
Abstract
The objectives of the study were to (1) characterize the dose-response relationship to the TXA2 analog, U46619 (0.01, 0.1, and 1 μmol/L) after global cerebral ischemia, (2) determine whether chronic 17β-estradiol (E2) replacement alters this relationship, and (3) determine if E2's mechanisms are transduced through cognate estrogen receptors. Rats were assigned to five groups (n = 6): placebo-implanted ovariectomized (OVX) females, OVX plus chronic E2 (CE), OVX plus acute E2 (AE), OVX plus chronic E2 plus the estrogen receptor inhibitor ICI 182,780 (CEI), and OVX plus acute E2 plus ICI 182,780 (AEI). Rats were anesthetized, intubated, cannulated (femoral artery and vein), fitted with a closed cranial window, and subjected to 15-min reversible forebrain ischemia (4-vessel occlusion, 4-VO) and 60 mins of reperfusion. Arterial blood gases, intrawindow pressure, and temperature were controlled. Vessel diameter was measured before and 5 mins after superfusion of each concentration of U46619. Compared with preischemic responses, contractile response to U46619 was depressed at all concentrations after ischemia in the OVX group. In the chronic E2 and acute E2 groups, contractile response to 1 μmol/L of U46619 was normalized to near baseline values. However, in the CEI and the AEI groups, postischemic vasoconstriction was similar to that observed in the OVX rats. We conclude that E2 targets the cerebral microvasculature to preserve postischemic pial artery reactivity and that the effect is receptor mediated. Restoration of normal constriction to vascular agonists may be an important mechanism by which E2 protects the vasculature and diminishes tissue damage after ischemia.
Introduction
Potential benefits of estrogen replacement therapy on stroke risk and recovery remain controversial. Data from the Women's Health Initiative suggest that estrogen replacement can increase risk for stroke (Wassertheil-Smoller et al, 2003), but definitive effects on recovery from stroke have yet to be established. The vasorelaxant properties of estrogen have been well studied in most vascular beds, including cerebral vessels (Geary et al, 2000; Pelligrino et al, 2000; Ospina et al, 2003), and are likely orchestrated via cyclooxygenase-1 induction of prostacyclin (Ospina et al, 2002) and/or by enhanced synthesis of nitric oxide (Geary et al, 2000; McNeill et al, 1999). Recent findings in humans indicate that increasing 17β-estradiol (E2) levels amplifies cerebral blood flow (CBF) (Slopien et al, 2003).
Cerebral ischemia results in vascular abnormalities, such as hyperemia, residual hypoperfusion (Uhl et al, 2000; Belayev et al, 2002; Pulsinelli et al, 1983), and microcirculatory dysfunction (Clavier et al, 1994; Cipolla et al, 1997; Pulsinelli et al, 1982). Striking endothelial defects in vasodilation and constriction have been described (Rosenblum, 1988; Dietrich, 1994; Rosenblum and Wormley, 1995; Cipolla et al, 1997; Watanabe et al, 2001). Platelets accumulate within the postischemic vasculature and contribute to hypoperfusion by mechanical obstruction and by secretion of the vasoconstrictors serotonin (5-HT) and thromboxane (TXA2) from internal granules (Tanahashi et al, 1999; Garcia et al, 1994). 17β-Estradiol partially preserves CBF after global (Pelligrino et al, 1998) and focal (Alkayed et al, 2000) ischemia from vascular occlusion. We have also shown that chronic treatment with E2 preserves dilation to endothelium-dependent and endothelium-independent agonists in pial arterioles in situ after near-complete forebrain ischemia (Watanabe et al, 2001); yet, it fails to restore normal endothelium-dependent vasoconstriction to 5-HT. Accordingly, we hypothesized that pial vessels might be similarly unresponsive to TXA2, which acts at vascular smooth muscle sites. Using an in vivo cranial window model, the objectives of this study were: (1) to characterize the dose-response relationship to the TXA2 receptor-dependent, stable analog U46619 (9,11-dideoxy-11 alpha,9 alpha-epoxymethano***prostaglandin F2 alpha) during reperfusion in ovariectomized females, (2) to determine whether chronic E2 replacement alters this relationship, and (3) to determine if E2's mechanisms are transduced through cognate estrogen receptors.
MATERIALS AND METHODS
Animal Preparation
General preparation: The Johns Hopkins Institutional Animal Care and Use Committee approved all animal protocols. Sexually mature female Wistar rats (250 to 300 g) were ovariectomized (OVX) under 1% halothane anesthesia, as previously described (Alkayed et al, 1998) at least 7 days before use. The rats were randomly assigned into the following five groups (n = 6 rats/group): placebo-implanted OVX females (OVX), OVX plus chronic E2 (CE), OVX plus acute E2 (AE), OVX plus chronic E2 plus the estrogen receptor inhibitor ICI 182,780 (CEI), and OVX plus acute E2 plus ICI 182,780 (AEI). Chronically implanted rats were treated with subcutaneous (dorsal neck) implants of either E2, 50 μg/kg, (21-day timed-release pellet; 1.2 μg/day for 21 days) or placebo (nitrocellulose carrier) for not less than 7 and not greater than 14 days after treatment. Previous work in our laboratory has shown that the slow-release pellets produce stable physiologic E2 levels between 7 and 21 days after implantation (Alkayed et al, 2000). To determine if responses to estrogen were preserved at both physiologic and supraphysiologic concentrations, rats in the acute E2-treated groups received intraperitoneal injection of 0.25 mg/kg E2 dissolved in sesame oil 1 h before initiation of ischemia.
Transient forebrain ischemia: Ischemia was induced by 4-vessel occlusion (4-VO), as previously described (Watanabe et al, 2001). Briefly, under 1% halothane anesthesia, vertebral arteries were permanently occluded bilaterally using electrocautery, and silastic vessel ties were loosely placed on both carotid arteries 24 h before induction of ischemia. In addition, a silk suture (1-0) was threaded posterior to the trachea and above the large cervical muscles, and the suture ends were fixed to the dorsal neck with tape. All wounds were closed and infiltrated with 1% bupivacaine to control immediate postoperative pain, and animals were recovered for 24 h. On the following day, anesthesia was induced with 4% halothane followed by 70% N2O and 30% O2. All rats were mechanically ventilated through an endotracheal tube to maintain physiologic parameters within normal limits. A femoral vein and artery were cannulated with PE 50 catheters for fluid infusion, for arterial blood pressure, and for blood gas monitoring. Rectal temperature was maintained between 36.5°C and 37.5°C with the use of a warming lamp and mat.
In all of the studies, 4-VO was induced by tightening both the carotid artery and the external suture ties (to reduce collateral blood flow from extracranial sources) that had been loosely placed 24 h before the experiments. Reduction in cortical blood flow was confirmed by visualizing blood flow stasis through vessels under the cranial window. After 15 mins of ischemia, the ties were released and CBF was reestablished.
Vascular diameter studies: Fifteen pial arterioles (six animals/group) were studied, as previously described (Littleton-Kearney et al, 2000; Watanabe et al, 2001). Briefly, a craniotomy was performed over the left parietal cortex with a cooled high-speed drill. The bone flap was removed, bone bleeding controlled, and a polypropylene ring (7 mm o.d.), equipped with inflow and outflow cannulae, was then cemented to the skull. The well formed by the ring was superfused with artificial cerebrospinal fluid (aCSF) that was pH, PCO2, PO2, and temperature controlled. The dura was carefully excised, to expose superficial pial vessels, and the window was sealed with a glass cover slip. Intrawindow pressure was maintained at 5 to 8 mm Hg and temperature was controlled with a warming lamp. After the window was fitted, fentanyl was administered (10 μg/kg), followed by a continuous intravenous infusion of 25 μg/kg h for anesthesia.
Pial arteriolar diameter was measured using a Zeiss compound microscope with fiberoptic epi-illumination and interfaced with a charge-coupled device camera. The camera was coupled to a high-resolution monitor, a VHS video recorder, and a graphic printer. For each rat, pial arteriolar measurements were obtained on 1 to 2 main arterioles and 2 to 3 daughter branches. Measurements on the daughter vessels were averaged with those of the parent vessel for analysis. Vessel diameters (resolution approximately 2 to 3 μm) were measured off-line and expressed as the percentage of the baseline diameter that had been determined prior to superfusion with each drug concentration.
Experimental protocols: In our first set of experiments, we sought to determine (1) the optimal concentration of U46619 for maximal change in vessel diameter and (2) the effects of ischemia on dose-response to U46619. After a 40-min recovery period after cranial window placement, we evaluated preischemic vasoconstriction response to three concentrations of U46619 (0.01, 0.1, and 1 μmol/L), which were administered in random order to control for loss of responsiveness because of tachyphylaxis. Arterial diameters were measured immediately before the drugs were superfused. The drugs were allowed to dwell in the window for 5 mins, and the pial arteriole diameter was remeasured. Responses to the U46619 were calculated as percent change from baseline diameter. After administration of each drug concentration, the window was superfused with warmed aCSF for 10 mins to clear out residual drug; then a new arteriolar baseline diameter was determined before the next drug infusion. After preischemic vessel measurements were completed, the animals were subjected to 15 mins of 4-VO, and response to U46619 was determined at 20 mins of reperfusion.
In the remaining experiments, we evaluated the effects of chronic E2 depletion, as well as acute and chronic E2 repletion using 1 μmol/L U46619, the dose that produced maximum change in vessel diameter. To determine if E2's actions are dependent on its receptor, the specific receptor antagonist ICI 182,780 was used (Tocris Cookson, Inc., Ellisvale, MO, USA). Based on our published work (Sawada et al, 2000) and our preliminary studies, we determined that intra-window superfusion of 10 μmol/L ICI 182,780 effectively blocked E2's vasodilation. ICI 182,780 (10 μmol/L) had no effect on baseline vessel reactivity, but effectively blocks E2-induced vasodilation in this system. Preischemic responses to U46619 were tested. ICI 182,780 was then superfused into the window for 10 mins (0.5 mL/min) and allowed to dwell throughout ischemia. Postischemic responses to U46619 were then re-evaluated.
Measurement of Physiologic Variables and Plasma Estrogen
All animals were weighed on an electronic scale before the study (Scout Pro, Pinebrook, NJ, USA). Arterial blood pressure was measured with Statham 23Db (Gulton-Statham Transducers Inc., Costa Mesa, CA, USA) pressure transducers and continuously recorded on a Gould-Brush polygraph (Modul Company, Danbury, CT, USA). Arterial blood samples were analyzed with a Radiometer electrode system (Chiron Diagnostics Corporation, Emeryville, CA, USA) for pH, PO2, and hemoglobin levels. Plasma E2 was measured in triplicate using a commercially available clinical radioimmunoassay kit (Coat-a-Count, Diagnostic Products Corp, Los Angeles, CA, USA) with a range of 0 to 3.600 pg/mL and a linear sensitivity of 8 pg/mL or greater.
Statistical analysis: All data are reported as means±s.d. Physiologic parameters and vessel diameters were evaluated using a two-way analysis of variance (ANOVA)—or Kruskal-Wallace ANOVA on ranks in cases where the data were not normally distributed. The post hoc comparisons were performed using a Newman-Keuls test. Significance was set at P≤0.05.
RESULTS
Physiologic Parameters
Physiologic variables were maintained within normal range throughout the experiments (Table 1). Intrawindow temperature was held constant at 37.6°C±0.08°C, and pressure was maintained at 5 to 8 mm Hg. As expected, E2 levels for the ovariectomized group were low (2.1±3.4 pg/mL). 17β-Estradiol levels in the chronic estrogen-treated group were within normal physiologic ranges for rat (102±51 pg/mL), while in the acute estrogen-treated groups, plasma E2 was supraphysiologic (761±4 pg/mL).
Physiologic data preischemia and at 60 mins of reperfusion
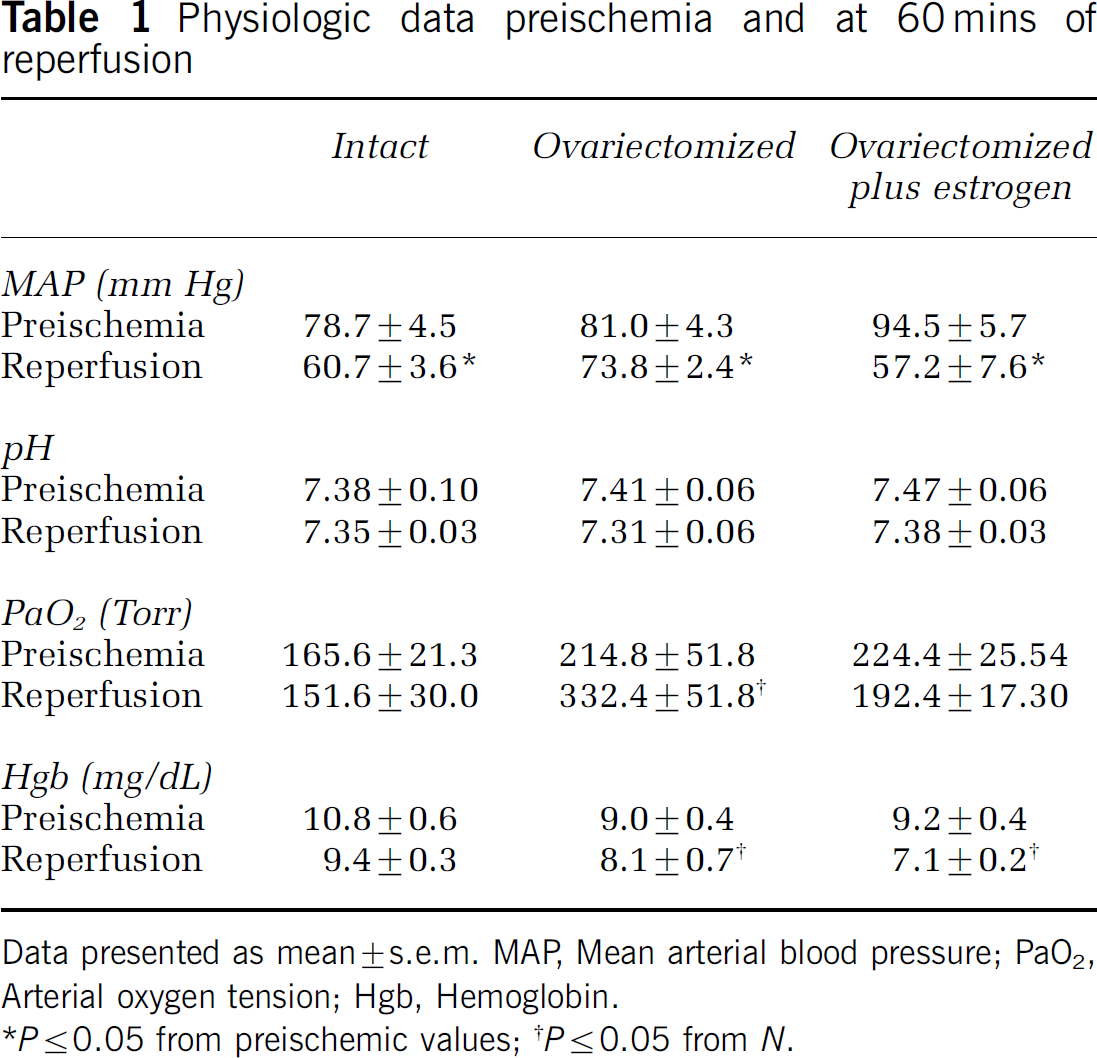
Data presented as mean ± s.e.m. MAP, Mean arterial blood pressure; PaO2, Arterial oxygen tension; Hgb, Hemoglobin.
*P≤0.05 from preischemic values; †P≤0.05 from N.
Pial arteriolar diameters ranged from 20.5 to 55.5 μm. Preischemic arteriolar diameters were similar between OVX (Figure 1A) and CE (Figure 1B) rats at all concentrations of U46619. Before ischemia, superfusion with 0.01, 0.1, and 1 μmol/L, and U46619 resulted in a brisk reduction of diameter (–13.6%±3.4%, –21.2%±5.3%, and –32.5%±2.5% of baseline diameters, respectively). In contrast, vasoconstriction was markedly depressed after 4-VO in response toa ll U46619 concentrations (Figure 1A). As shown in Figure 1B, chronic E2 pretreatment dose-dependently restored vasoconstriction after ischemia. 17β-Estradiol depletion diminished pial artery responsiveness to 1 μmol/L U46619 (–18.8%±9.3% of baseline diameter, Figure 2), while chronic E2 repletion returned postischemic constriction to U46619 (–33.2%±5.7%) to control values (–34.1%±2.4%).
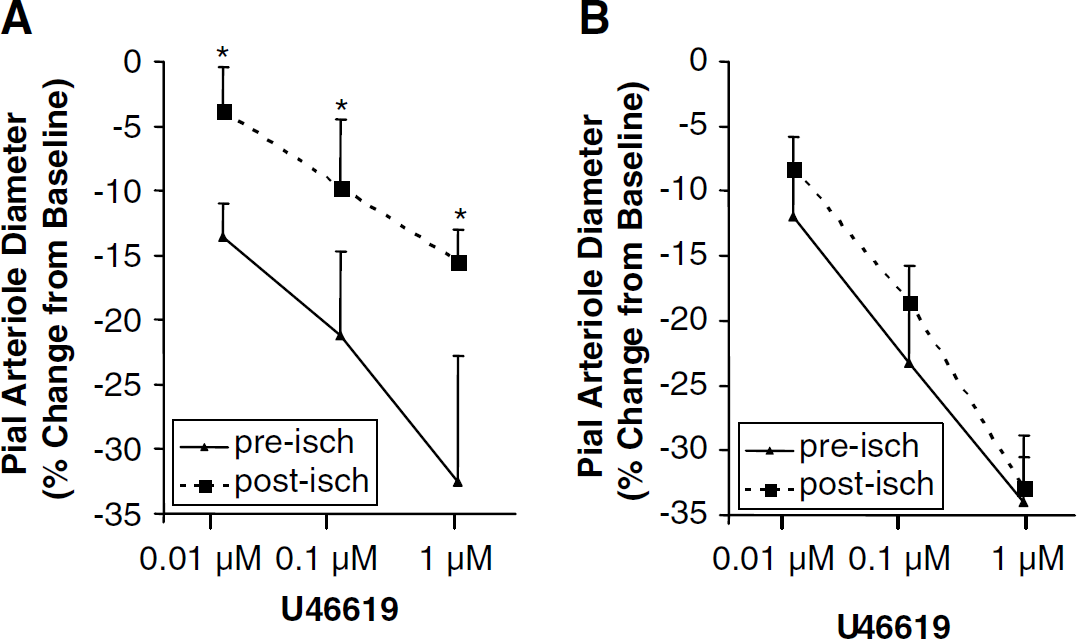
Pial artery sensitivity to the vasoconstrictor U46619 (0.01, 0.1, 1 μm): (
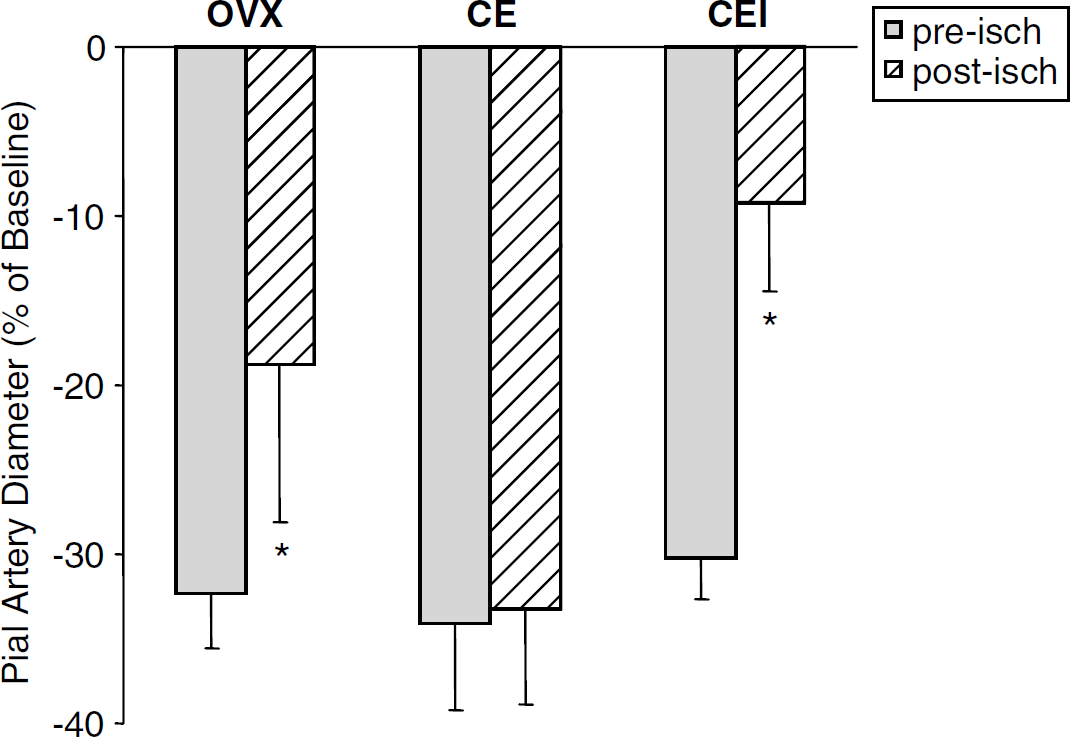
Percent change in baseline diameter of pre- and postischemic pial artery in response to U46619 (1 μm) in ovariectomized (OVX) rats, chronic estrogen-treated (CE) rats, and chronic estrogen-treated rats with pial vessels exposed to ICI 182,780 (CEI). Intrawindow superfusion with ICI 182,780 reverses the protection conferred by chronic estrogen replacement (mean±s.d., *P<0.05 from CE).
Because E2 restored postischemic U46619-induced pial artery vasoconstriction, we sought to determine if E2's vasoprotection involved the estrogen receptor. In the chronic E2-treated rats, superfusion of ICI 182,760 sharply reduced postischemic vasoconstriction to –9.11%±5.2% of baseline diameter in a manner similar to that observed in the OVX (from –32.3%±3.2% to –18.8%±9.3% of baseline, Figure 2).
Likewise, acute E2 treatment restored normal postischemic constriction to U46619. However, superfusion of ICI 182,780 lowered constriction from –30.3%±2.4% to –9.9%±5.2% (Figure 3).
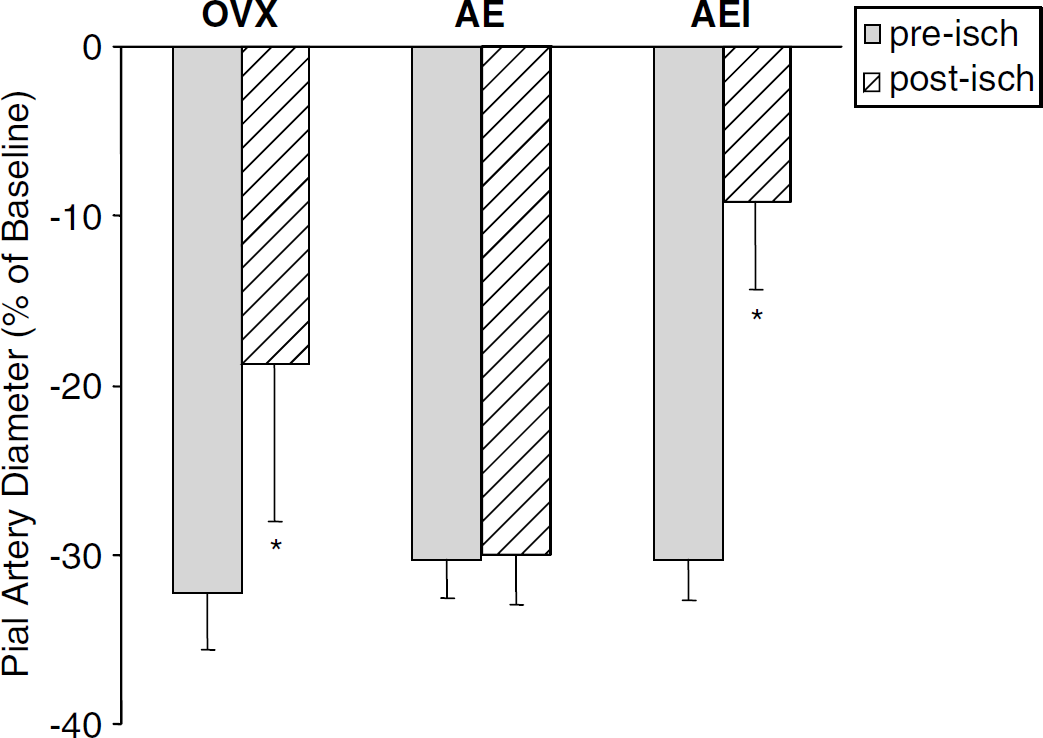
Percent change in baseline diameter of pre- and postischemic pial artery in response to U46619 (1 μm) in ovariectomized (OVX) rats, acute estrogen-treated (AE) rats, and acute estrogen-treated rats with pial vessels exposed to ICI 182,780 (AEI). Intrawindow superfusion with ICI 182,780 reverses the protection conferred by acute estrogen replacement (mean±s.d., *P≤0.05 from AE).
DISCUSSION
In this study, we report three novel findings in the intact postischemic cerebral microvascular network. First, pial arterioles are refractory to dose-dependent stimulation with the TXA2 receptor-mediated agonist U46619 during early reperfusion. Second, both acute and chronic E2 repletion normalizes vasoconstriction to this agonist. Third, the mechanism by which E2 acts likely involves interaction with the estrogen receptor. The functional consequences for brain could include mitigation of deleterious hyperemia that follows cerebral ischemia and potentiates ischemic damage (Yamagami et al, 1998) and autoregulatory failure. Cipolla et al (2000) suggest that loss of microvascular autoregulatory capacity and reactivity exacerbate edema and thus intensify cerebral injury once circulation is restored. We speculate that restoration of pial vascular reactivity to vasoconstrictor agents may diminish postischemic hyperemic damage and preserve tissue viability.
Depressed postischemic cerebrovascular dilation is present after global cerebral ischemia (Clavier et al, 1994; Rosenblum and Wormley, 1995; Busija et al, 1996), but less is known about the effects of transient ischemia on microcirculatory response to vasoconstrictors. Our previous studies and those of others show that global ischemic brain injury can attenuate contractile responses to 5-HT (Watanabe et al, 2001; Fadyukova et al, 2004) and to arterial hypercapnia (Nelson et al, 1992). Further, myogenic tone is reduced after focal ischemia (Cipolla and Curry, 2002). In the present study, we observed a marked postischemic pial arterial insensitivity to U46619. Our data do not show the specific mechanism by which postischemic vasoconstriction to U46619 is diminished. However, our data are in agreement with that of others who report postischemic diminution of myogenic reactivity and tone to 5-HT in pressurized middle cerebral arteries after focal stroke (Cipolla et al, 2000, 1997). The loss of response to U46619 that we observed in the present study may reflect impaired myogenic reactivity that results from altered intracellular signaling or enhanced synthesis of thromboxane leading to increased receptor interaction. In that U46619 is a receptor-mediated agonist, the occupation of binding sites by thromboxane may be reflected by diminished contractile responsiveness to U46619.
Alternatively, the relative insensitivity to U46619 in postischemic pial vessels in the estrogen-depleted rats may be a consequence of ongoing Interleukin-1β (IL-1β)-induced vasodilator prostanoid synthesis. Several reports indicate that ischemia rapidly stimulates marked expression of the inflammatory mediator IL-1β in brain (Minami et al, 1992; Buttini et al, 1994) and in cerebral vessels (Zhang et al, 1998). Recent data confirm a relationship between IL-1β, amplification of PGE2 synthesis, and reduction of cerebral vascular tone (Ospina et al, 2003; Ospina et al, 2004). Other studies indicate that cerebral ischemia and reperfusion increases expression of COX-2 with subsequent increased synthesis of the vasodilator PGE2 (Catley et al, 2003). The postischemic upregulation of COX-2 is rapid and occurs at as early as 30 mins of reperfusion (Planas et al, 1995), a time course well within the window of observation of our current study.
We originally hypothesized that E2 would not alter postischemic vessel responsiveness to U46619, an endothelium-independent vasoconstrictor. Surprisingly, we observed the opposite effect and found that estrogen in acute supraphysiologic or chronic physiologically relevant concentrations were equally effective. The restorative effect appears to be estrogen-receptor mediated, as ICI 182,780 reversed E2 effects. It is possible that the mechanism may involve a genomic receptor, as supraphysiologic concentrations of estrogen could elicit a nonspecific response. However, this seems unlikely due to the rapidity with which the acutely administered estrogen restored postischemic contractile response, strongly suggesting the involvement of a nonnuclear estrogen receptor. In fact, estrogen has been reported to exert its biologic effects via nontranscriptional, rapid-acting mechanisms or through a membrane-associated receptor that is upregulated after cerebral ischemia (Toran-Allerand et al, 2002; Toran-Allerand, 2004).
The effects of E2 on the postischemic cerebrovascular function are not well understood. To our knowledge, this is the first study to show that E2 can modify postischemic contractile responses to U46619. It is possible that E2 modulates dynamic signaling between the endothelium and vascular smooth muscle. In previous work with the same model, E2 failed to restore postischemic contractile response to 5-HT (Watanabe et al, 2001). However, 5-HT has been shown to induce contraction via endothelial-mediated mechanisms (Rosenblum and Nelson, 1988), while U46619 acts at the level of vascular smooth muscle and requires both extracellular and intracellular calcium (Liu et al, 1997; Moffatt and Cocks, 2004). Therefore, our current data suggest that E2 modulates signaling within vascular smooth muscle, perhaps via a calcium-mediated mechanism. Clearly, more work is necessary to determine the precise mechanisms by which E2 restores postischemic contractile response at the level of vascular smooth muscle.
In conclusion, E2 targets the cerebral microvasculature to preserve postischemic pial artery reactivity. The present study, showing restoration of cerebrovascular reactivity to endothelium-independent vasoconstrictors, together with previous work that showed improved reactivity to endothelium-dependent vasodilators, indicates that estrogen acts to enhance vascular regulation over a dynamic range to a broad spectrum of vasoactive stimuli. This restoration may represent an important mechanism by which E2 protects the vasculature and diminishes tissue damage after ischemia.
Footnotes
Acknowledgements
The authors thank Tzipora Sofare, MA, for her editorial assistance in preparing this manuscript.