electrocoagulation. Mice were killed 7 hours, 24 hours, or 7 days after MCAO by an isoflurane overdose.
Quantification of infarct volume
Brains were removed immediately after mice were killed, rinsed in cold phosphate-buffered saline (PBS), cut into 2-mm-thick coronal sections. Brain sections were incubated for 30 minutes at room temperature in a solution of 2% 2,3,5-triphenyltetrazolium chloride (TTC) in PBS, and then fixed for 30 minutes with 4% paraformaldehyde in PBS. The infarct border in each brain slice was outlined and the area quantified using a NIH image 6.1 software (National Institutes of Health, Bethesda, MD, U.S.A.). To correct for brain swelling, the infarct area was determined by subtracting the area of undamaged tissue in the left hemisphere from that of the intact contralateral hemisphere. The infarct volume was calculated by integration of infarct areas for all slices of each brain (Swanson et al., 1990).
Histochemical analyses
In situ terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL) was used to detect the occurrence of DNA fragmentation after ischemic injury using methods described previously (Noshita et al., 2001). Briefly, cryosections were rehydrated, fixed with 4% formaldehyde, and incubated with deoxynucleotide triphosphate, biotinylated deoxyuridine triphosphate, and terminal deoxynucleotidyl transferase. The conjugates were viewed with fluorescein isothiocyanate conjugate-avidin, and images were acquired using a confocal microscope (LSM510; Carl Zeiss Inc., Thornwood, NY, U.S.A.). Additional brain sections were stained with hematoxylin and eosin, or were immunostained with antibodies against glial fibrillary acidic protein (GFAP) (Chemicon, Temecula, CA, U.S.A.) using methods described previously (Lee et al., 2002). Images of TUNEL staining and immunofluorescence were acquired using a Zeiss LSM510 confocal microscope.
Primary neuronal cultures and quantification of cell survival
Primary hippocampal cell cultures were established from E18 rat embryos using methods described previously (Mattson et al., 1997). Dissociated cells were seeded onto polyethyl-eneimine-coated plastic dishes or 22-mm2 glass coverslips, and maintained in neurobasal medium containing B-27 supplements, 2-mmol/L L-glutamine, 0.001% gentamicin sulfate, and 1-mmol/L HEPES (pH 7.2). The neurons in these cultures are vulnerable to chemical hypoxia and apoptosis induced by staurosporine (Mattson et al., 1997, 1998). Potassium cyanide was prepared as a 500x stock in water, whereas diazoxide, 5HD, and staurosporine were prepared as 500x stocks in dimethyl sulfoxide. Neuron survival was quantified by counting undamaged neurons in premarked microscope field before and at indicated time points after exposure to experimental treatments using methods described previously (Mattson et al., 1997). Neurons that died in the intervals between examination points were usually absent, and the viability of the remaining neurons was assessed by morphologic criteria. Neurons with intact neurites and soma with a smooth round appearance were considered viable, whereas neurons with fragmented neurites and vacuolated soma were considered nonviable. Analyses were performed without knowledge of the culture treatment history.
Assessments mitochondrial membrane potential and cytochrome c release
The fluorescent probe tetramethylrhodamine ethyl ester (TMRE) (Molecular Probes, Eugene, OR, U.S.A.) was used as an indicator of mitochondrial membrane potential using methods described previously (Krohn et al., 1999; Scaduto and Grotyohann, 1999). Briefly, cells were treated with diazoxide or 5HD in Locke buffer for 30 minutes at 37°C. Cells were then incubated for 20 minutes in the presence of 100-nmol/L TMRE, washed three times in fresh culture medium, and confocal images of cellular TMRE fluorescence were acquired using a Zeiss 510 confocal laser scanning microscope (543-nm excitation and 585-nm emission). The average pixel intensity in individual cell bodies was determined using the software supplied by the manufacturer (Zeiss); all images were coded and analyzed without knowledge of experimental treatment history of the cultures. To evaluate the subcellular localization of cytochrome c, we used confocal imaging of cells doubled-labeled with Mitotracker Red CMX Ros (Molecular Probes) and a cytochrome c antibody using methods similar to those described previously (Cheng et al., 2001). After experimental treatment, cells were incubated with 100-nmol/L Mitotracker Red CMX Ros for 30 minutes at 37°C (the dye is taken up by mitochondria where it forms thiol conjugates with peptides, and is thereby trapped in the mitochondria), washed with PBS, and fixed with 4% paraformaldehyde in PBS at 37°C for 30 minutes. Fixed cells were permeabilized with 0.1% Triton X-100 for 5 minutes at 4°C, followed by a 2-hour incubation at room temperature in blocking solution (2% normal goat serum, 0.1% Triton X-100 in PBS, pH = 7.4) containing 10 μg/mL primary monoclonal cytochrome c antibody (PharMingen, San Diego, CA, U.S.A.). After washing, cells were incubated for 2 hours in PBS containing fluorescein isothiocyanate-conjugated goat antimouse immunoglobulin G (1:100; Molecular Probes). Cells were then imaged in dual-scan mode on a Zeiss CLSM 510 confocal microscope using a 40x water immersion objective (numerical aperture = 1.4). The excitation and emission wavelengths for Mitotracker CMX Ros were 510 and 590 nm, respectively and for fluorescein isothiocyanate conjugate were 488 and 510 nm, respectively.
Assessments of Bax association with mitochondria and Bcl2 protein levels
In order to evaluate the subcellular localization of Bax, we used confocal imaging of cells doubled-labeled with Mitotracker Red and a Bax antibody using methods similar to those described for localization of cytochrome c. Bcl2 protein levels in the mitochondrial fraction of cell homogenates were assessed by immunoblot analysis using methods identical to those described in our previous studies (Cheng et al., 2001).
RESULTS
Activation of mitochondrial ATP-sensitive potassium channels decreases focal ischemic brain injury
In light of data showing that activation of Mito-KATP channels can protect the heart against ischemic injury (O'Rourke, 2000), we examined the effect of the Mito-KATP opener diazoxide on focal ischemic brain injury in adult male C57BL/6 mice. Previous studies have shown that diazoxide is 1,000 times more potent in opening Mito-KATP (half maximal opening, 0.5 μmol/L) than plasma membrane potassium channels (half maximal opening, 1 mmol/L) (Szewczyk and Marban, 1999; Grover and Garlid, 2000). Diazoxide was administered intravenously in a bolus dose of 5 mg/kg 30 minutes before permanent occlusion of the left middle cerebral artery. The dose of diazoxide was chosen based on previous studies documenting the efficacy of this dose in activating Mito-KATP and reducing cell death in a model of myocardial infarction (Nakai and Ichihara, 1994). Control mice received an equivalent volume of vehicle (0.2% dimethyl sulfoxide). Mice were killed 7 or 24 hours after MCAO, and infarct volume was quantified by analysis of brain sections stained with TTC. Infarct volume was dramatically decreased at both the 7- and 24-hour poststroke time points in mice treated with diazoxide compared with vehicle-treated controls (Figs. 1 and 2A). Measurement of CBF showed that diazoxide caused a small (20% to 40%) increase in cerebral blood flow during the 30-minute period before the onset of ischemia but, as expected, had no effect on the near-complete elimination of CBF caused by permanent MCAO (Fig. 3).
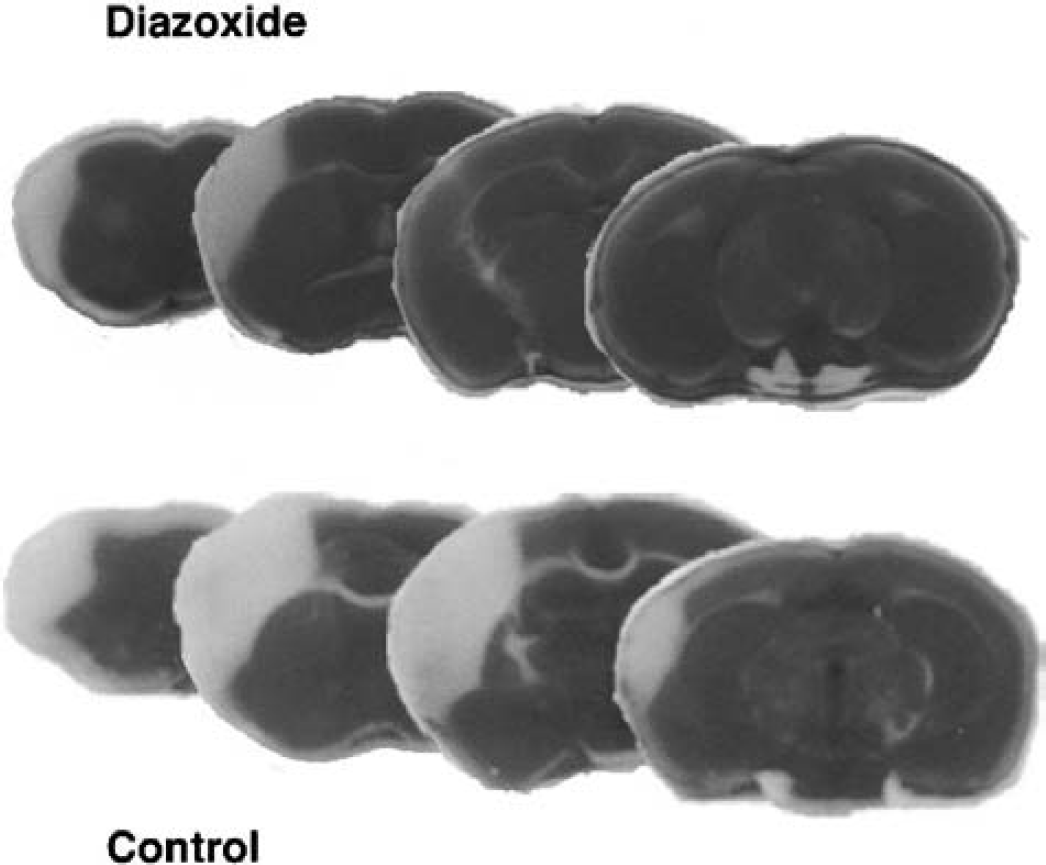
Diazoxide reduces infarct volume in mice subjected to permanent focal cerebral ischemia. Mice were administered 0.2% dimethyl sulfoxide in saline (vehicle control) or diazoxide (5 mg/kg) intravenously 30 minutes before permanent occlusion of the middle cerebral artery. Mice were killed 24 hours later and brains were removed, sectioned, and stained with TTC. Representative brain sections from a control mouse and a diazoxide-treated mouse are shown.
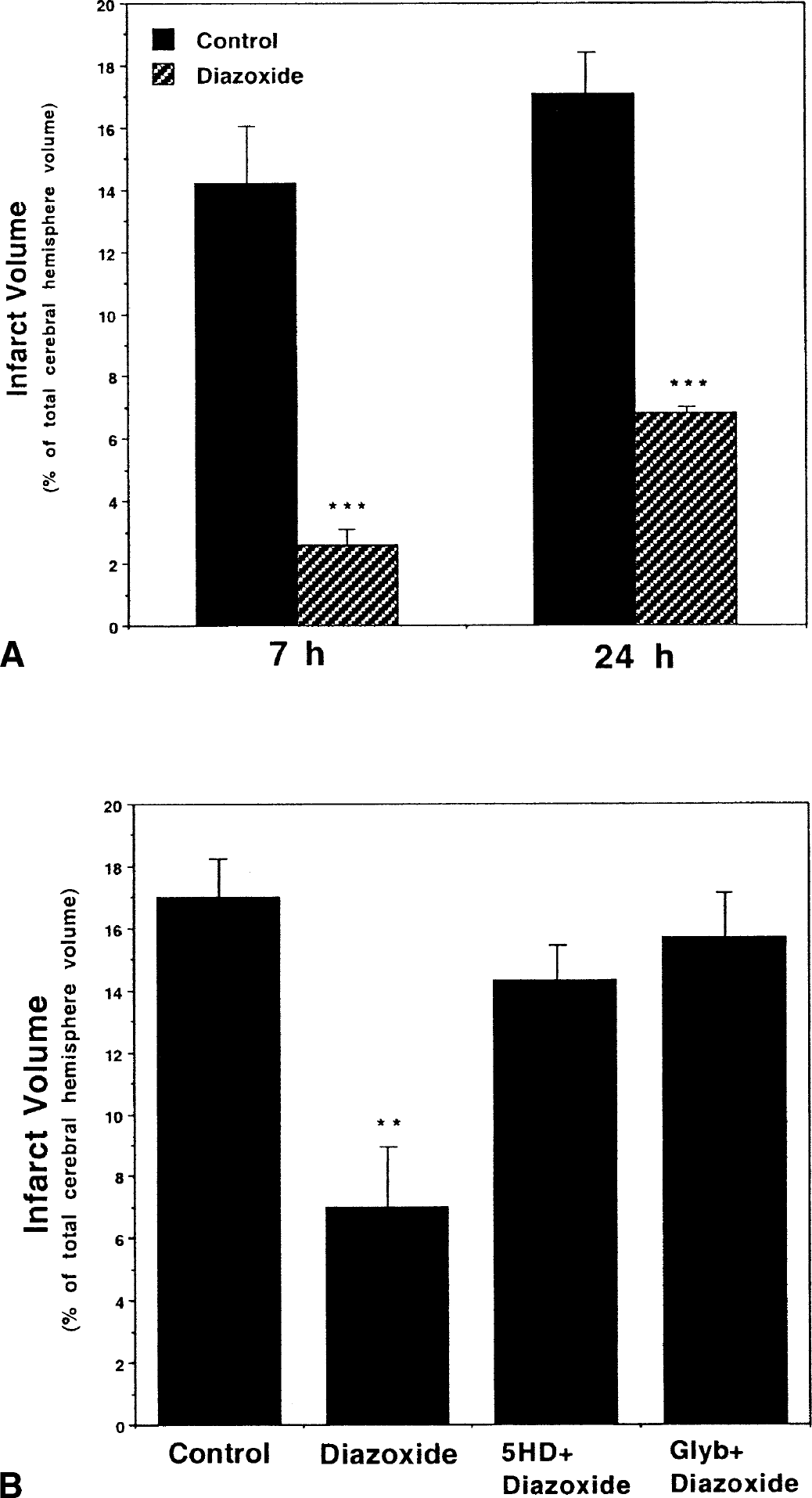
Activation of Mito-KATP channels protects against focal ischemic brain injury.
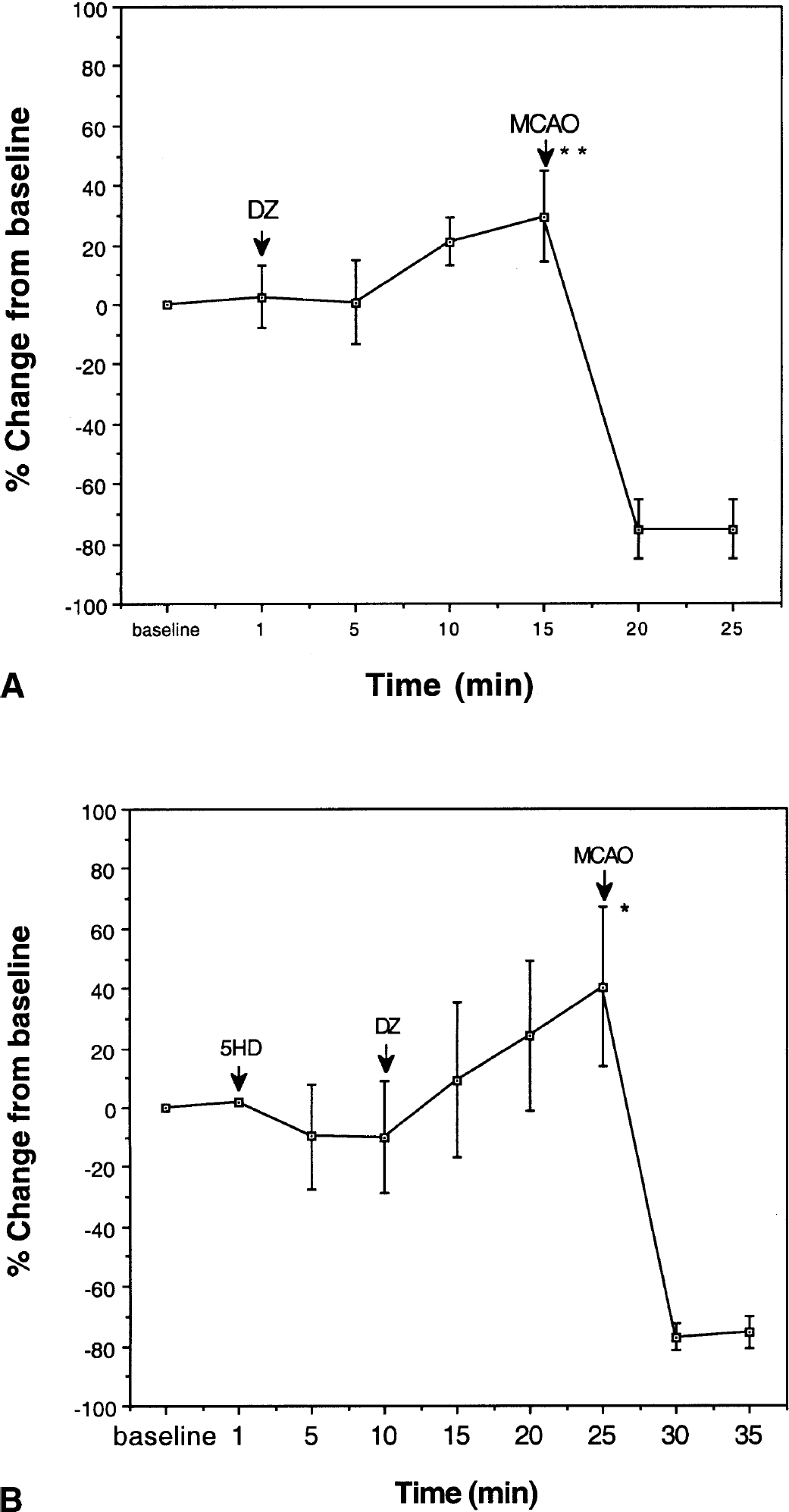
Diazoxide (DZ) increases cerebral blood flow under nonischemic conditions by a mechanism independent of Mito-KATP. Regional cerebral blood flow (rCBF) was determined in the territory of the middle cerebral artery (see Materials and Methods). The rCBF was measured in the preischemic period before and after intravenous administration of DZ and 5HD at the indicated time points and after middle cerebral artery occlusion (MCAO) (A and B). The rCBF was increased by DZ and unaffected by 5HD. Note that, regardless of drug treatment, rCBF decreased to negligible levels after MCAO.
Diazoxide can activate potassium channels in both mitochondrial and plasma membranes. To determine whether the mechanism whereby diazoxide reduces is chemic brain injury involves mitochondrial potassium channels, we used 5HD, an agent that blocks Mito-KATP channels but not plasma-membrane KATP channels (Sato et al., 2000). Mice were administered 5HD by intravenous infusion (20 mg/kg) 15 minutes before the intravenous administration of diazoxide (5 mg/kg). The left middle cerebral artery was occluded 30 minutes after diazoxide administration. Mice were killed 24 hours later and cerebral infarct volume was quantified. 5HD almost completely abolished the protective effect of diazoxide in this stroke model (Fig. 2B), had no effect on cerebral blood flow, and did not modify the increase in blood flow induced by diazoxide (Fig. 3). In another experiment, glibenclamide was infused into the left lateral ventricle of mice 20 minutes before the intravenous administration of diazoxide. The left middle cerebral artery was occluded 30 minutes after diazoxide administration, mice were killed 24 hours later, and cerebral infarct volume was quantified. Glybenclamide blocked the ability of diazoxide to decrease infarct volume (Fig. 2B), further supporting a role for potassium channels in the neuroprotective effect of diazoxide.
Increasing evidence suggests that neurons undergo apoptosis after cerebral ischemia, and that such neuronal deaths may be triggered by DNA damage (States et al., 1996; Jin et al., 1999; Kim et al., 2001). We therefore stained brain sections from control and diazoxide-treated ischemic mice using the TUNEL method to detect cells with damaged nuclear DNA. The number of TUNEL-positive cells was markedly reduced in the ischemic hemisphere in mice treated with diazoxide compared with vehicle-treated control mice (Fig. 4A). Based on findings of previous studies (Chen et al., 1997), it is likely that the majority of TUNEL-positive cells are neurons and that the decreased numbers of TUNEL-positive cells in diazoxide-treated mice reflect neuroprotection. Brain sections from control and diazoxide-treated ischemic mice were immunostained with an antibody against GFAP to label astrocytes. In vehicle-treated mice subjected to ischemia, there was an increase in GFAP immunoreactivity in the periinfarct area, but a marked depletion of GFAP-positive cells within the infarct area (Fig. 4B). The periinfarct area in the cerebral cortex of diazoxide-treated mice exhibited numerous GFAP-positive cells, suggesting that activation of Mito-KATP protects astrocytes against ischemic injury. However, it is also possible that diazoxide induced astrocytosis or increased GFAP expression.
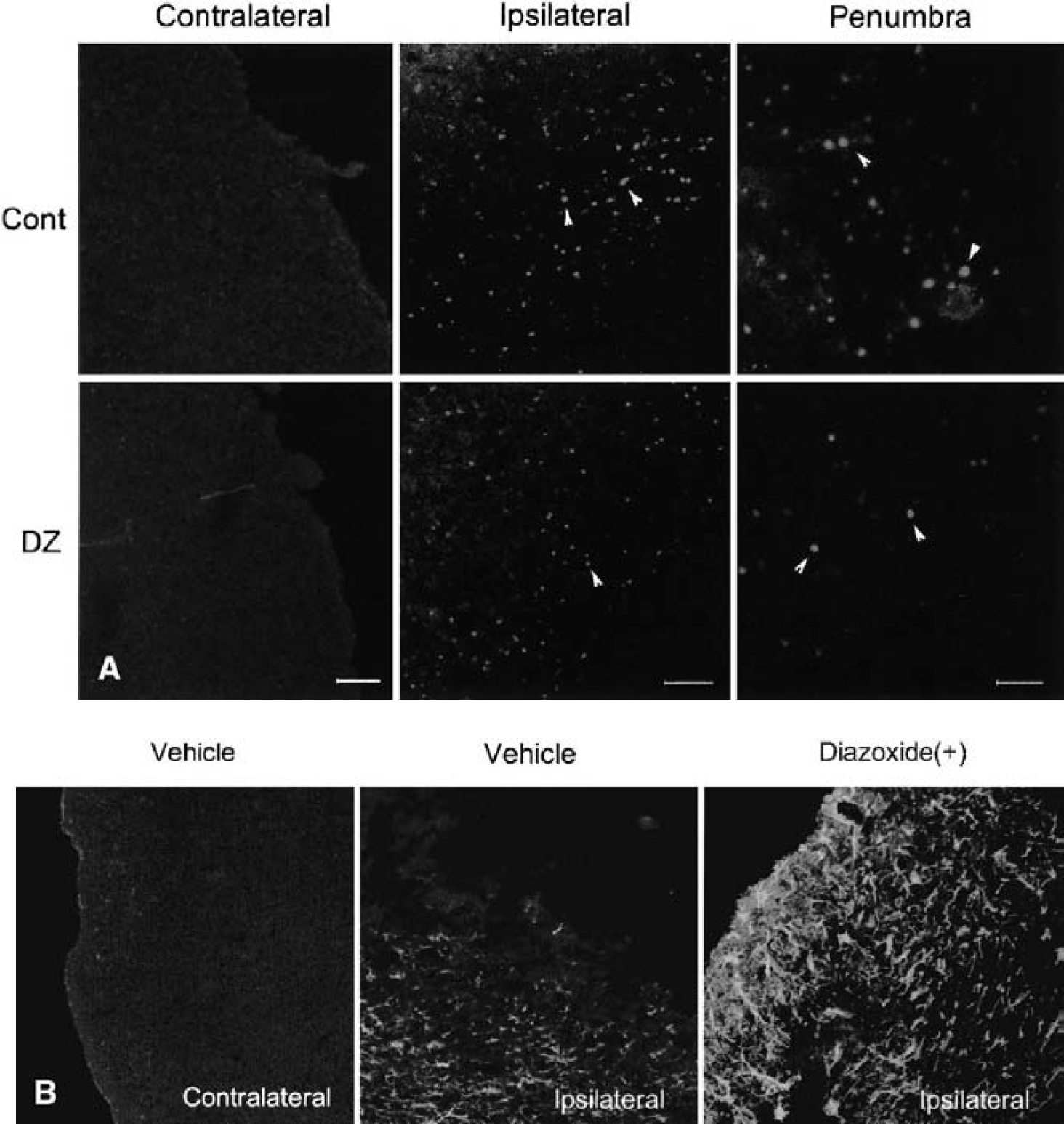
Confocal laser micrographs showing TUNEL staining
Activation of mitochondrial ATP-sensitive potassium channels protects cultured neurons against hypoxic and apoptotic cell death
These results suggest that activation of Mito-KATP can protect neurons in the brain against ischemic injury. However, because of the cellular complexities inherent in vivo, indirect effects of diazoxide on glial cells or the vasculature cannot be ruled out. We therefore determined whether diazoxide can exert a direct cytoprotective effect on neurons. Cultured hippocampal neurons were pretreated for 30 minutes with diazoxide or vehicle (0.2% dimethyl sulfoxide), and were then subjected to chemical hypoxia by exposure to potassium cyanide. Whereas more than 90% of the neurons were killed by cyanide (30 μmol/L) in vehicle-treated cultures, less than 60% of the neurons were killed in cultures pretreated with diazoxide (Figs. 5A and 5B). The neuroprotective effect of diazoxide against chemical hypoxia was significantly attenuated when cultures were exposed to 5HD before exposure to diazoxide (Fig. 5B). To determine if activation of Mito-KATP can protect neurons against apoptosis, we examined the effects of diazoxide on cell death induced by the bacterial alkaloid staurosporine, an agent known to induce apoptosis of neurons (Kruman et al., 1998; Krohn et al., 1999; Poppe et al., 2001). Diazoxide treatment significantly protected neurons against staurosporine-induced death, and 5HD blocked this neuroprotective effect of diazoxide (Fig. 5C).
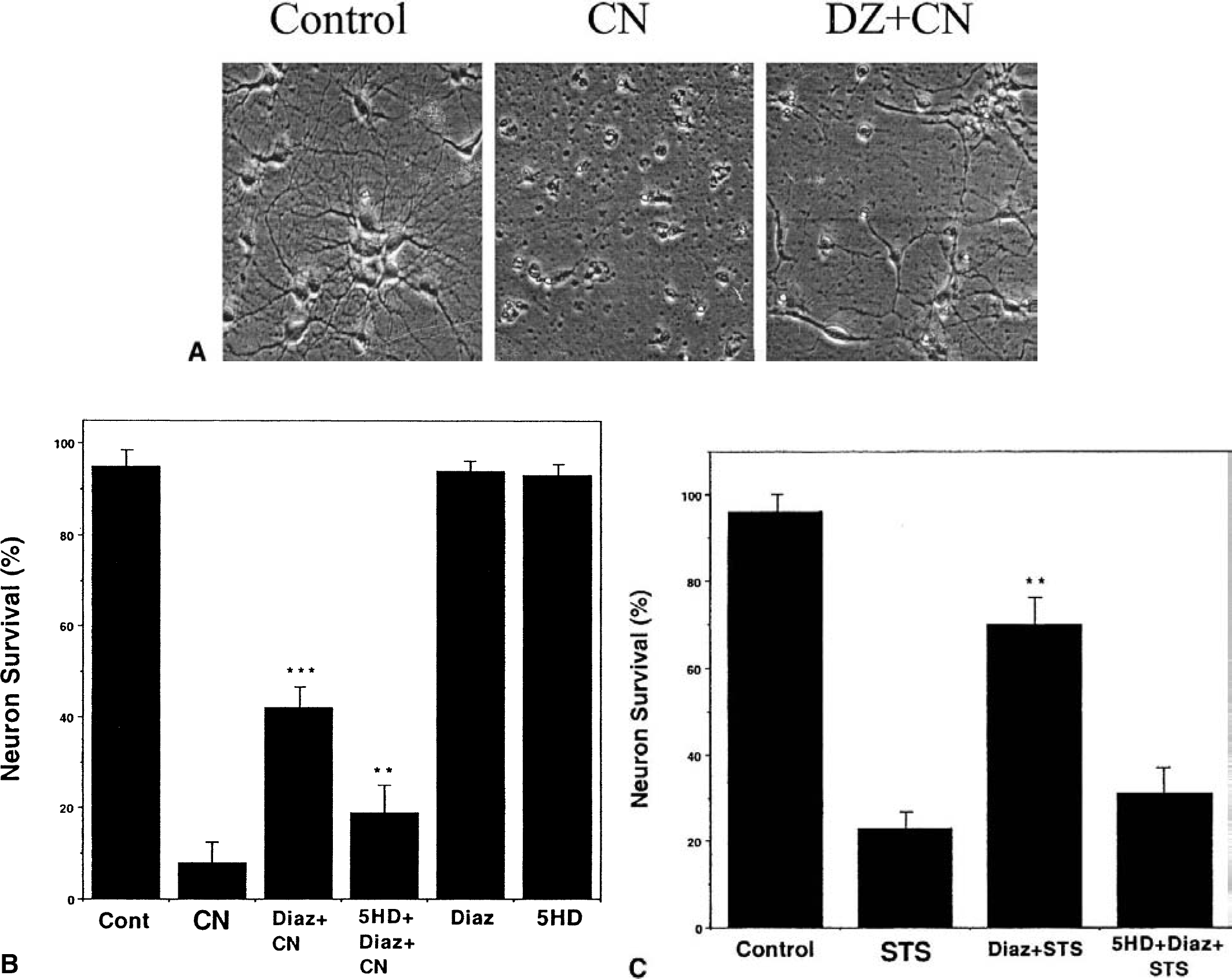
Activation of Mito-KATP protects cultured neurons against hypoxia-induced cell death and apoptosis.
Activation of mitochondrial ATP-sensitive potassium channels depolarizes mitochondria and prevents cytochrome c release in neurons exposed to hypoxic and apoptotic insults
Changes in mitochondrial membrane permeability and release of cytochrome c are thought to be pivotal events in neuronal apoptosis (Budd et al., 2000), and recent findings suggest that these events occur in many neurons that die after a stroke (Mattson et al., 2000). To determine whether Mito-KATP modulate apoptosis-related events in mitochondria, we first determined the effects of diazoxide and 5HD on mitochondrial membrane potential in cultured hippocampal neurons using the fluorescent probe TMRE. Diazoxide caused a decrease in mitochondrial transmembrane potential and 5HD blocked this effect of diazoxide, indicating that activation of Mito-KATP depolarizes mitochondria in cultured hippocampal neurons (Fig. 6).
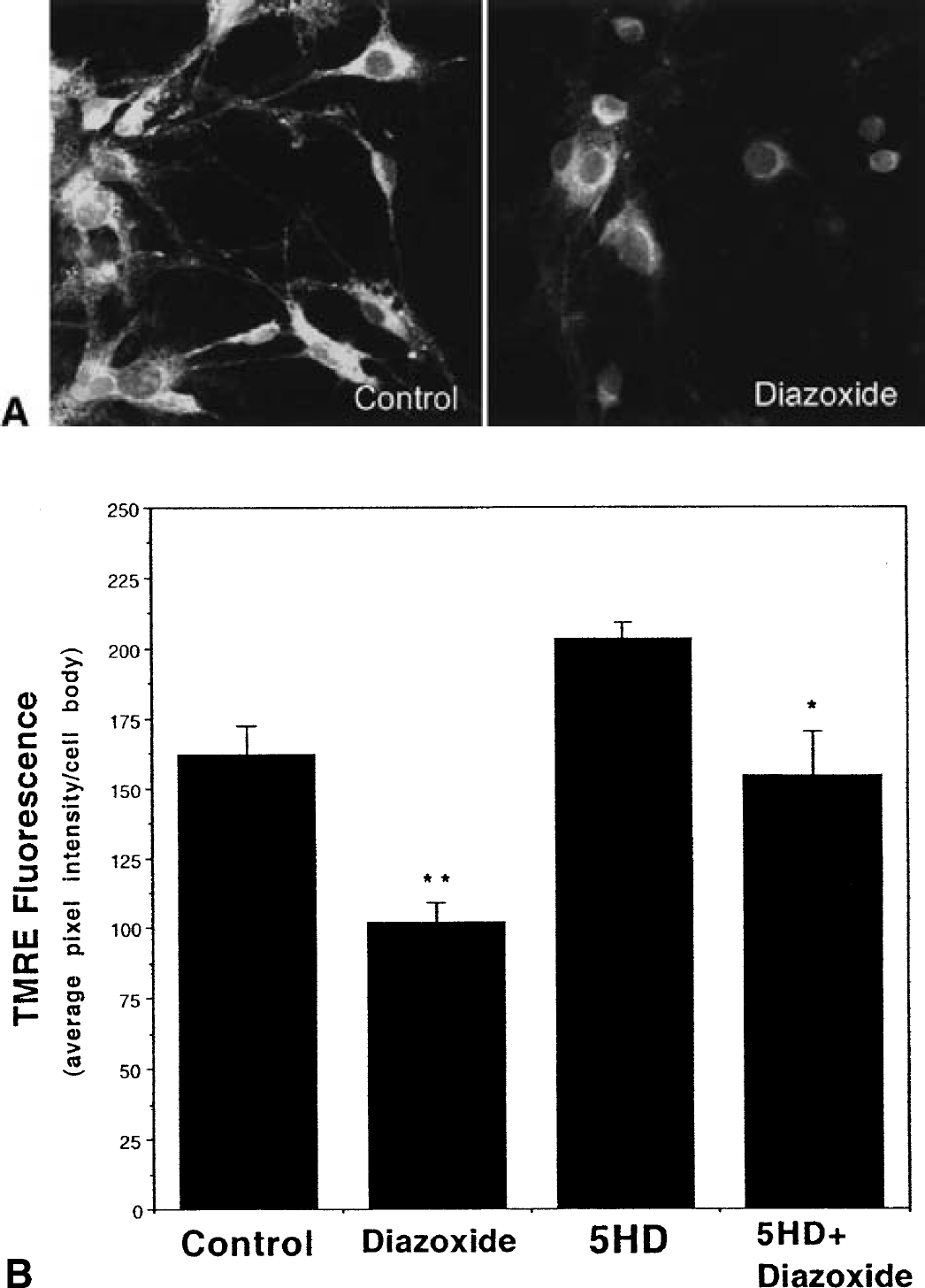
Activation of Mito-KATP depolarizes mitochondria in cultured hippocampal neurons.
We next determined the effects of activation of Mito-KATP on release of cytochrome c from mitochondria of neurons exposed to hypoxic and apoptotic insults. Cultures were pretreated with diazoxide alone or in combination with 5HD and exposed to cyanide or staurosporine. Exposure of neurons to cyanide resulted in the release of cytochrome c from mitochondria in nearly all neurons during a 6-hour exposure period (Fig. 7). Diazoxide treatment greatly decreased the number of neurons that had released cytochrome c from their mitochondria during the 6-hour exposure period, and 5HD blocked this effect of diazoxide (Fig. 7). Staurosporine induced the release of cytochrome c from the mitochondria of essentially all neurons during a 3-hour exposure period (Fig. 8). Diazoxide decreased the number of neurons in which cytochrome c was released from mitochondria, and 5HD significantly attenuated the effect of diazoxide (Fig. 8). These results suggest that activation of Mito-KATP protects neurons against apoptosis by preventing cytochrome c release from the mitochondria.
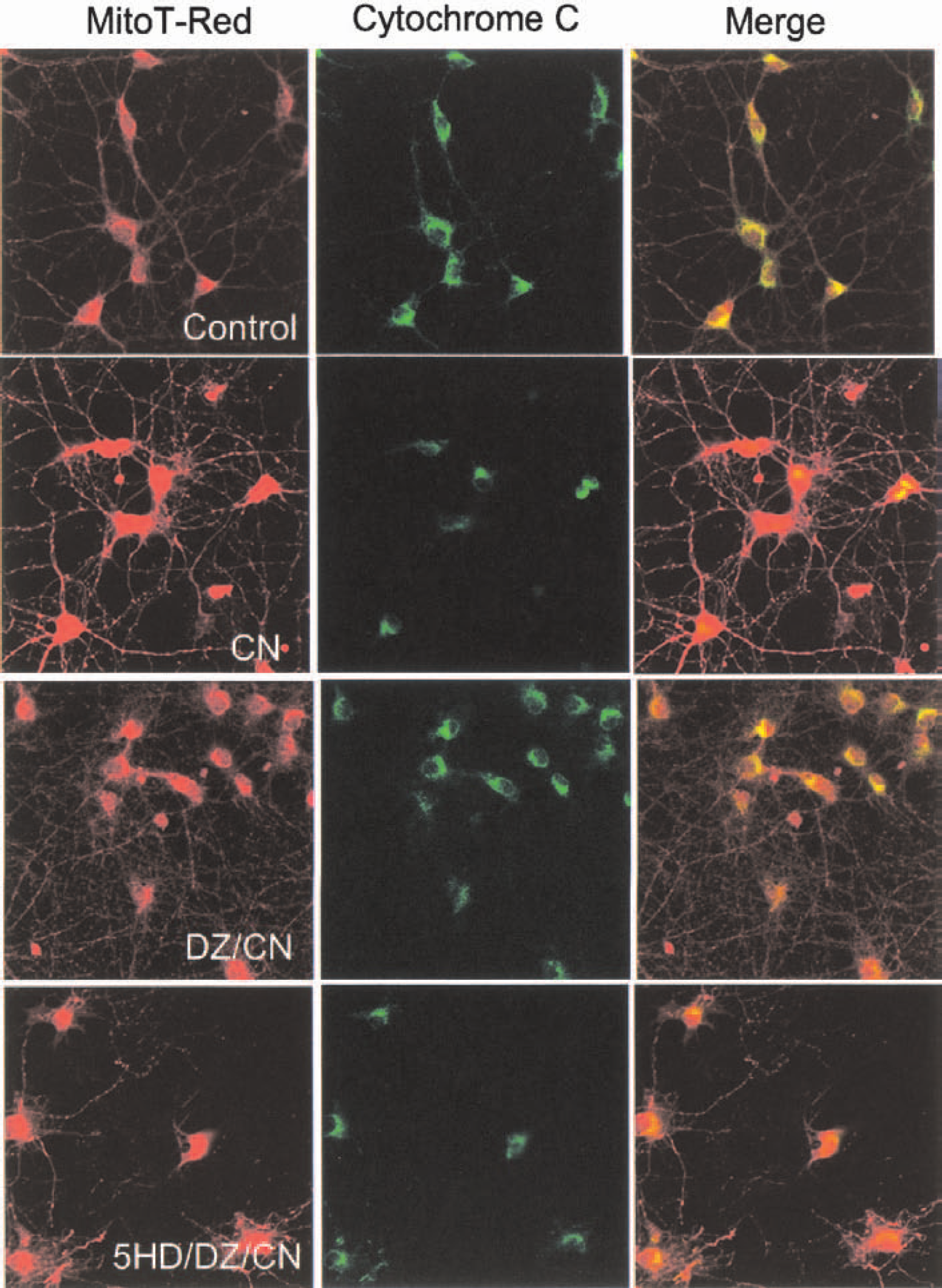
Activation of Mito-KATP prevents cytochrome c release from mitochondria in hippocampal neurons subjected to hypoxia. Cultures were pretreated for 30 minutes with vehicle (Control), 30-μmol/L diazoxide (DZ), or 100-μmol/L 5HD plus 30-μmol/L DZ. Cultures were then exposed to 30-μmol/L cyanide (CN) for 6 hours and were then double-labeled with Mitotracker Red and a cytochrome c antibody. Confocal images of Mitotracker Red fluorescence (red), cytochrome c immunoreactivity (green), and the merged image (yellow indicates sites of co-localization) are shown. Note that CN induced release of cytochrome c from mitochondria, DZ prevented cytochrome c release from many neurons, and 5HD blocked the effect of DZ.
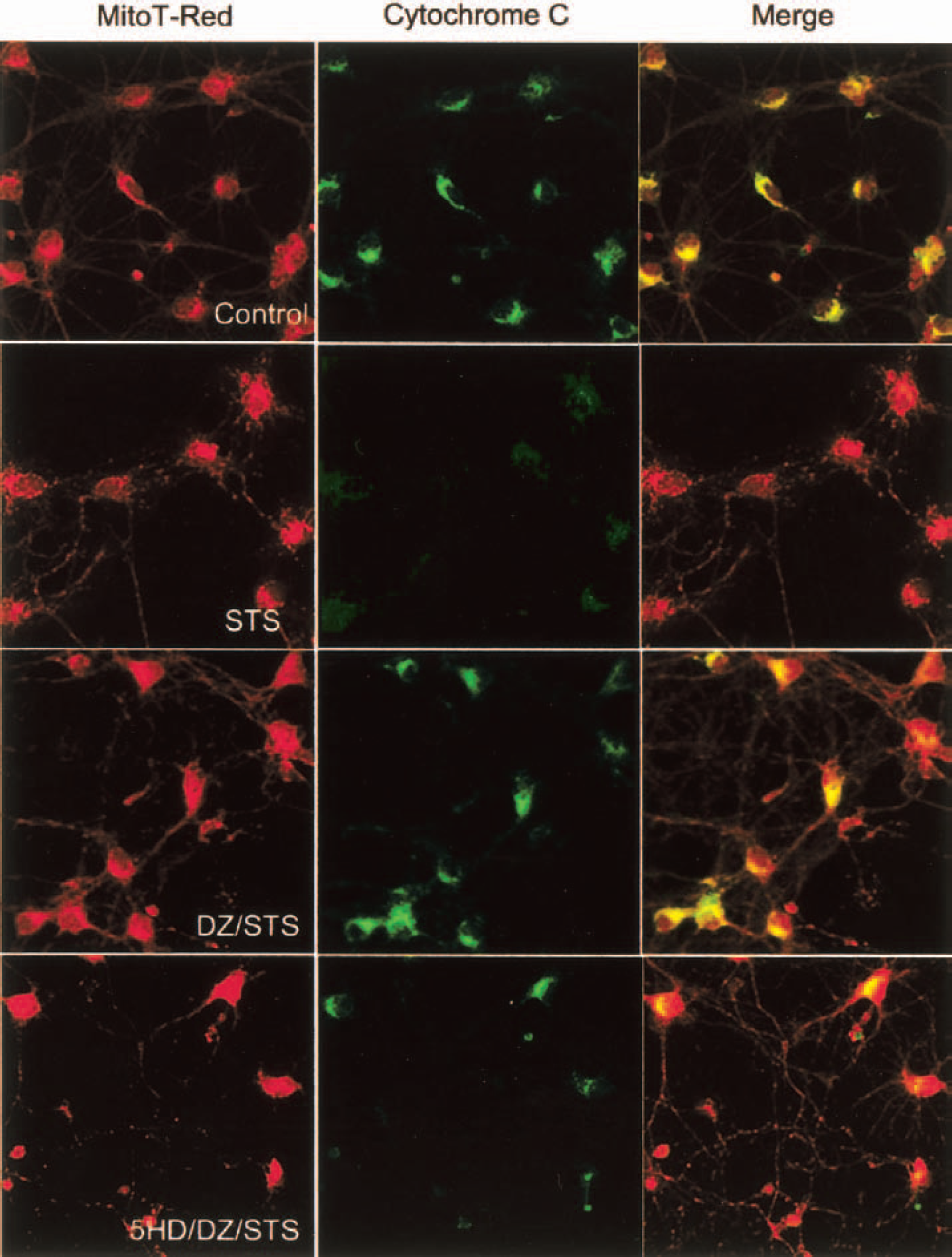
Activation of Mito-KATP prevents cytochrome c release from mitochondria in hippocampal neurons subjected to an apoptotic signal. Cultures were pretreated for 30 minutes with vehicle (Control), 30-μmol/L diazoxide (DZ), or 100-μmol/L 5HD plus 30-μmol/L DZ. Cultures were then exposed to 200-nmol/L staurosporine (STS) for 6 hours and double-labeled with Mitotracker Red and a cytochrome c antibody. Confocal images of Mitotracker Red fluorescence (red), cytochrome c immunoreactivity (green), and the merged image (yellow indicates sites of colocalization) are shown. Note that STS induced release of cytochrome c from mitochondria, DZ prevented cytochrome c release from many neurons, and 5HD blocked the effect of DZ.
Evidence that activation of mitochondrial ATP-sensitive potassium channels inhibits Bax translocation to mitochondria and association of Bcl2 with mitochondria
Previous studies of cardiac cells have provided evidence that preconditioning ischemia upregulates the expression of the antiapoptotic protein Bcl2 (Maulik et al., 1999) and may also decrease levels or change the subcellular localization of the proapoptotic protein Bax (Nakamura et al., 2000). Because it is believed that the actions of Bcl2 family members at mitochondrial membranes may determine whether a neuron lives or dies (Martin, 2001), we performed experiments aimed at establishing the effects of diazoxide on Bax and Bcl2. Confocal image analysis of cultured hippocampal neurons double-labeled with Mitotracker Red and an antibody against Bax indicated that Bax was diffusely distributed in the cytoplasm in untreated neurons and in neurons exposed to diazoxide (Fig. 9). Exposure of neurons to staurosporine resulted in colocalization of Bax and Mitotracker Red, indicating translocation of Bax to the mitochondria; this translocation of Bax was abolished by diazoxide treatment (Fig. 9). Levels of Bcl2 protein in the mitochondrial fraction were increased in hippocampal cells exposed to diazoxide (Fig. 9). These findings suggest that activation of Mito-KATP results in suppression of Bax translocation to mitochondria while increasing the amount of Bcl2 associated with mitochondria.
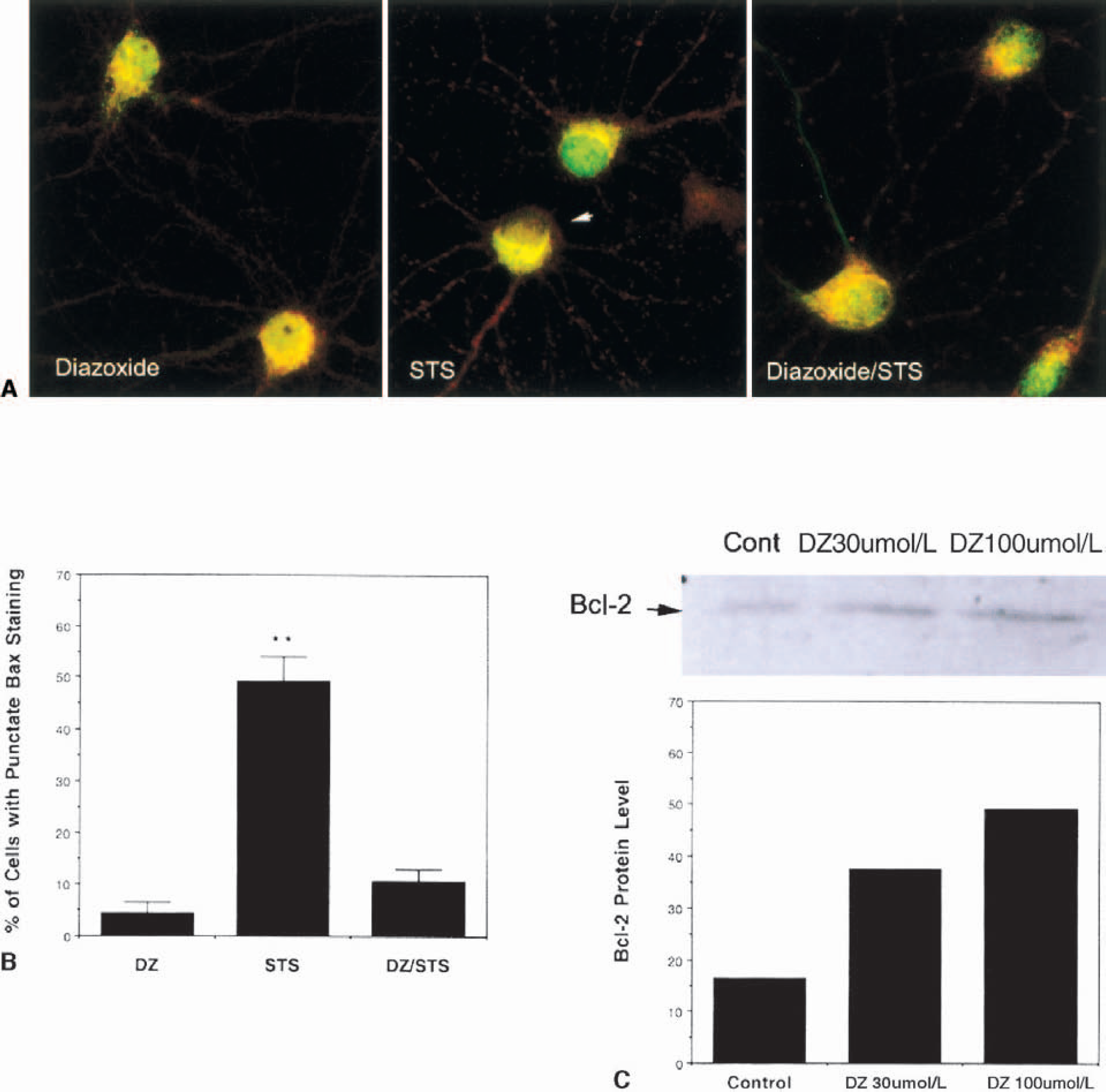
Evidence that activation of Mito-KATP inhibits Bax translocation to mitochondria and increases Bcl2 levels. (
DISCUSSION
The pharmacologic and electrophysiologic characteristics of Mito-KATP have been established in previous studies of cardiac cells (Grover and Garlid, 2000). Consistent with the presence of similar Mito-KATP in neurons, we found that diazoxide at concentrations below those that activate plasma-membrane potassium channels depolarizes mitochondrial membranes in neurons, and that 5HD blocks this action of diazoxide. Although the genes that encode Mito-KATP channel proteins have not been cloned, the partial purification and reconstitution of brain Mito-KATP was recently reported; the reconstituted channels were activated by diazoxide and inhibited by 5HD (Bajgar et al., 2001). Interestingly, the latter study provided evidence that the levels of Mito-KATP proteins in neurons are six to seven times greater than in cardiac cells, suggesting a prominent role for these channels in neuronal physiology. We observed that diazoxide is very effective in reducing ischemic infarct size in mice subjected to permanent occlusion of the middle cerebral artery. The neuroprotective actions of diazoxide in vivo and in cell culture were largely abolished by 5HD, strongly suggesting that the effects of diazoxide were mediated by activation of Mito-KATP. Several observations suggest that the protective effects of diazoxide in the present study were not due to a vascular action of this drug. First, previous studies have shown that effects of diazoxide on blood pressure and flow are due to activation of plasma-membrane potassium channels (Richer et al., 1990). Second, we found that although diazoxide increased CBF, this vascular effect was not blocked by 5HD; thus, preischemic blood flow changes were dissociated from infarct size. Third, we used a permanent focal ischemia model in which blood flow was essentially eliminated in all mice regardless of drug treatment. Finally, diazoxide directly protected cultured neurons against chemical hypoxia in a 5HD-inhibitable manner. It was recently reported that administration of diazoxide to newborn piglets enhances functional recovery after transient global cerebral ischemia (Domoki et al., 1999). Thus, activation of Mito-KATP may protect neurons against different kinds of ischemic insults.
Events that occur in mitochondria are increasingly recognized as determinants of whether a neuron lives or dies when challenged with adverse conditions. Previous studies of experimental cell culture and animal models relevant to stroke have documented several changes in mitochondria that occur in response to ischemia. These changes include an increase in oxyradical production (Keller et al., 1998; Murakami et al., 1998), membrane depolarization or hyperpolarization (Budd et al., 2000), and release of cytochrome c (Ouyang et al., 1999; Sugawara et al., 1999; Noshita et al., 2001). Although some studies have suggested an association between mitochondrial membrane depolarization and release of cytochrome c leading to apoptosis (Kruman and Mattson, 1999; Budd et al., 2000), more recent studies have clearly established a dissociation between mitochondrial membrane depolarization and cell death (Krohn et al., 1999). In some cell death paradigms relevant to stroke, hyperpolarization rather than depolarization of the mitochondrial membrane may induce cytochrome c release (Poppe et al., 2001). Our data showing that diazoxide depolarizes mitochondrial membranes but inhibits cytochrome c release and cell death in neurons exposed to apoptotic insults suggest that mitochondrial depolarization can be associated with neuroprotection.
Although the physiologic signals that activate Mito-KATP and the mechanisms whereby activation of Mito-KATP protects neurons against ischemic injury are unknown, data from studies of myocardial ischemia suggest possibilities. Preconditioning ischemia in the heart may activate Mito-KATP by inducing the production of reactive oxygen species, resulting in the activation of protein kinase C, which phosphorylates proteins that promote Mito-KATP opening (Sato et al., 1998; Wang et al., 2001a). It has also been suggested that protein kinase C mediates activation of Mito-KATP by diazoxide (Wang et al., 2001b). Activation of Mito-KATP, in turn, may induce a stress response involving protein kinase C and mitogen-activated protein kinases (Baines et al., 1999; Takashi et al., 1999). A similar preconditioning mechanism may be operative in neurons because previous studies have shown that activation of protein kinase C and mitogen-activated protein kinases can protect neurons against conditions relevant to ischemic brain injury, including oxidative stress, excitotoxins, and hypoxia (Maiese et al., 1996; Han and Holtzman, 2000; Skaper et al., 2001).
We have shown that activation of Mito-KATP by diazoxide increases the association of Bcl2 with mitochondria while preventing Bax translocation to mitochondria. Previous studies have provided convincing evidence that Bcl2 and Bax exert their respective antiapoptotic and proapoptotic actions by actions at the level of mitochondrial membranes (Martin, 2001). The mechanisms involved in mitochondrial localization and membrane interactions of Bcl2 family members is poorly understood, and it is therefore unclear how activation of Mito-KATP might affect these processes. In addition to direct effects of Mito-KATP activation on mitochondria, their activation may also increase resistance of neurons to ischemic injury indirectly by activating Akt kinase or NF-κB, both of which have been linked to increased resistance of neurons to apoptosis and excitotoxicity (Gary and Mattson, 2001; Mattson and Camandola, 2001; Yano et al., 2001). The latter signaling pathways (Akt and NF-KB) have been shown to induce the expression of antiapoptotic proteins, including Bcl2 and antioxidant enzymes (Mattson et al., 1997; Camandola and Mattson, 2000; Gary and Mattson, 2001; Kang et al., 2001). Studies of heart cells suggest that preconditioning may involve a similar antiapoptotic pattern of gene expression (Shimizu et al., 2001). The available data are therefore consistent with a model in which the activation of Mito-KATP and resultant mitochondrial membrane depolarization induce a controlled stress response in neurons that results in upregulation of genes that encode neuroprotective proteins.
Our findings suggest that diazoxide and related agents that activate Mito-KATP are candidates for use in clinical trials in human stroke patients. Diazoxide has been used in the clinical setting for more than 30 years, primarily for the treatment of patients with acute and severe hypertension (Breslin, 1969), and is still used to manage hypertension during pregnancy (Lowe and Rubin, 1992). The direct neuroprotective action of diazoxide, in combination with its ability to enhance CBF at doses that cause only a modest reduction in peripheral blood pressure (Fig. 1) (Dieguez et al., 1980), suggest that this drug may be particularly effective in preventing neuronal damage and restoring blood flow after a stroke. However, for diazoxide to be beneficial in human stroke patients, it must be effective when given within a defined postischemic period, which remains to be established.