
Abstract
Exposure of brain microvascular endothelial cells (BMEC) to human immunodeficiency virus-1 (HIV-1) Tat protein can decrease expression and change distribution of tight junction proteins, including claudin-5. Owing to the importance of claudin-5 in maintaining the blood–brain barrier (BBB) integrity, the present study focused on the regulatory mechanisms of Tat-induced alterations of claudin-5 mRNA and protein levels. Real-time reverse-transcription-polymerase chain reaction revealed that claudin-5 mRNA was markedly diminished in BMEC exposed to Tat. However, U0126 (an inhibitor of mitogen-activated protein kinase kinase1/2, MEK1/2) protected against this effect. In addition, inhibition of the vascular endothelial growth factor receptor type 2 (VEGFR-2) by SU1498, phosphatidylinositol-3 kinase (PI-3 K) by LY294002, nuclear factor-κB (NF-κB) by peptide SN50, and intracellular calcium by BAPTA/AM partially prevented Tat-mediated alterations in claudin-5 protein levels and immunoreactivity patterns. In contrast, inhibition of protein kinase C did not affect claudin-5 expression in Tat-treated cells. The present findings indicate that activation of VEGFR-2 and multiple redox-regulated signal transduction pathways are involved in Tat-induced alterations of claudin-5 expression. Because claudins constitute the major backbone of tight junctions, the present data are relevant to the disturbances of the BBB in the course of HIV-1 infection.
Introduction
The cerebral microvascular endothelium constitutes the principal component of the blood–brain barrier (BBB). The morphologic basis of the BBB integrity is the formation of tight junctions, which are composed of specific junctional proteins, including claudin-5 (Huber et al, 2001; Pardridge, 2002). Considerable experimental evidence indicates BBB damage in human immunodeficiency virus-1 (HIV-1) infection (McArthur et al, 1992; Petito and Cash, 1992; Giovannoni et al, 1998). Such alterations were shown in pathologic studies (Rhodes and Ward, 1991; Krebs et al, 2000), CSF studies (Singer et al, 1994), and by dynamic magnetic resonance imaging (Avison et al, 2002). Disruption of the BBB appears to be critical for HIV-1 trafficking into the brain (Wu et al, 2000) and the development of HIV-1-associated central nervous system (CNS) complications. It has been shown that disruptions of the BBB are more frequent in AIDS patients with dementia as compared with nondemented AIDS patients or seronegative controls (Power et al, 1993). In addition, HIV-1 encephalitis was associated with the disruption of endothelial cell junctions and mononuclear cell infiltration (Dallasta et al, 1999; Boven et al, 2000).
Central nervous system manifestations of HIV-1 infection frequently do not correlate with the viral load. Therefore, it has been hypothesized that specific soluble factors released by HIV-infected cells can contribute to the pathogenesis of HIV-1 infection. One of such potential mediators is Tat protein, which has a crucial role in HIV-1 gene expression and replication (Nath and Geiger, 1998; Ansari et al, 1999). Tat can be released actively from HIV-1-infected cells (Chang et al, 1997), easily cross cell membranes (Vives et al, 2003), and interact with a variety of cell surface receptors, including vascular endothelial growth factor receptor type 2 (VEGFR-2). Elevated levels of Tat protein and mRNA were shown in brains of AIDS patients (Hudson et al, 2000; Valle et al, 2000).
In addition to its neurotoxic influence, Tat can evoke profound vascular effects (Acheampong et al, 2002; Kim et al, 2003; Khan et al, 2003). For example, Tat induced oxidative stress and disturbances in redox balance in brain microvascular endothelial cell (BMEC) and the brain tissue were associated with activation of redox-responsive transcription factors and inflammatory genes (Pu et al, 2003; Toborek et al, 2003; Lee et al, 2004). Furthermore, our studies indicate that alterations of expression and distribution of selected tight junction proteins, including claudin-5, may be one of the major cytotoxic effects of Tat (András et al, 2003). Such effects may affect the BBB permeability and can be critical for HIV-1 entry into the brain. Claudin-5 plays an important role in maintaining the integrity of brain endothelial cells because of its involvement in both paracellular sealing and membrane domain differentiation (Furuse et al, 1998; Morita et al, 1999; Tsukita and Furuse, 2000). Indeed, the size-selective loosening of tight junctions was observed in claudin-5 knockout mice (Nitta et al, 2003). It also has been proposed that the ratio of claudin-5 to other claudins can regulate the tightness of the BBB (Liebner et al, 2000a, b). Thus, because of the importance of claudin-5 in the regulation of the BBB integrity, the present study focused on the regulatory mechanisms of Tat-induced alterations of claudin-5 expression.
Materials and methods
Primary Cultures of Brain Microvascular Endothelial Cells
Brain microvascular endothelial cells were isolated from the forebrains of 2-week-old Wistar rats and cultured as previously described (Deli et al, 1997; András et al, 2003). Brain microvascular endothelial cells were cultured in Dulbecco's modified Eagle's medium with 20% plasma-derived bovine serum (PDBS, Animal Technologies Inc., Tyler, TX, USA), 40 μg/mL endothelial cell growth supplement (BD Biosciences, Bedford, MA, USA), 100 μg/mL heparin, 2 mmol/L glutamine, 5 μg/mL insulin, 5 μg/mL human transferrin, 5 ng/mL Na-selenite (Insulin-Transferrin-Sodium Selenite Media Supplement, Sigma, St Louis, MO, USA), and 25 μg/mL gentamicin. Cultures were incubated at 37°C in a humidified atmosphere of 5% CO2. Brain microvascular endothelial cells can dedifferentiate very rapidly and lose their specific characteristics as early as after the first passage. Therefore, only primary cultures were used in the present study.
Tat Treatment and Inhibitors of Specific Signaling Pathways
Human immunodeficiency virus-1 Tat protein is encoded by a gene consisting of two exons. The first exon encodes the neurotoxic and membrane interactive domains. Therefore, the present study was performed on recombinant Tat protein derived from the first exon, which encodes for the first 72 amino acids (Tat1–72). Tat was synthesized and purified as previously described by Ma and Nath (1997). The functional properties of Tat were confirmed by its ability to activate the β-galactosidase gene in an HIV long terminal repeat-β-galactosidase plasmid that had been transfected into HeLa cells (Ma and Nath, 1997).
To exclude the possibility that Tat-induced effects were secondary to endotoxin contaminants of the Tat preparation, Tat solutions were immunoabsorbed with Tat anti***sera conjugated to protein-A beads (Magnuson et al, 1995), and the supernatant was tested for proinflammatory activity.
Because Tat strongly binds to serum proteins, BMEC were treated with Tat in serum-free media. All experiments were performed on confluent endothelial cell cultures (normally 3 to 4 days after seeding). Similar experimental design was used in our previously published papers (András et al, 2003; Pu et al, 2003; Toborek et al, 2003; Lee et al, 2004). Tat concentrations used in the present study are consistent with the literature data (Bonavia et al, 2001; Prendergast et al, 2002; Speth et al, 2002). In addition, evidence indicates that pathologic concentrations of Tat in HIV-infected patients can reach the range of nanograms per milliliter of serum (Xiao et al, 2000).
To study specific mechanisms of Tat-induced alterations of claudin-5 expression, the following inhibitors of signal transduction pathways were used (Figure 1): SU1498 (1 μmol/L, an inhibitor of VEGFR-2), ZM336372 (0.5 μmol/L, an inhibitor of c-Raf), U0126 (10 or 20 μmol/L, an inhibitor of mitogen-activated protein kinase kinase1/2 (MEK1/2)), LY294002 (100 nmol/L, an inhibitor of phosphatidylinositol-3 kinase (PI-3 K)), SN50 (2 μmol/L, an inhibitor of nuclear factor-κB (NF-κB) translocation) or BAPTA/AM (5 μmol/L, an intracellular calcium chelator). In addition, we used 1-(5-isoquinolinesulfonyl)-2-methyl-piperazine dichloride (H-7; an inhibitor of protein kinase C (PKC) and cAMP-dependent protein kinase (PKA), 10 μmol/L), and calphostin-C (CPC; an inhibitor of PKC, 10 nmol/L). SU1498, ZM336372, U0126, LY294002, SN50, and BAPTA/AM were purchased from Calbiochem (San Diego, CA, USA); CPC and H-7 were from Sigma (St. Louis, MO, USA).
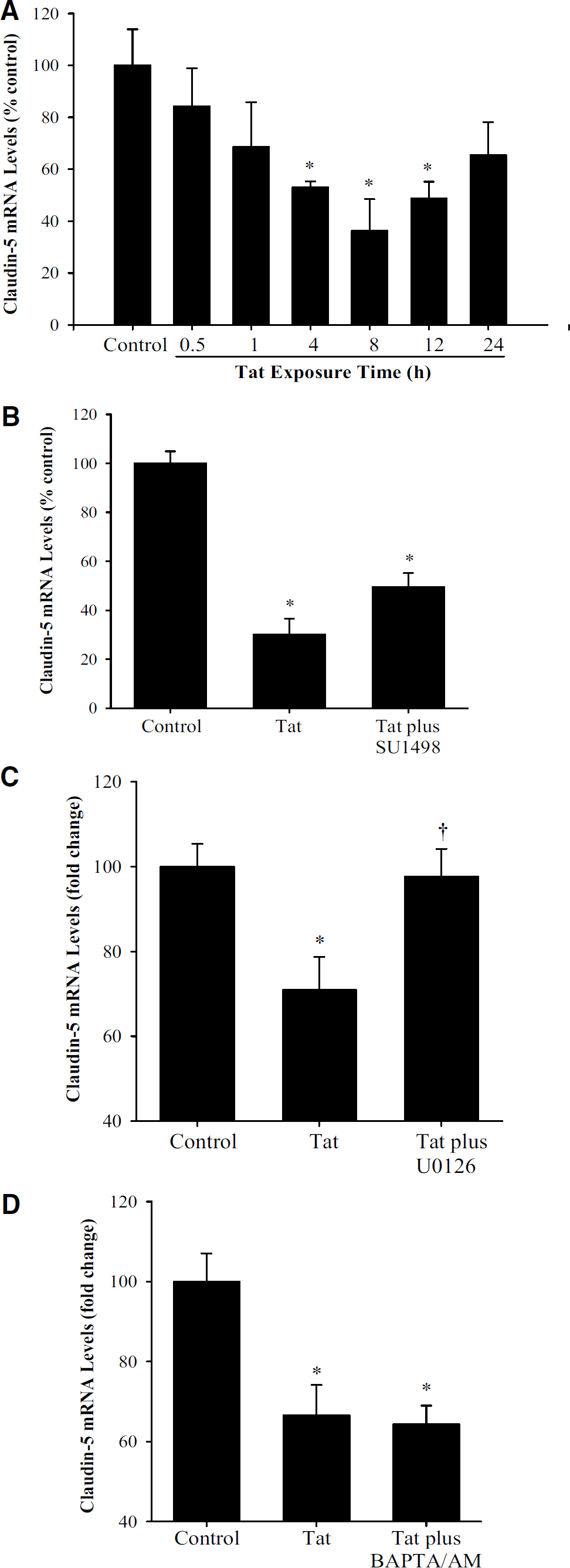
The involvement of specific signaling pathways in Tat-mediated alterations of claudin-5 mRNA levels in brain microvascular endothelial cells. (
Antibodies
Rabbit polyclonal anti-claudin-5 antibody was purchased from Zymed Laboratories (San Francisco, CA, USA) and mouse monoclonal anti-phosphotyrosine antibody was obtained from Upstate Group (Lake Placid, NY, USA). The following antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA): mouse monoclonal anti-pERK1/2 antibody, rabbit polyclonal anti-Akt antibody as well as HRP-conjugated anti-mouse and anti-rabbit secondary antibodies. Rabbit polyclonal anti-ERK1/2 and mouse monoclonal anti-phospho-Akt (Ser473) antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA). FITC and Texas Red-conjugated anti-mouse as well as anti-rabbit secondary antibodies were purchased from Santa Cruz Biotechnology and Chemicon International (Temecula, CA, USA).
Real-Time Reverse-Transcription-Polymerase Chain Reaction
Quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) was used to study the claudin-5 mRNA level changes. Specific rat claudin-5 primers were designed using Primer Express 2.0 and the following sequences were used: claudin-5 forward, 5'-CAC GGG AGG AGC GCT TTA T-3’ and claudin-5 reverse, 5'-GGC ACC GTT GGA TCA TAG AAC T-3'. In addition, the following primers were used to detect β-actin as a housekeeping gene: β-actin forward, 5'-AGG CCA ACC GTG AAA AGA TG-3’ and β-actin reverse, 5'-ACC AGA GGC ATA CAG GGA CAA-3'.
Total RNA was isolated using RNeasy kit (Qiagen, Valencia, CA, USA), treated by DNase I to remove possible genomic DNA contamination, and then reverse transcribed into cDNA using the Reverse Transcription System (Promega, Madison, WI, USA). Real-time PCR was conducted on the ABI Prism 7000 system (Applied Biosystems, Foster City, CA, USA) using 100 ng cDNA (2 μL of RT product), 12.5 μL SYBR Green PCR Master Mix (2 x), 300 nmol/L of forward and reverse primers in a total volume of 25 μL. The following thermocycling conditions were used: 95°C for 10 mins, followed by 95°C for 15 secs and 60°C for 60 secs for 40 cycles. The standard curve was generated by plotting the threshold cycle (Ct) versus the log concentration of the serial dilutions of one specific cDNA sample served as a calibrator. All samples were prepared in duplicate, their concentrations were calculated based on the standard curve and normalized according to β-actin mRNA level.
Immunoblotting
Homogenates of cultured BMEC were prepared in lysis buffer (20 mmol/L Tris-HCl, pH 7.4, 150 mmol/L NaCl, 0.5% Triton X-100, 0.1 mg/mL phenylmethylsulfonyl fluoride, 0.5% Nonidet 40, 1 mmol/L EDTA, 2.5 μg/mL leupetin, 1 μg/mL pepstatin A, 1 μg/mL aprotinin). After centrifugation at 15000 g for 15 mins, the supernatants were collected and protein concentrations were determined using the Bradford protein assay. Samples (20 μg protein/lane) were separated on SDS-polyacrylamide gel, blotted onto nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA, USA) and incubated with the respective antibodies. For visualization of detected proteins, immunoblots were analyzed using an ECL Western blot detection kit (Amersham Biosciences, Piscataway, NJ, USA).
Immunofluorescence Microscopy
Brain microvascular endothelial cells cultured on 35 mm dishes were fixed with ethanol for 30 mins at 4°C. After washing with phosphate-buffered saline (PBS) and blocking with 3% bovine serum albumin (BSA)-PBS for 30 mins, samples were incubated overnight at 4°C with primary antibody. The excess of primary antibody was removed, slides were washed with PBS, and incubated with Texas Red or FITC-conjugated secondary antibody for 2 h at room temperature. After washing with PBS, slides were mounted in aqueous mounting media and covered with coverslips. Specimens were evaluated under an epifluorescence Nicon Eclipse E600 microscope and the images were captured using a Spot CCD camera system.
Statistical Analysis
Routine statistical analysis of data was completed using SigmaStat 2.0 (SPSS Inc., Chicago, IL, USA). One-way ANOVA was used to compare responses among treatments. Treatment means were compared using Bonferroni's least significant difference procedure. P<0.05 was considered significant.
Results
We hypothesize that disturbances of cellular redox status may be the underlying mechanism of Tat-induced vascular toxicity. Therefore, to study the mechanisms of Tat-mediated alterations of claudin-5 expression, we concentrated on redox-responsive signal transduction pathways, such as the VEGFR-2/Ras/ERK1/2 pathway, the PI-3K/Akt/NF-κB pathway, and calcium-dependent signaling. The dose and time-dependent effects of Tat on claudin-5 expression were presented previously (András et al, 2003).
Tat Exposure Diminishes Claudin-5 mRNA Levels
To investigate whether Tat-induced alterations of claudin-5 expression is regulated at the transcriptional level, claudin-5 mRNA levels were determined using real-time RT-PCR in BMEC exposed to 100 nmol/L Tat for up to 24 h. As indicated in Figure 1A, a 4-h exposure to Tat resulted in a significant decrease in claudin-5 mRNA levels. These levels reached the minimal levels in cells treated with Tat for 8 h and returned to the normal values after a 24-h Tat exposure.
Tat-induced decrease in claudin-5 mRNA level was prevented by pretreatment with U0126, a specific inhibitor of MEK1/2 (Figure 1C). However, pretreatment with the VEGFR-2 inhibitor SU1498 or calcium chelator BAPTA/AM did not affect Tat-induced alterations of claudin-5 mRNA levels (Figures 1B and 1D, respectively). In all experiments presented in Figures 1B-1D, BMEC were exposed to Tat for 8 h.
VEGFR-2 and the Ras/ERK1/2 Pathway are Involved in Tat-Induced Downregulation of Claudin-5 Protein Expression
Evidence suggests that Tat-induced cellular effects can be mediated, at least in part, by VEGFR-2. Therefore, tyrosine phosphorylation of VEGFR-2 was assessed to evaluate if’ Tat can interact with this receptor in BMEC. In addition, to better visualize Tat-induced changes, the nuclei of control and Tat-treated cells were stained with Hoechst dye. As illustrated in Figure 2A, exposure to Tat resulted in marked alterations in VEGFR-2 tyrosine phosphorylation pattern, indicating the involvement of this receptor.
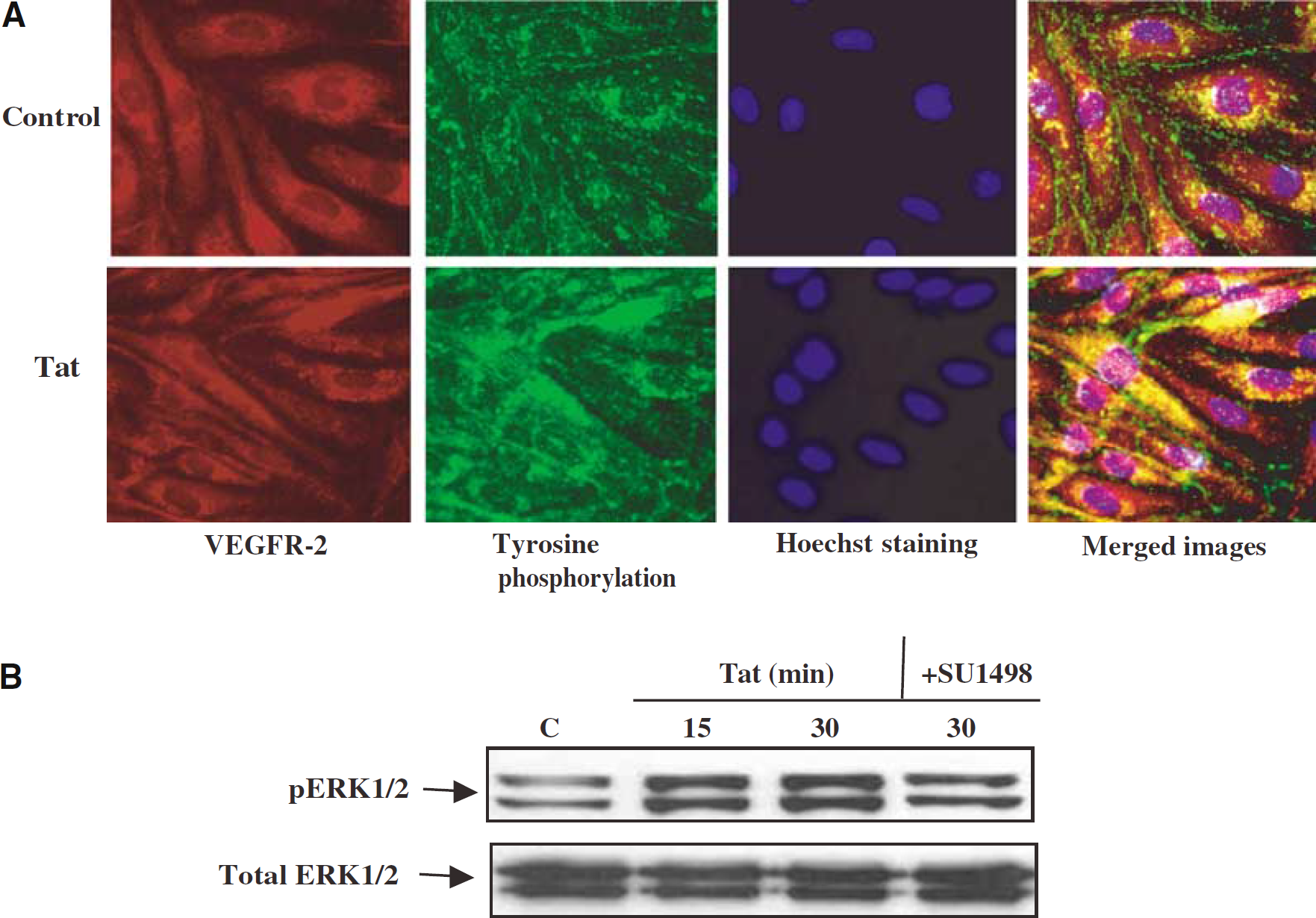
(
We hypothesized that activation of VEGFR-2 may lead to stimulation of the Ras/c-Raf/ERK1/2 pathway in BMEC. Therefore, activation of extracellular signal regulated kinase 1/2 (ERK1/2) was determined in control and Tat-treated BMEC by immunoblotting with antibodies for the active, dually phosphorylated ERK1/2. Figure 2B depicts that exposure to Tat resulted in a time-dependent increase in phosphorylated ERK1/2. In addition, inhibition of the VEGFR-2 by SU1498 attenuated Tat-induced phosphorylation of ERK1/2, confirming that this effect is mediated via activation of VEGFR-2 (Figure 2B).
A possible involvement of VEGFR-2 and the Ras/c-Raf/ERK1/2 pathway in Tat-mediated alterations of claudin-5 expression was studied in a series of Western blots and immunofluorescence analyses. As shown in Figures 3A and 7C, a functional inhibition of VEGFR-2 by SU1498 partially blocked Tat-induced alterations of claudin-5 protein expression. Similarly, a selective c-Raf inhibitor ZM336372 markedly attenuated a decrease in claudin-5 protein in Tat-treated BMEC as determined by Western blot (Figure 3B) and immunostaining (Figure 7D). Finally, a blockage of ERK1/2 activation by the MAPK kinase 1/2 (MEK1/2) inhibitor U0126 effectively prevented Tat-induced downregulation of claudin-5 protein expression, indicating the important role of the ERK1/2 signaling in this effect (Figures 3C and 7E).
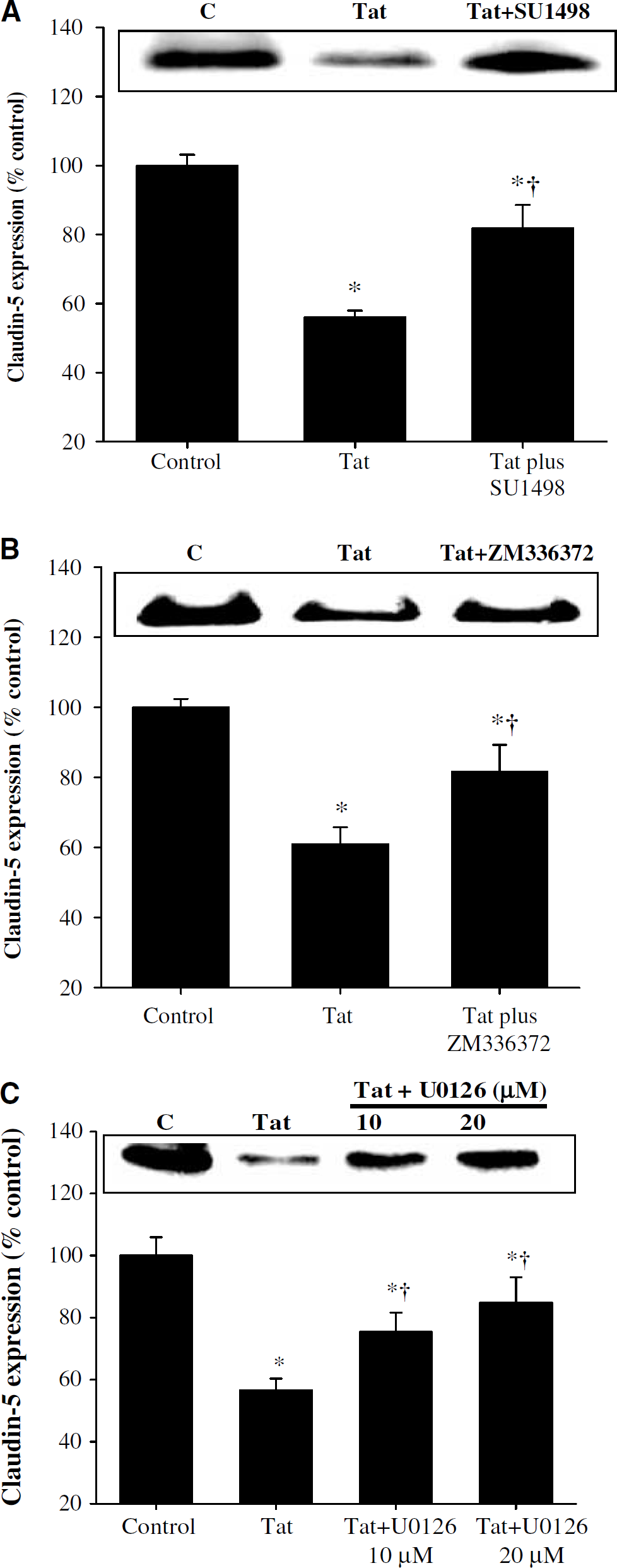
The involvement of the VEGFR-2/Ras/ERK1/2 signaling in Tat-induced alterations of claudin-5 expression in brain microvascular endothelial cells. Confluent cultures were treated with vehicle (control) or 100 nmol/L Tat for 24 h and claudin-5 protein levels were detected by Western blot. In addition, selected cultures were pretreated for 15 mins with: (
(
Tat-Induced Stimulation of the PI-3K/Akt/NF-κB Pathway Contributes to Decreased Expression of Claudin-5 Protein
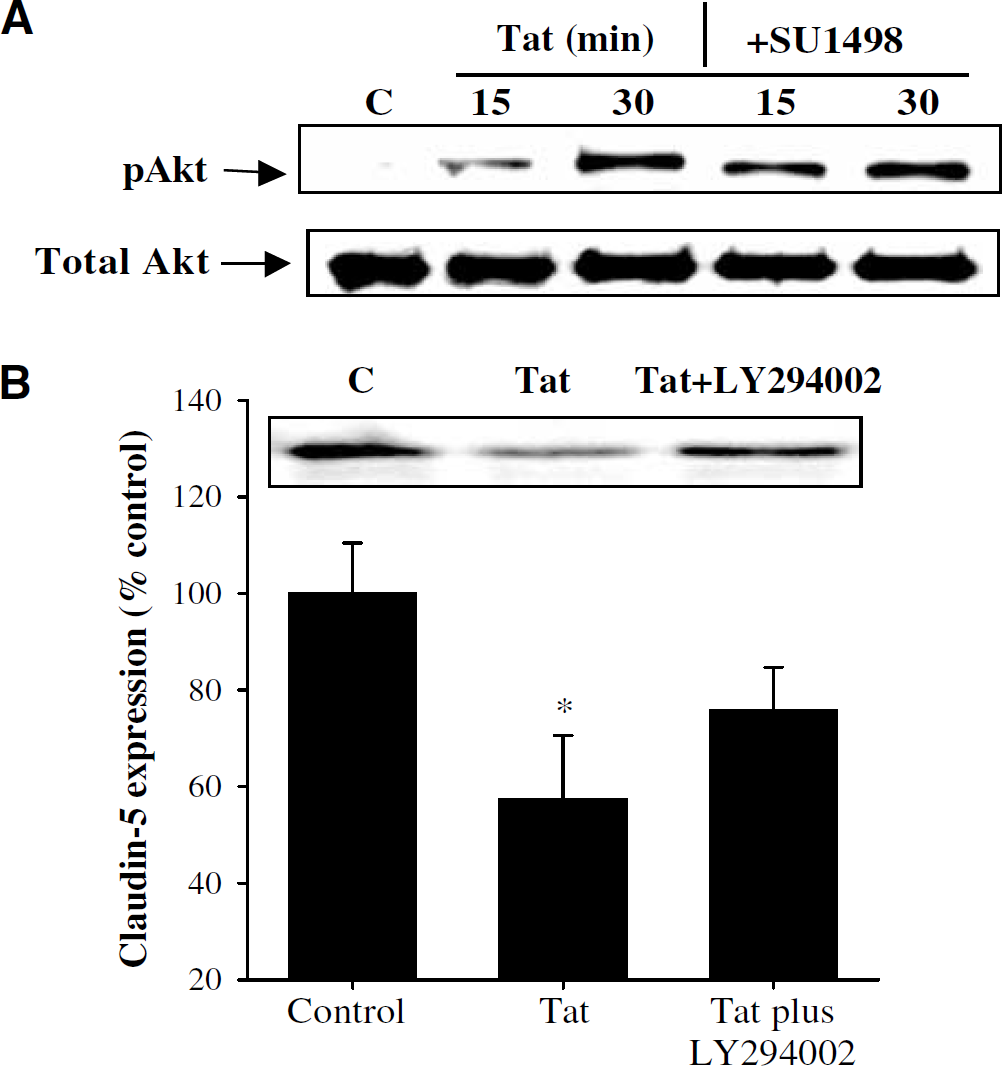
The PI-3K/Akt/NF-κB pathway is a classical redox-regulated signaling mechanism; therefore, its involvement in Tat-induced alterations of claudin-5 expression also was investigated in the present study. To elucidate the regulatory effects of Tat on the PI-3K/Akt/NF-κB signaling, BMEC were incubated with 100 nmol/L Tat, and phosphorylation of Akt, a target of PI-3 K, was determined by Western blot. As illustrated in Figure 4A, Akt phosphorylation was apparent after 15 mins of Tat exposure with a further increase after a 30-min treatment. However, Tat-mediated activation of the PI-3K/Akt signaling appeared to be independent on stimulation of VEGFR-2. Indeed, VEGFR-2 inhibitor SU1498 had no effect on Tat-mediated Akt activation (Figure 4A). In parallel experiments, inhibition of PI-3 K by LY294002 also did not affect Tat-simulated activation of ERK1/2 (data not shown). Thus, no evident crosstalk occurs between Tat-activated VEGFR-2/Ras/ERK1/2 and the PI-3K/Akt signaling pathways in BMEC.
As illustrated in Figures 4B (Western blot) and 7F (immunoreactivity), Tat-induced stimulation of the PI-3K/Akt pathway is involved in alterations of claudin-5 expression. Specifically, pretreatment with the PI-3 K inhibitor LY294002 protected against decreased claudin-5 protein levels in Tat-treated BMEC.
Stimulation of the PI-3K/Akt pathway can lead to the activation of NF-κB DNA-binding activity. Indeed, using double immunofluorescence studies, we detected that treatment with Tat can induce translocation of NF-κB from the cytoplasm into the nuclei of BMEC (Figure 5A). Activation of NF-κB also appeared to be involved in Tat-mediated downregulation of claudin-5 expression. As shown in Figures 5B and 7G, inhibition of NF-κB translocation into the nuclei by treatment with SN50 markedly attenuated Tat-induced alterations of claudin-5 protein expression.
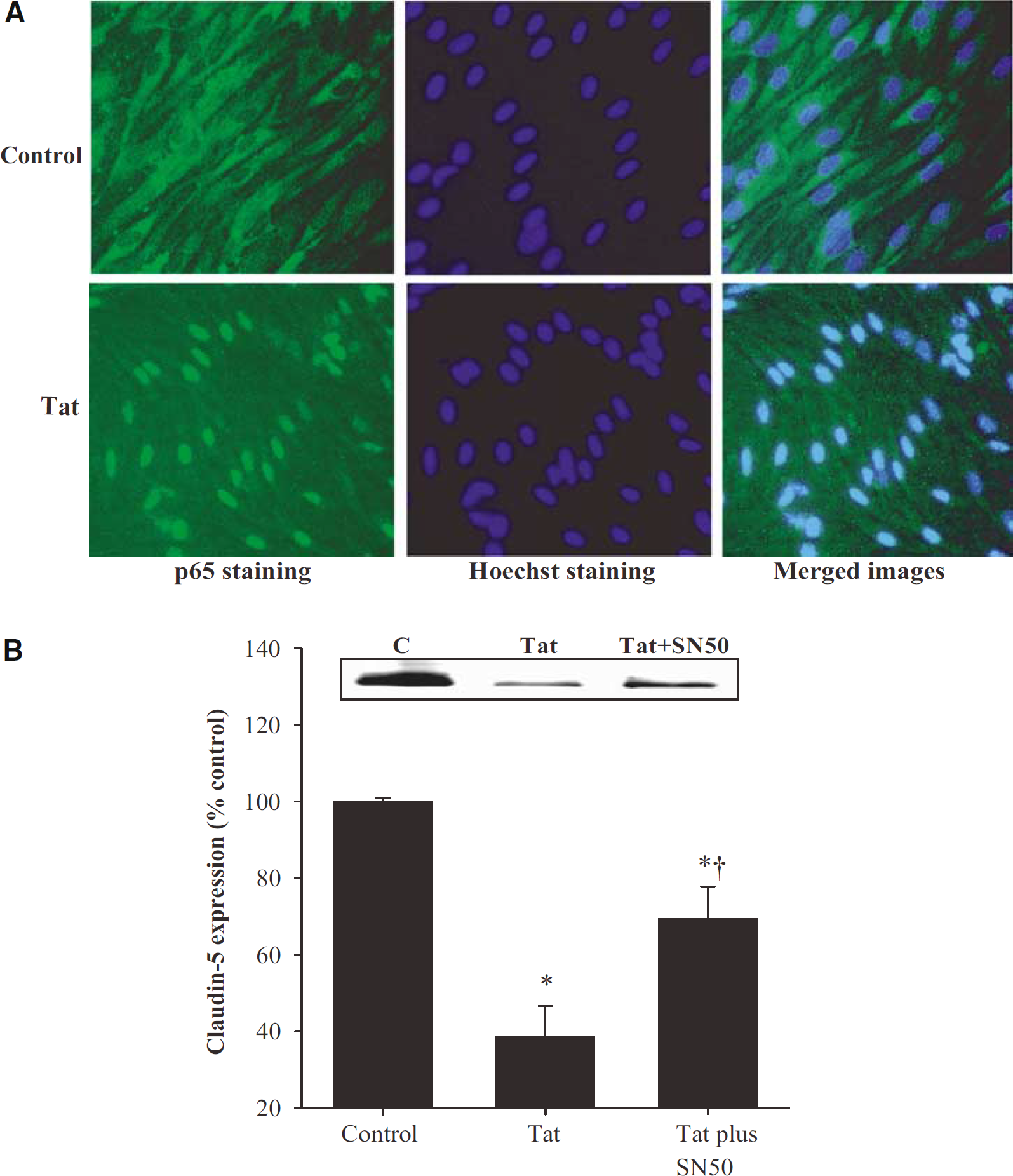
(
Chelation of Intracellular Calcium Inhibits HIV-1 Tat-Evoked Alterations of Claudin-5 Protein Expression
Treatment with Tat can increase intracellular free calcium levels (Haughey and Mattson, 2002). Therefore, we tested the hypothesis that intracellular calcium also may be involved in the Tat-induced downregulation of claudin-5 expression. As illustrated in Figures 6 (Western blot) and 7 H (immunoreactivity), pretreatment with an intracellular calcium chelator BAPTA/AM efficiently blocked Tat-mediated decrease in claudin-5 protein expression. It should be pointed out that these protective effects were at the posttranscriptional level because exposure to BAPTA/AM did not affect Tat-induced downregulation of claudin-5 mRNA levels (Figure 1D).
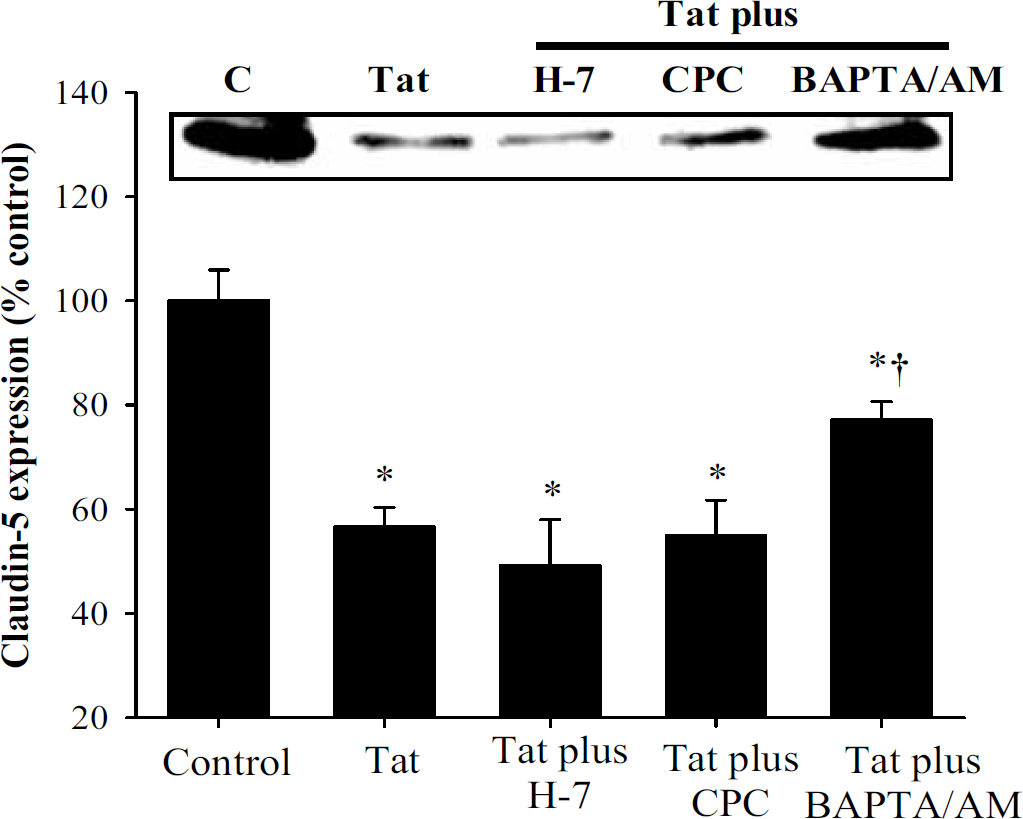
The involvement of protein kinase C (PKC) and intracellular calcium in Tat-induced alterations of claudin-5 expression in brain microvascular endothelial cells. Confluent cultures were treated with vehicle (control) or 100 nmol/L Tat for 24 h and claudin-5 protein levels were detected by Western blot. In addition, selected cultures were pretreated with H-7 (10 μmol/L, 15 mins preexposure), calphostin C (CPC; 10 μmol/L, 15 min preexposure), or BAPTA/AM (5 μmol/L, 5 mins preexposure), followed by treatment with 100 nmol/L Tat for 24 h. The bar graph is the combined densitometry data from four independent experiments and the blot is the representative image from these experiments. *Significantly different as compared with control. †Values in the group Tat plus BAPTA/AM are significantly different as compared with those in the Tat group.
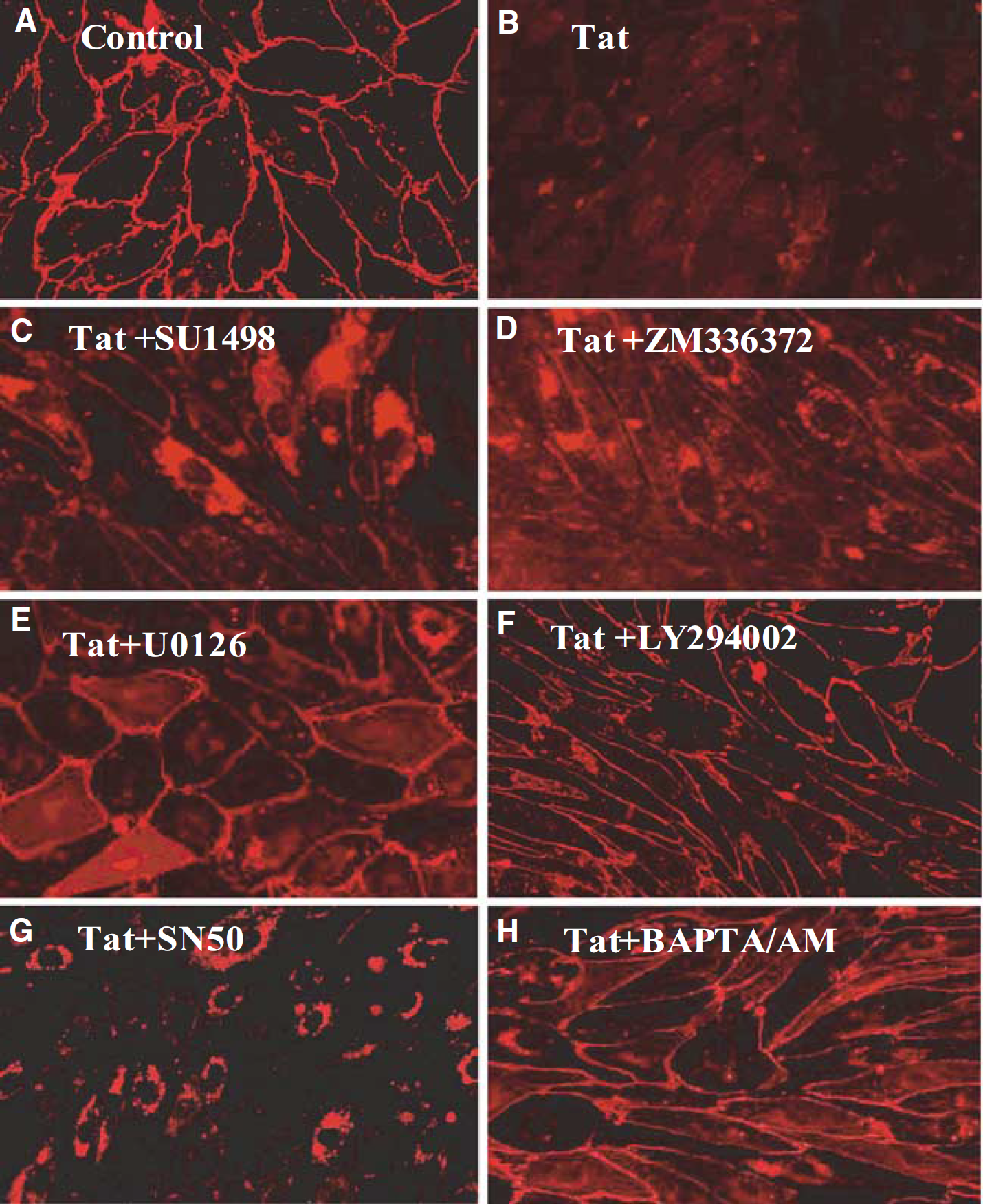
The involvement of specific signaling pathways in Tat-mediated alterations of claudin-5 immunoreactivity in brain microvascular endothelial cells. Confluent cultures were treated with (
An increase in intracellular calcium can regulate PKC activity. In addition, PKC was suggested to be involved in PMA-mediated decrease in claudin-5 immunoreactivity in choroid plexus (Lippoldt et al, 2000). However, pretreatment of BMEC with two different PKC inhibitors, H-7 and calphostin-C, did not affect Tat-mediated alterations of claudin-5 expression in BMEC (Figure 6).
Discussion
Evidence indicates that exposure to Tat can increase BMEC permeability in vitro and in vivo (Oshima et al, 2000; Arese et al, 2001; Kim et al, 2003; Pu et al, 2003), suggesting the disruption of the BBB integrity. However, signaling pathways induced by this protein in BMEC are poorly understood. In particular, the mechanisms involved in Tat-induced changes of tight junction protein expression are not known. The results of the present study show that Tat can stimulate several redox-regulated signal transduction pathways including the Ras/ERK1/2, PI-3K/Akt/NF-κB, and calcium signaling. In addition, activation of these signaling pathways provides the regulatory mechanisms of Tat-mediated alterations of claudin-5 expression (Figure 8). Exposure to Tat induces a transient decrease in claudin-5 mRNA levels, reaching minimal levels after an 8-h treatment (Figure 1). The recovery of claudin-5 mRNA levels after a 24-h exposure to Tat suggests a self-defense mechanism of BMEC in an attempt to restore the BBB integrity. In contrast, a decrease in claudin-5 protein expression is persistent and the levels of this tight junction protein almost completely disappear in BMEC treated with Tat for 24 h (András et al, 2003).
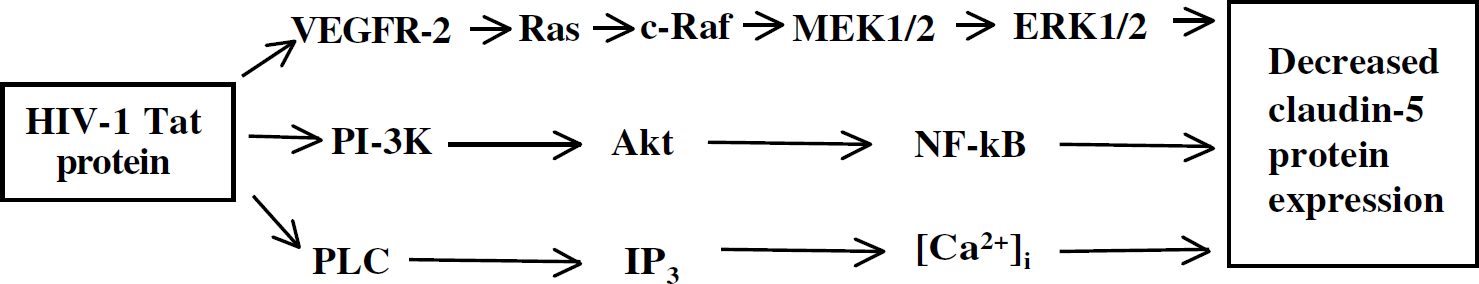
Redox-responsive signaling pathways that can participate in Tat-mediated alterations of claudin-5 expression in brain microvascular endothelial cells.
In the present study, we indicated that Tat could induce the Ras/ERK1/2 signaling through activation of VEGFR-2 (Figure 2B). These results are supported by the literature data, which show the importance of VEGFR-2 in Tat-mediated cellular effects. Specifically, VEGFR-2 was proposed to serve as a high-affinity receptor for Tat in endothelial cells (Albini et al, 1996) and regulate Tat-mediated effects on cell growth and angiogenic activity. In fact, Tat-stimulated angiogenesis was attenuated by a cotreatment with anti-VEGFR-2 antibody (Albini et al 1996; Barillari et al, 1999). It was shown that Tat can interact with VEGFR-2 through the cysteine-rich and basic domains encoded by the first exon of tat gene (Mitola et al, 2000). In contrast, the C-terminal domain of Tat encoded by the second exon is not required for VEGFR-2 activation (Mitola et al, 2000). This is an important fact, because the form of Tat peptide encoded by the first exon (amino acids 1–72) was used in the present study.
Tat-mediated activation of the Ras/ERK1/2 pathway via VEGFR-2 appears to be specific, because inhibition of VEGFR-2 had no effect on Tat-induced activation of Akt (Figure 4A). However, it should be pointed out that blocking VEGFR-2 by SU1498 offered only a partial inhibition of ERK1/2 phosphorylation (Figure 2B), indicating that other Tat-mediated signaling pathways also may be involved in this process. The form of Tat (1–72) used in the present study can interact with other cell surface receptors, such as chemokine receptors CCR2, CCR3 (Albini et al, 1998), CXCR4 (Xiao et al, 2000; Poggi et al, 2004), and VEGFR-1 (Mitola et al, 1997). In addition, Tat can be internalized by cells through the interaction of its basic domain with cell surface heparan-sulfate proteoglycans (Rusnati et al, 1999). Thus, Tat may affect signaling mechanisms and cellular metabolism without interacting with cell surface receptors.
The ERK1/2 pathway is known to be involved in multiple cascades of serine/threonine and tyrosine phosphorylation reactions that mediate diverse cellular responses to growth factors, physical stress, and cytokines (Kyriakis and Avruch, 2001). In agreement with the results of the present study, it was reported that Tat can induce ERK1/2 activation in vascular endothelial cells (Rusnati et al, 2001). However, the novelty of the current study is to show that the VEGFR-2/Ras/ERK1/2 signaling can participate in Tat-mediated alterations of claudin-5 expression. This involvement appears to be complex, for example, inhibition of VEGFR-2 by SU1498 had no effect on Tat-induced alterations of claudin-5 mRNA levels (Figure 1B) but partially protected against changes in claudin-5 protein expression (Figure 3A). In contrast, inhibition of ERK1/2 phosphorylation by U0126 protected against both Tat-induced diminished claudin-5 mRNA and protein levels (Figures 1C and 3C, respectively). In strong support of the importance of the ERK1/2 signaling in Tat-induced cell toxicity, it was shown that Tat-evoked permeability changes can be mediated through a MAPK and tyrosine kinase-dependent pathway (Oshima et al, 2000). Evidence also indicates that activation of the ERK1/2 can stimulate HIV-1 replication by phosphorylation of Tat and other HIV-1 proteins and enhancing the infectivity of HIV-1 virions in vitro (Yang and Gabuzda, 1999). Thus, Tat-mediated activation of ERK1/2 may not only participate in alterations of claudin-5 expression and endothelial cell permeability but also contribute to increased viral infectivity, generating a self-intensifying mechanism of cell toxicity.
lts of the present study show that Tat can activate the PI-3K/Akt signaling in BMEC (Figure 4A), a pathway which is well known to lead to activation of NF-κB (Thomas et al, 2002). Indeed, exposure of BMEC to Tat resulted in a marked activation of NF-κB as illustrated by nuclear translocation of this transcription factor (Figure 5A). These findings are confirmed by the literature data that documented Tat-mediated activation of the PI-3K signaling in PC12 cells (Milani et al, 1996). In addition, Tat-induced activation of NF-κB was shown in a variety of cell types, including brain endothelial cells (Toborek et al, 2003).
Several lines of evidence indicate that the PI-3K/Akt/NF-κB pathway can be involved in Tat-mediated cell toxicity. For example, it was shown that blocking PI-3 K resulted in inhibition of Tat-induced apoptosis of brain endothelial cells (Kim et al, 2003). In addition, the role of NF-κB activation in Tat-mediated inflammatory responses is well documented (Toborek et al, 2003; Lee et al, 2004). However, the novelty of the present study is to show a connection between the PI-3K/Akt/NF-κB signaling pathway and Tat-induced alterations in claudin-5 expression. Several different mechanisms can be responsible for these effects. Activation of PI-3 K was shown to be involved in induction of Snail expression (Peinado et al, 2003), a transcriptional repressor for occludin and claudin-3, 4, and 7 (Ikenouchi et al, 2003). Snail can bind directly to the E-boxes of the promoter regions of claudin/occludin genes, repressing their activity. However, the specific effects of Snail on claudin-5 expression are not known.
In this study, inhibition of NF-κB translocation into the nuclei also resulted in attenuation of Tat-mediated alterations of claudin-5 protein levels (Figures 5B and 7G). Therefore, we suggest that activation of this transcription factor may serve as a negative regulator of claudin-5 expression. Such a potential mechanism is supported by the literature data, which showed that NF-κB can inhibit expression of occludin, a tight junction protein associated with claudin-5 (Wachtel et al, 2001). Thus, activation of NF-κB can not only induce inflammatory responses in the brain endothelium but also may directly affect the BBB integrity by interference with tight junction protein expression.
Tat-induced cell toxicity has been linked to an increase in intracellular calcium level through its release from the inositol triphoshate (IP3) sensitive pool (Hegg et al, 2000; Haughey and Mattson, 2002). Increased calcium ions can trigger several signaling cascades, including the ERK1/2 and PKC pathways. Therefore, a possible involvement of intracellular calcium in Tat-mediated changes in claudin-5 expression was also examined in this study. A pretreatment with intracellular calcium chelator BAPTA/AM markedly prevented Tat-induced alterations of claudin-5 expression (Figures 6 and 7(H). However, it should be pointed out that these effects were observed only at the protein level and BAPTA/AM did not affect Tat-mediated changes in claudin-5 mRNA expression (Figure 1D). In addition, blockage of PKC activity with two different inhibitors, H-7 and calphostin-C, did not influence Tat-mediated alterations of claudin-5 expression (Figure 6). These were unexpected results, because PKC signaling was suggested to be involved in diminished claudin-5 immunoreactivity in choroid plexus in response to phorbolester treatment (Lippoldt et al, 2000). However, different experimental models and treatment factors can be responsible for the discrepancy of these results.
In summary, the present study shows that Tat can decrease claudin-5 expression both at the transcriptional and posttranscriptional levels. In addition, several signal transduction mechanisms, including the VEGFR-2/Ras/ERK1/2, PI-3K/Akt/NF-κB, and calcium signaling pathways, are involved in Tat-mediated alterations of claudin-5 levels. The effects appear to be specific, because inhibition of PKC did not affect Tat-mediated downregulation of claudin-5 expression. Tat-induced alterations of claudin-5 levels may constitute a critical mechanism of the BBB disturbances in the course of HIV-1 infection.