
Abstract
Neuronal apoptosis inhibitory protein (NAIP/BIRC1), the inhibitor of apoptosis protein (IAP) family member, suppresses neuronal cell death induced by a variety of insults, including cell death from ischemia and stroke. The goal of the present study was to develop an efficient method for identification of compounds with the ability to upregulate endogenous NAIP and to determine the effects on these compounds on the cellular response to ischemia. A novel NAIP-enzyme-linked immunosorbent assay (ELISA)-based in vitro drug-screening system is established. Use of this system identified an antagonist of dopamine D4 receptor, termed L-745,870, with a potent NAIP upregulatory effect. L-745,870-mediated NAIP upregulation in neuronal and nonneuronal cultured cells resulted in decreased vulnerability to oxidative stress-induced apoptosis. Reducing NAIP expression via RNA interference techniques resulted in prevention of L-745,870-mediated protection from oxidative stress. Further, systemic administration of L-745,870 attenuated ischemia-induced damage of the hippocampal CA1 neurons and upregulated NAIP expression in the rescued hippocampal CA1 neurons in a gerbil model. These data suggest that the NAIP upregulating compound, L-745,870, has therapeutic potential in acute ischemic disorders and that our NAIP-ELISA-based drug screening may facilitate the discovery of novel neuroprotective compounds.
Introduction
Several studies have demonstrated that the mechanisms of ischemia-induced neuronal cell death fall on the continuum between necrosis and apoptosis (Raghupathi et al, 2000; Graham and Chen, 2001). While relatively more interest has been focused on prevention of cell necrosis after ischemic insults, apoptosis, with its ordered series of events, including caspase activation (Graham and Chen, 2001) and/or the perturbation of calcium homeostasis (Love, 1999), may represent a more suitable target for modulation for the goal of preventing cell death. This may be particularly true for neuronal damage after cerebral ischemia (Nitatori et al, 1995).
Recent studies have demonstrated that various antiapoptotic proteins, including Bcl-2, neuronal apoptosis inhibitory protein (NAIP, also called baculoviral IAP repeat-containing 1 (BIRC1)), and X-linked inhibitor of apoptosis (XIAP), are upregulated in neurons after ischemia (Graham and Chen, 2001; Krajewski et al, 1995; Xu et al, 1997, 1999). Among these antiapoptotic proteins, NAIP, the founding member of the inhibitor of apoptosis protein (IAP) family, was identified in the course of the positional cloning of the gene responsible for spinal muscular atrophy (SMA) (Roy et al, 1995). In actuality, the primary genetic defect in SMA results from mutations in an adjacent gene, survival motor neurons (SMN). However, patients with the most severe form of SMA have large deletions that encompass both the SMN and NAIP genes. These data suggest that NAIP may also play a role in neuronal viability (Gavrilov et al, 1998; Hsieh-Li et al, 2000; Monani et al, 2000).
Indeed, overexpression of NAIP by adenovirusmediated gene transfer reduces ischemic cell damage in the rat hippocampus (Xu et al, 1997). Further, ectopic NAIP expression rescues motor neurons after peripheral nerve axotomy (Perrelet et al, 2000) and preserves nigrostriatal dopaminergic function in the intrastriatal 6-hydroxydopamine (6-OHDA) rat model of Parkinson's disease (Crocker et al, 2001). Moreover, NAIP promotes motor neuron survival through the intracellular signaling of glial cell-derived neurotrophic factor (GDNF) (Perrelet et al, 2002). Finally, unlike other IAP proteins and Bcl-2 family proteins, NAIP exerts a unique antiapoptotic activity against oxidative stresses. These findings suggest that NAIP plays an important role in the protection of the neuronal cells from apoptotic insults, and that upregulation of endogenous NAIP may represent a therapeutic approach for prevention of oxidative stress-induced neuronal cell damage.
Therefore, the goal of the present study was to develop an efficient method for identification of compounds with the ability to upregulate endogenous NAIP and to determine the effects of these compounds on the cellular response to ischemia.
Materials and methods
Chemicals
A total of 953 compounds, including 3-[(4-[4-chlorophenyl]piperazine-1-yl)methyl]-[1H]-pyrrolo[2,3-b] pyridinetrihydrochloride (L-745,870) listed in ‘Neurochemicals, Signal Transduction Agents, Pharmacological Probes and Biochemicals compounds; Tocris Cookson Ltd’, was purchased from Tocris Cookson Ltd (Bristol, UK) and subjected to drug-screening experiments. Drug concentrations were tested in a range from 1 to 100 μmol/L. All other cytotoxins including menadione, H2O2, α-naphthoquinone, 2,3-dimethoxy-1,4-naphthoquinone (DMNQ), actinomycin D, staurosporine, cis-platinum, okadaic acid, oligomycin, and etoposide were purchased from Sigma-Aldrich (St Louis, MO, USA).
Antibodies
Two independent anti-human NAIP antibodies, IB9 and ME1, were generated. IB9, a mouse monoclonal antibody, was raised using an epitope of the human NAIP C-terminal peptide (amino acids 841 to 1052), and a polyclonal antibody, ME1 (1:3,000 dilution), was obtained by immunizing rabbits with recombinant NAIP peptide (amino acids 256 to 587). ME1 recognizes the BIR3 region of NAIP and crossreacts with the mouse and gerbil NAIP proteins (data not shown). Other antibodies used in this study included rabbit polyclonal anti-XIAP antibody (#AF822; 1:1,000; R&D Systems, Minneapolis, MN, USA), rabbit polyclonal anti-cIAP-1 antibody (#AF818; 1:1,000; R&D Systems), rabbit polyclonal anti-cIAP-2 antibody (#AF817; 1:1,000; R&D Systems), rabbit polyclonal anti-Survivin antibody (#NB500-201; 1:1,000; Novus Biologicals, Littleton, CO, USA), rabbit polyclonal anti-Bcl-2 antibody (#SC-783; 1:1,000; Santa Cruz Biotechnology, CA, USA), rabbit polyclonal anti-Bcl-xL antibody (#SC-634; 1:1,000; Santa Cruz Biotechnology), and mouse monoclonal anti-β-tubulin antibody (#SC-5274; 1:50,000; Santa Cruz Biotechnology).
Cell Lines and Culture Conditions
The human monocyte-derived cell line, THP-1, was cultured in RPMI-1640 (Invitrogen, Carlsbad, CA, USA), while HeLa and SH-SY5Y neuroblastoma cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen). All culture media contained penicillin (50 IU/mL) and streptomycin (50 μg/mL) and were supplemented with 10% fetal calf serum. SH-SY5Y cells, which were seeded with a density of 4 × 105 cells/well in six-well plates, were differentiated by treatment with all-trans-retinoic acid (Tocris Cookson) at a concentration of 10 μmol/L.
NAIP-ELISA and Screening of NAIP Upregulating Compounds
NAIP-neuronal apoptosis inhibitory protein-enzyme-linked immunosorbent assay (ELISA)-based screening with THP-1 cells was conducted to identify compounds that induced endogenous NAIP expression. THP-1 cells were cultured in 24-well plates at a density of 2 × 105 cells/well for 24 h, followed by incubation in the presence of tested compounds for either 24 or 72 h. Cells were then lysed with NP40 buffer (50 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl, 1% NP40, and protein inhibitor cocktail (Complete; Roche Diagnostics, Indianapolis, IN, USA)). Aliquots of extracts were subjected to the NAIP-ELISA assay, with 1B9 as primary antibody conjugated to the ELISA plate and with ME1 as secondary antibody for quantification of NAIP immunoreactivity. Assays were conducted according to standard ELISA procedure (Crowther, 1995).
Western Blot Analysis
The extracts from cultured cells were electrophoretically separated on 5% to 20% SDS-polyacrylamide gels and transferred onto polyvinylidene difluoride (PVDF) membranes (Bio-Rad Laboratories, Hercules, CA, USA). Membranes were then incubated with the indicated primary antibodies in TBST buffer (50 mmol/L Tris-HCl; pH 7.4, 150 mmol/L NaCl, 0.1% (w/v) Tween-20) for 2 h, after incubation with the peroxidase-linked secondary anti-rabbit IgG (#NA934; Amersham Pharmacia Biotech, Uppsala, Sweden) or anti-mouse IgG (#NA931; Amersham Pharmacia Biotech) antibody for 1 h. Signals were detected using ECL Plus (Amersham Pharmacia Biotech).
Cell Viability Assay
Approximately 1 × 105 cells, which were either pretreated with 10 μmol/L of L-745,870 for 24 h or left untreated, were plated on a 96-well plate and incubated for 5 h at 37°C. The appropriate amounts of the cytotoxins, including free radical generating compounds (DMNQ: 120 μmol/L, menadione: 20 to 80 μmol/L, α-naphthoquinone: 60 μmol/L and H2O2: 30 μmol/L), kinase inhibitor (staurosporine: 1 nmol/L), ATPase inhibitor (oligomycin: 100 μmol/L), DNA-damaging reagent (cis-platinum: 100 μmol/L), phosphatase inhibitor (okadaic acid: 100 nmol/L), and topoisomerase II inhibitors (actinomycin D: 1 μmol/L and etoposide: 100 μmol/L), were added, and the preparation was allowed to incubate for another 1 to 10 h. The medium was then replaced with fresh medium containing 10% (v/v) alamarBlue (AccuMed International, Westlake, OH, USA), followed by incubation for an additional 4 h at 37°C. Cell numbers were counted fluorometrically as per the alamarBlue Assay instructions.
Flow-Cytometric Analysis of Apoptotic/Necrotic Cell Death
Quantification of the apoptotic/necrotic cell death was performed by flow cytometry in conjunction with the MEBCYTO Apoptosis Kit (MBL, Nagoya, Japan). In brief, HeLa cells that were pretreated with or without L-745,870 were plated on a six-well plate (3 × 105 cells/well). After 4 h of incubation, menadione (or H2O2) was added to the cells, followed by incubation for an additional 4 h (or 40 mins for H2O2). The cells were washed once with phosphate-buffered saline (PBS) and suspended in fresh medium for Annexin V-FITC and propidium iodide (PI) labeling, which was performed according to the manufacturer's instructions (MBL). The cells were sorted and analyzed by FACScan (Beckton Dickinson, San Jose, CA, USA). Cell damage was classified according to the extent of staining of AnnexinV-FITC and PI (Quadrant analysis). Quadrants were comprised of the upper left (UL; AnnexinV-/PI +), upper right (UR; AnnexinV + /PI +), low left (LL; AnnexinV-/PI-), and low right (LR; AnnexinV + /PI-). LL, LR, and UR represent the cells in normal state, early stage of apoptosis, and late stage of apoptosis/necrosis, respectively.
RNA Interference
Neuronal apoptosis inhibitory protein RNA interference (RNAi) was achieved by expressing the hairpin-forming short RNA molecules generated from a portion of the 3'UTR of the human NAIP/BIRC1 gene. Briefly, 19-nucleotide-long inverted repeats, 5'-GTCAACTCCCCT - CCCCTTG-3' (sense) and 5'-CAAGGGGAGGGGAGTT GAC-3' (antisense), which were intervened with the 9-nucleotide spacer (TTCAAGAGA), were inserted downstream of the U6 promoter of pSilencer 1.0-U6 vector (Ambion, Austin, TX, USA), generating pSilencer 1.0-U6-NAIP. HeLa cells, which were either pretreated with L-745,870 for 24 h or left untreated, were cotransfected with 10 μg of pSilencer 1.0-U6-NAIP and 1 μg of pMACS Kk. II (Miltenyi Biotec, Bergisch Gladbach, Germany) using FuGENE6 (Roche Diagnostics). After 48 h, the transfected cells were magnetically enriched and cultured for 24 h. NAIP expression and cell viability were analyzed by Western blotting and by the alamarBlue Assay, respectively.
Generation of Forebrain Ischemic Model in Gerbils
All animal experimental procedures were performed in accordance with the guidelines of the Tokai University School of Medicine Committee on Animal Care and Use. Twelve-week-old Male Mongolian gerbils (Clea Japan Inc., Tokyo, Japan), weighing 70 to 80 g, were used. Animals were anesthetized with halothane (4%) in a mixture of N2O/O2 (70:30) initially, and halothane was gradually decreased to 2% for maintenance of anesthesia during surgery. Under anesthesia, a femoral artery catheter was placed to monitor mean arterial blood pressure, and rectal and temporal muscle temperatures were monitored and maintained at 37°C ± 0.5°C via a heating pad and radiant heat during and after surgery. Forebrain ischemia was induced by bilateral common carotid artery occlusion (BCCAO) using 3-mm sugita-aneurysm clips (Kirino and Sano, 1984). After 10 mins of occlusion, the aneurysmal clips were removed to allow reperfusion, and complete reperfusion of the arteries was verified by direct visual observation. Sham-operated gerbils underwent identical surgery with the exception of the BCCAO procedure. Under these experimental conditions, in which a relatively severe ischemic condition was used, all animals survived until fixation.
To investigate the in vivo effect of L-745,870 (Tocris Cookson Ltd) on ischemia-induced cell death, the compound was dissolved in physiologic saline and was administered to gerbils at a dose of 7 mg/kg (n = 6), 70 mg/kg (n = 7), 140 mg/kg (n = 7), or 210 mg/kg (n = 6). We have administered the compound to the animals 1 h before ischemic surgery to ensure the protection of neurons from the oxidative stress-induced cell death. This experimental protocol was designed based on the in vitro experimental data in which the pretreatment with the compound effectively protects cultured cells from the menadione-induced insults (refer to Figures 1 to 4). Administration of the compound was performed intragastrically via oral cannula under anesthesia. Vehicle-treated animals (n = 7) received physiologic saline alone. Sham-operated animals (n = 3) were administered with a compound dose of 210 mg/kg.
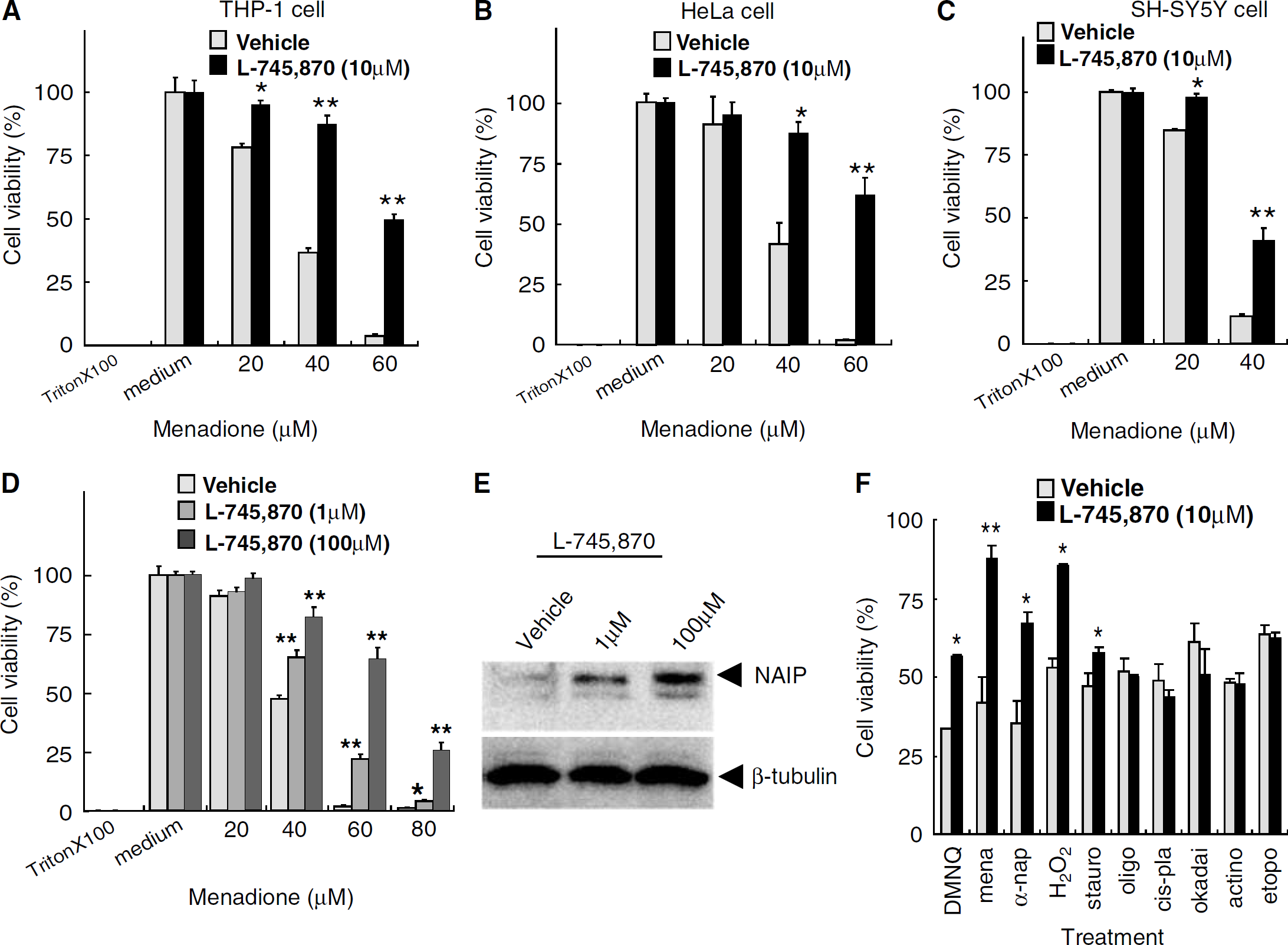
The effect of L-745,870 on oxidative stress-induced cell death. Cell viability after menadione treatment in (
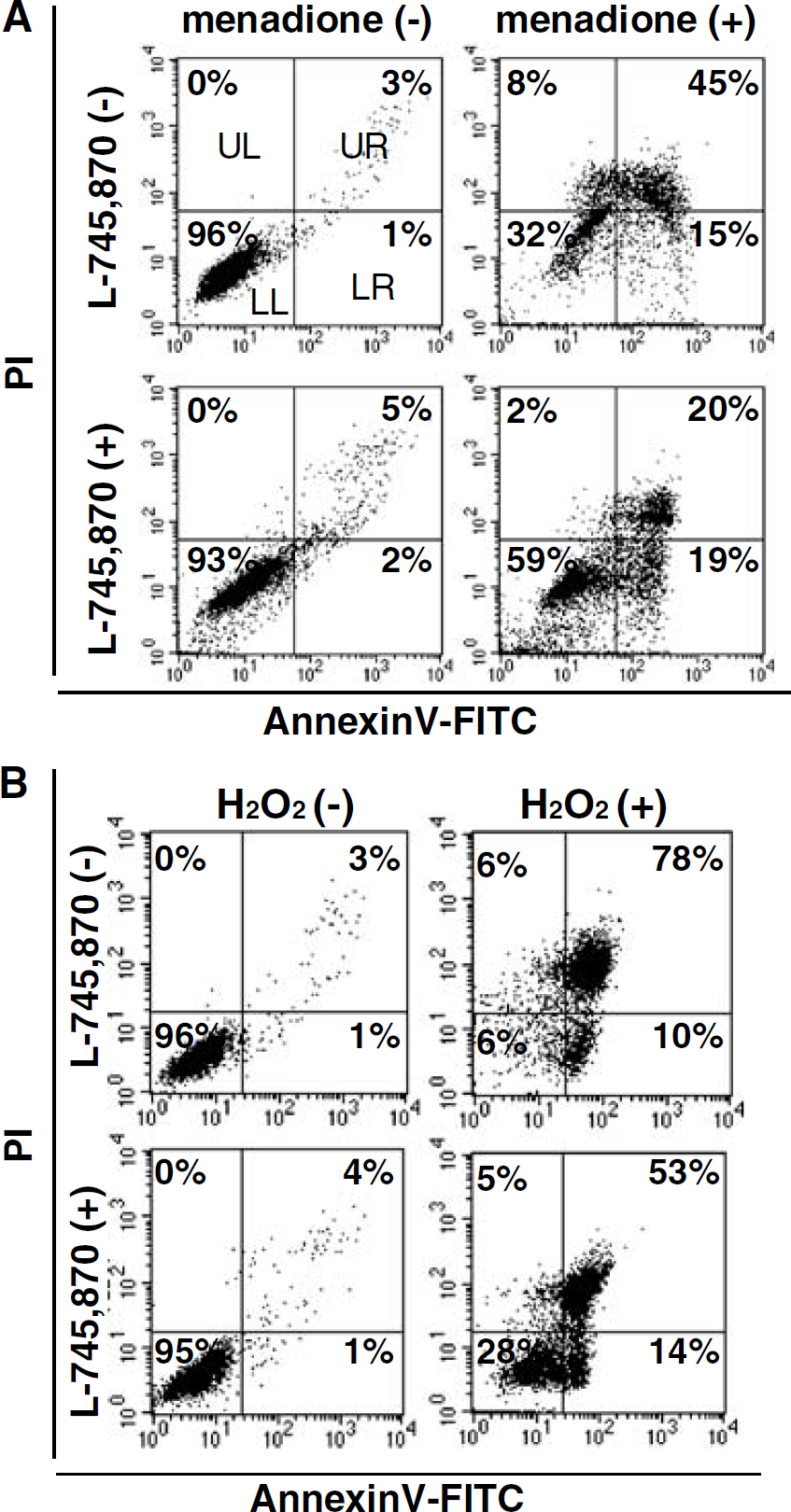
Effect of the neuronal apoptosis inhibitory protein (NAIP) upregulating compound (L-745,870) on the oxidative stress-induced apoptotic cell death, and a flow-cytometric analysis of the apoptotic and/or necrotic cell death in HeLa cells. HeLa cells that were precultured with vehicle or L-745,870 (10 μmol/L) for 24 h were challenged with (
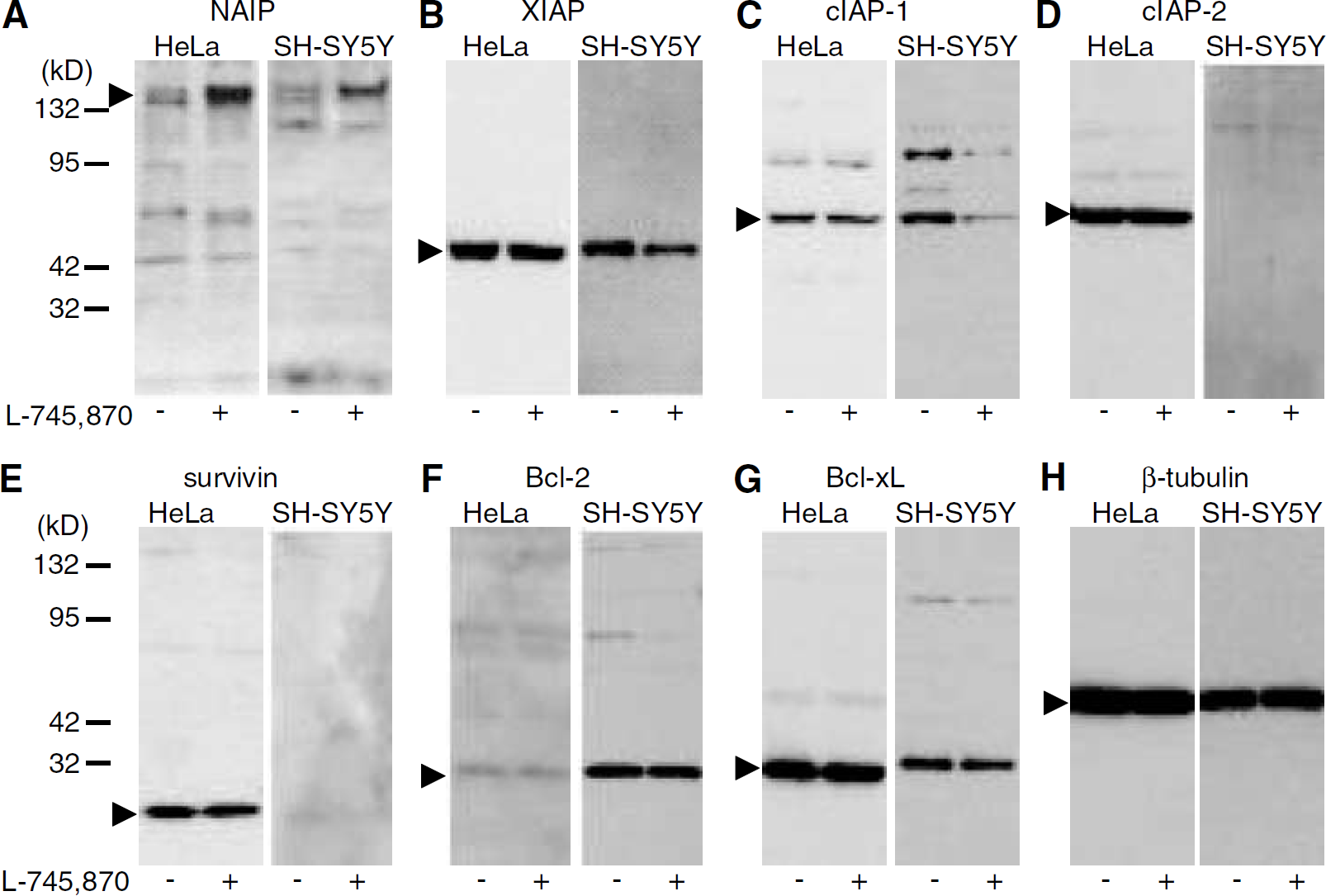
Western blot analysis of antiapoptotic proteins in HeLa and differentiated SH-SY5Y cells after treatment with L-745,870 (+, 10 μmol/L) or vehicle (–) for 24 h. Seven antiapoptotic proteins, including (
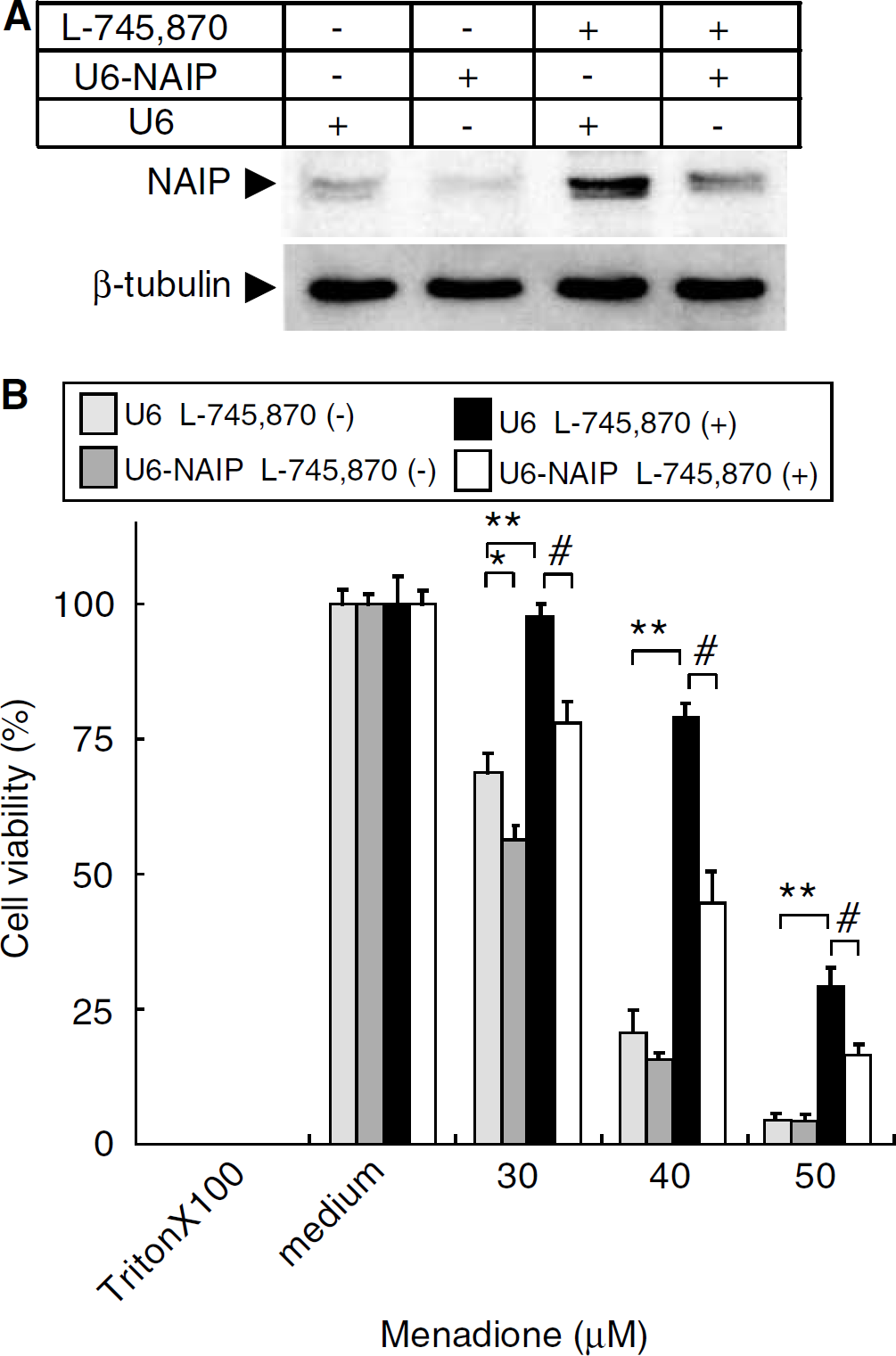
Effect of inhibition of neuronal apoptosis inhibitory protein (NAIP) expression on menadione-induced cell death in HeLa cells. Neuronal apoptosis inhibitory protein RNAi was achieved by ectopically expressing the small interference RNA (siRNA) corresponding to a portion of the 3'-UTR of the NAIP gene using the pSilencer 1.0-U6 system. (
Measurement of Regional Cerebral Blood Flow
To analyze the effect of the compound on regional cerebral blood flow (CBF), a subset of animals underwent measurement of CBF at 2 and 24 h after administration of L-745,870 (210 mg/kg) or vehicle. Cerebral blood flow was measured by the hydrogen clearance method via a platinum wire electrode stereotaxically inserted into the right hippocampus using coordinates of 2 mm posterior and 2 mm lateral to the bregma, and 2.5 mm below the brain surface in a flat cranial presentation, as previously reported (Osuga et al, 2000).
Histopathology
After 3 days of reperfusion, animals were anesthetized with 4% halothane and perfused with 60 mL of 4% paraformaldehyde in phosphate buffer (pH 7.4) via a catheter placed in the heart. The brains were removed, fixed in 10% formalin for 10 days, and embedded in paraffin. Paraffin sections were sliced at a thickness of 7 μm for histopathologic and immunohistochemical evaluation. Neuronal cell density of the CA1 subfield of the hippocampus, that is, the number of intact CA1 pyramidal neurons per 1 mm linear length of pyramidal cell layer, was measured by counting 7 μm sections stained with hematoxylin and eosin from 3 to 7 independent animals in a double-masked manner.
Immunohistochemistry
Brain sections were subjected to NAIP and XIAP immunohistochemistry with polyclonal anti-human NAIP (ME1) and XIAP antibodies (R&D Systems Inc.), respectively, and stained using the Vectastain elite ABC kit (Vector Laboratories Inc., Burlingame, CA, USA) according to the manufacturer's instructions. In brief, after deparaffinization, the sections were washed with 0.01 mol/L PBS (pH 7.2) for 5 mins and were incubated with anti-NAIP (5 μg/mL) or anti-XIAP (5 μg/mL) antibody overnight at 4°C. The sections were rinsed three times with PBS containing 0.05% Triton X-100 for 10 mins, incubated with biotinylated secondary antibody for 3 h, and then incubated with avidin-biotin-peroxidase complex for 1 h at room temperature. Finally, the sections were treated with 0.5% 3,3'-diaminogenzidine (DAB) and 0.01% H2O2 in Tris-HCl buffer (pH 7.5), and the DAB reaction products were observed under a microscope.
Statistical Analysis
All data in this study are presented as mean ± s.e. Data were analyzed for significance using Student's t-test for pair-wised comparisons or ANOVA followed by Scheffe's test for multiple comparisons between groups (Statview 5.0 software; SAS, Cary, NC, USA). A P-value <0.05 was considered as reaching statistical significance.
Results
Identification of NAIP Upregulating Compounds
To identify compounds with the ability to induce NAIP expression, 953 compounds (Tocris) were screened using NAIP-ELISA. Compounds were arbitrarily categorized as ‘downregulator’ (n = 16) if the resulting NAIP level was less than 70% of the normal endogenous level (i.e., ~12 ng/mL), or ‘upregulator’ (n = 30) if the resulting NAIP level was more than 200% of the normal endogenous NAIP level (Table 1).
Screening of 953 Tocris compounds by NAIP-ELISA, and viability assays after challenging with menadione-induced oxidative stress in THP-1 cells
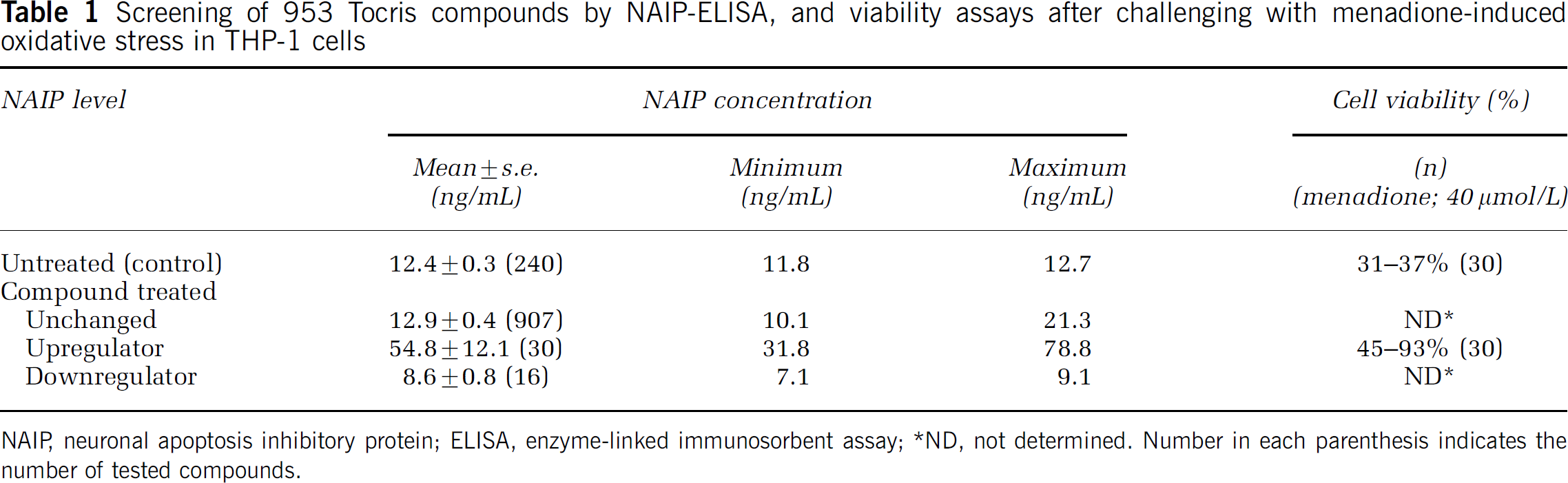
NAIP, neuronal apoptosis inhibitory protein; ELISA, enzyme-linked immunosorbent assay;
ND, not determined. Number in each parenthesis indicates the number of tested compounds.
To examine whether the 30 identified NAIP upregulators suppressed menadione-induced cell death, THP-1 cell viability assays were performed in cells pretreated with menadione followed by the addition of each NAIP upregulator. All 30 compounds exerted a protective effect against menadione-induced cell death with variable degrees (Table 1). The compound that exerted the most potent protective effect was L-745,870, a dopamine D4 receptor antagonist (Figure 1A). This compound was used for subsequent experiments.
L-745,870 Protects a Variety of Cultured-Cells Against Menadione-Induced Cell Death
To determine whether the protective effect of L-745,870 on THP-1 cells was specific to cell type, cell viability studies were also conducted in HeLa and SH-SY5Y (differentiated neuroblastoma by all-trans-retinoic acid treatment) cells after exposure to the compound. L-745,870 protected both cell lines from menadione-induced cell death (Figures 1A, THP-1; B, HeLa; and C, SH-SY5Y). Further, the dose-dependent increase in cell viability correlated with a concomitant increase in NAIP level in HeLa cells (Figures 1D and 1E).
L-745,870 Selectively Protects Cells Against Oxidative Stress-Induced Apoptosis
To gain insight into the suppression of the cell death with L-745,870, HeLa cells pretreated with L-745,870 were challenged with various apoptosis-inducing stimuli and chemical cell-stressors. L-745,870 specifically suppressed cell death induced by oxidative stressors, including cell death in response to menadione, hydrogen peroxide (H2O2), DMNQ, and α-naphthoquinone, but did not protect against cell death induced by nonoxidative stressors (e.g., staurosporine, oligomycin, cis-platinum, okadaic acid, actinomycin D, and etoposide) (Figure 1F).
To confirm the antiapoptotic effect of L-745,870 against oxidative stress-induced cell death, flow cytometric analysis of cell treated with L-745,870 and menadione and double-stained with Annexin V-FITC and PI was performed. Half of the population of the menadione-treated cells that were not exposed to L-745,870 were distributed in the LR quadrant (early stage of apoptosis) in an early phase of incubation (60 mins) but subsequently shifted to the UR quadrant (apoptosis/necrosis) during the late phase of incubation (4 h) (data not shown). These data suggest that cell death in response to menadione is the result of apoptosis rather than necrosis. L-745,870 treatment reduced the apoptotic cell population in the UR quadrant from 45% to 20% and increased the LL and LR quadrant populations from 32% to 59% and 15% to 19%, respectively (Figure 2A). L-745,870 treatment also reduced the apoptotic cell population in the UR quadrant from 78% to 53% and increased the normal cells in the LL quadrant from 6% to 28% in the cells exposed to H2O2 for 40 mins (Figure 2B). These results suggest that L-745,870 specifically protects cells from apoptosis induced by oxidative stress.
L-745,870 Specifically Upregulates NAIP
To examine whether L-745,870 specifically induces NAIP expression level among the antiapoptotic proteins, expression levels of the IAP family (XIAP, cIAP-1, cIAP-2, and survivin) and Bcl-2 family (Bcl-2 and Bcl-XL) proteins were assessed in HeLa and SH-SY5Y cells by Western blotting. L-745,870 specifically elevated the endogenous level of NAIP (Figure 3A) but had no effect on the levels of other antiapoptotic proteins (Figures 3B-3G). This observation was consistent with results obtained by the DNAChip analysis (8,300 genes, including all of the known antiapoptotic proteins with the exception of the NAIP/BIRC1 gene: the Atlas Plastic Arrays analysis; Beckton Dickinson), in which no upregulation of the antiapoptosis relating genes was observed (data not shown). Slight decreases in the levels of XIAP and cIAP-1 were noted only in differentiated SH-SY5Y cells (Figures 3B and 3C), although the physiologic significance for this small effect may not be significant. These results indicate that L-745,870 selectively enhances endogenous NAIP levels.
L-745,870 Inhibits Oxidative Stress-Induced Apoptosis Via NAIP Upregulation
To investigate whether the elevation of the endogenous NAIP level by L-745,870 is responsible for the protection against oxidative stress-induced apoptosis, NAIP gene expression was suppressed using NAIP-RNAi. Transfection of HeLa cells with NAIP-U6 resulted in a marked decrease in endogenous NAIP levels (Figure 4A) and a concomitant increase in susceptibility to menadione (Figure 4B). Further, L-745,870-treated/NAIP-U6 transfected HeLa cells showed higher susceptibility to menadione than cells with NAIP-U6 treatment alone (Figure 4B). These results indicate that the increased endogenous NAIP level by L-745,870 likely mediates its ability to protect cells from oxidative stress-induced apoptosis.
Systemic Administration of L-745,870 Results in a Transient Upregulation of NAIP in Gerbil Hippocampal CA1 Neurons
L-745,870 administration had no effect on CBF (control/vehicle; 103.9 ± 13.2 mL/100 g/min, 2 h after administration; 100.3 ± 12.0 mL/100 g/min, and 24 h after administration; 98.9 ± 13.2 mL/100 g/min; control versus 2 h; P>0.1, control versus 24 h; P>0.1), blood pressure, and heart rate in gerbils (data not shown), which were consistent with results from a previous study (Patel et al, 1999). However, administration of L-745,870 (210 mg/kg) resulted in increased NAIP-immunoreactivity in the hippocampal CA1 neurons at 2 h (Figures 5A and 5B). Further, the increase in NAIP-immunoreactivity peaked at 24 h and then gradually returned to baseline at 72 h after administration (Figures 5A-5D). Results of the Western blotting were consistent with those obtained by the immunohistochemical studies (Figure 5E).
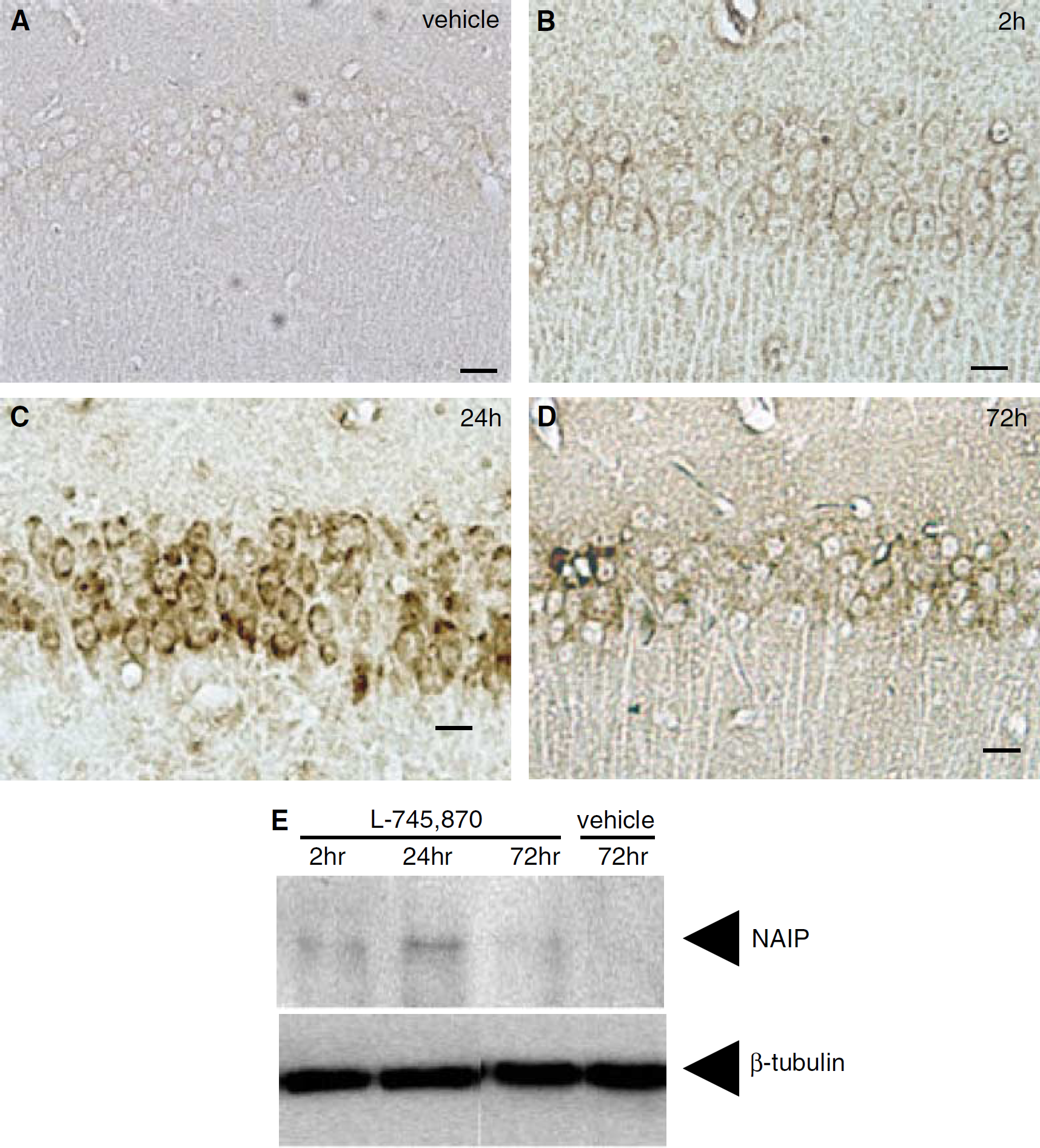
Upregulation of neuronal apoptosis inhibitory protein (NAIP) by L-745,870 in CA1 neurons. Adult gerbils were treated with vehicle (
Administration of L-745,870 Attenuates Ischemia-Induced neuronal Cell Death in the Hippocampal CA1 Region
To determine whether administration of L-745,870 exerted neuroprotective against ischemic insults in vivo, gerbil forebrain ischemic models were established by transient BCCAO, which induces a selective loss of CA1 pyramidal cells in the hippocampus (Kirino and Sano, 1984). Because the time course of changes in NAIP expression occurred over a 72 h period after administration of L-745,870, the experimental protocol used induction of a relatively severe ischemic condition (10 min BCCAO), followed by the analysis of delayed neuronal loss in the CA1 region over the subsequent 72 h. At 72 h after the ischemic insult, vehicle treated animals showed widespread hippocampal CA1 neuron death with very few surviving neurons present in the pyramidal layer (Figures 6A and6B). Further, a large number of glial cells were also observed in this area (Figures 6A and 6B). However, these animals showed little or no cell loss in the CA3 region or dentate gyrus (data not shown).
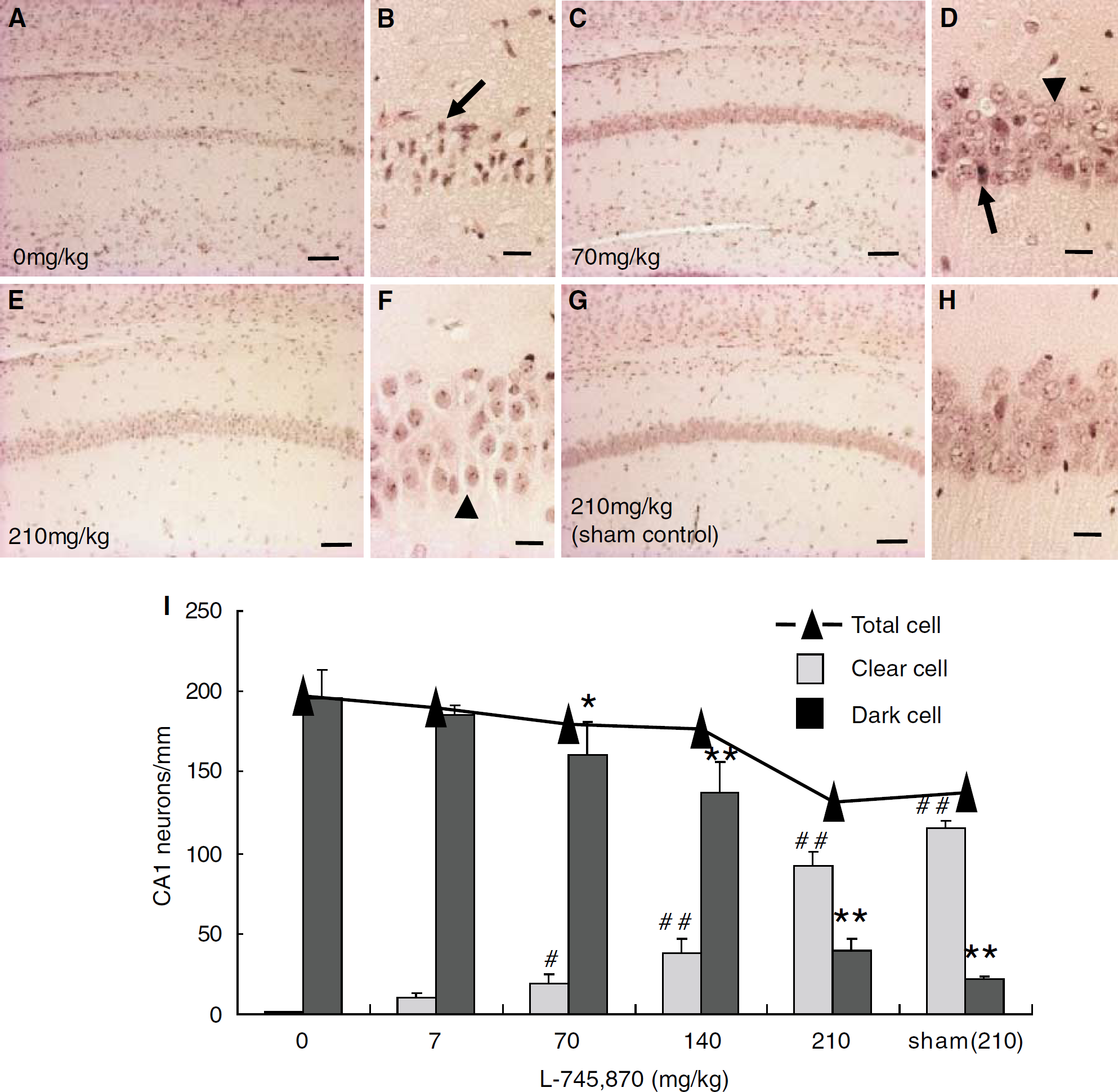
Effects of L-745,870 on ischemia-induced neuronal cell death in gerbils. Adult gerbils were treated with L-745,870 and then subjected to forebrain ischemia (bilateral common carotid artery occlusion (BCCAO) for 10 mins;
Administration of L-745,870 at 60 mins before the ischemic surgery exerted significant neuroprotective effects against ischemic insults in a concentration-dependent manner. A decreased dosage of L-745, 870 (7 mg/kg) showed the most modest protection in the hippocampus CA1 neurons (data not shown), whereas medium to high dosages (70 and 210 mg/kg) exhibited prominent protective effects (Figures 6C-6F). The quantitative scoring of the neuronal cell damage was consistent with these results (Figure 6I). Moreover, the highest dose of L-745,870 (210 mg/kg) was not associated with any CA1 neuron toxicity (Figures 6G and 6H). Further, these protective effects were significant even at 5 days after reperfusion in this experimental condition, despite the fact that progressive cell death in CA1 neurons was observed (data not shown). Together, these results indicate that L-745,870 inhibits progression of the ischemia-induced CA1 neuron death.
Hippocampal CA1 Neurons Rescued by the Administration of L-745,870 Show Strong NAIP-Immunoreactivity
To investigate whether L-745,870-mediated hippocampal CA1 neuronal protection was associated with elevation of neuronal NAIP levels, immunohistochemical and Western blot analyses of endogenous NAIP were performed in CA1 neurons using an NAIP antibody. After 72 h of reperfusion, the CA1 neurons showed significant enhancement of the NAIP-immunoreactivity in the rescued neurons in a dose-dependent manner (Figures 7A-7F). Results of the Western blotting were consistent with results obtained by immunohistochemical studies (Figure 7H). In contrast, L-745,870 produced no detectable upregulation of XIAP-immunoreactivity in the rescued hippocampus (Figures 7G and 7H). These results show that L-745,870 selectively upregulates NAIP in vivo, which represent a potential mechanism by which L-745,870 exerts a neuroprotective effect against ischemia in hippocampal CA1 neurons.
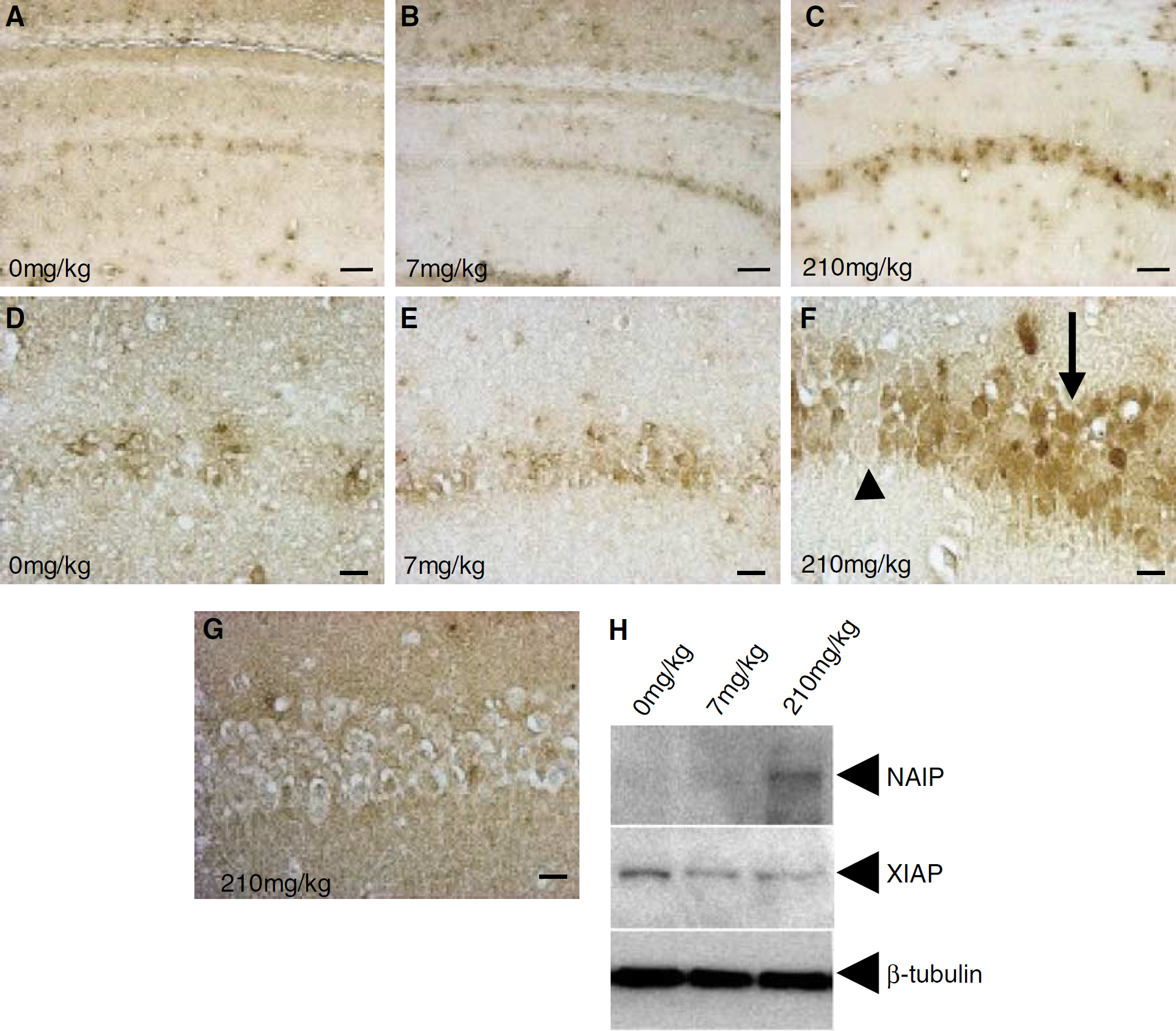
Effect of L-745,870 on expression of neuronal apoptosis inhibitory protein (NAIP) in CA1 neurons of ischemic gerbils. Adult gerbils were treated with L-745,870 and then subjected to forebrain ischemia (bilateral common carotid artery occlusion (BCCAO) for 10 mins;
Discussion
Neuronal apoptosis inhibitory protein (BIRC1) is the founding member of the IAP family of proteins, and has been shown to inhibit apoptosis of neurons and other types of the cell in vitro and in vivo. Increases in NAIP, by either viral-mediated NAIP gene transfer or by enhancement of endogenous levels, results in the attenuation of ischemic neuronal cell death (Xu et al, 1997). Further, ectopic NAIP expression enhances rescue of motor neurons from peripheral nerve axotomy (Perrelet et al, 2000) and leads to preservation of nigrostriatal dopaminergic neurons in the intrastriatal 6-OHDA rat Parkinson's disease model (Crocker et al, 2001). Neuronal apoptosis inhibitory protein also contributes to motor neuron survival through intracellular signaling of GDNF (Perrelet et al, 2002). Ischemic neuronal injury is associated with excessive generation of reactive oxygen species (ROS) and oxidative stress in the brain (Chan, 1994; Mattson et al, 2001; Friedlander, 2003), and the present study showed that NAIP suppresses neuronal cell death by exerting an antiapoptotic function against oxidative stress.
Most tissues and cells, with the exception of hematopoietic tissues, express NAIP in very low levels (Yamamoto et al, 1999). Thus, upregulation of endogenous NAIP in neuronal cells may represent a potent therapeutic strategy for prevention of neurodegeneration. Among the 30 NAIP upregulating compounds identified, two compounds, L-745,870 (dopamine D4 receptor antagonist) and bromocriptine (dopamine D2 receptor agonist; data not shown), exerted particularly prominent protection against neurodegeneration in an ischemia gerbil model. A previous study showed that bromocriptine protected neuronal cells from oxidative stress-induced apoptosis (Schapira, 2002), while another study showed that a number of other dopamine D2 receptor agonists attenuated neuronal cell death under ischemic conditions (Liu et al, 1995). L-745,870 was originally identified as a specific antagonist for the dopamine D4 receptor and as a drug candidate for antipsychotic treatment because of its excellent oral bioavailability and brain penetration (Patel et al, 1997). The present study showed the novel finding that L-745,870 specifically elevated the endogenous NAIP level and enhanced neuronal cell resistance to oxidative stress-induced apoptotic cell death and ischemic neurodegeneration.
The dopamine D4 receptor is a G-protein-coupled receptor that shares sequence homology with the D2 and D3 receptors and is classified as a member of the dopamine D2-like receptors group (Baldessarini, 1997). However, the action of L-745,870 in the present study is likely not mediated via dopaminergic receptors, because the antiapoptotic properties of L-745,870 against oxidative stress-induced cell death were observed in both differentiated dopaminergic SH-SY5Y cells, in which several types of dopamine receptors are expressed (Kamakura et al, 1997), and in nonneuronal cells, which do not express these receptors. Indeed, recent reports suggest that the dopamine agonists, bromocriptine and pergolide, act as free radical scavengers (Yoshikawa et al, 1994; Sam and Verbeke, 1995; Grünblatt et al, 1999) and exert their neuroprotective effects in nonreceptor-mediated fashions (Uberti et al, 2002). The present finding that L-745,870 may exert its neuroprotective via increases in NAIP, either by increasing its expression or stabilization, may provide a mechanism by which all of these effector molecules exert their effects.
The present study showed that L-745,870 upregulated NAIP but not other antiapoptotic proteins. Further, L-745,870 specifically protected both neuronal and nonneuronal cultured cells from apoptosis induced by several oxidative stressors, including DMNQ, menadione, α-naphthoquinone, and H2O2. This increase in NAIP likely mediates the protective effect of L-745,870 because reduction of NAIP expression with RNAi inhibited the neuroprotective effect. Recent studies have shown that NAIP suppresses caspase-dependent and -independent apoptosis (Deveraux et al, 1997, 1998; Roy et al, 1997; Seshagiri and Miller, 1997; Takahashi et al, 1998; Maier et al, 2002), while we previously showed that the antiapoptotic effect of NAIP was mediated by the caspase-3-independent pathway (Sakai et al, unpublished). Further studies to characterize the molecular mechanisms of NAIP upregulation and subsequent inhibition of oxidative stress-induced cell death would be of benefit.
It is notable that L-745,870 slightly but consistently downregulates the levels of both XIAP and cIAP-1 in differentiated SH-SY5Y cells. Recent studies have shown that a group of the ring finger-containing members of IAP family protein, such as XIAP and cIAP, can function as ubiquitin protein ligases, and regulates the levels of not only their target proteins but also themselves through ubiquitylation (Yang et al, 2000; Salvesen and Duckett, 2002). Thus, it is possible that L-745,870 may affect the stability of these IAP proteins via regulating the proteasome-dependent protein degradation. Equally likely is that this compound directly or indirectly modulates the expression of these genes and/or proteins in SH-SY5Y cells. Further studies will be needed to clarify the molecular mechanism underlying this inhibitory effect. Nevertheless, as the downregulation of XIAP and cIAP-1 is expected to exert the opposite effect to antiapoptosis, the decreases in XIAP and cIAP-1 observed in this study may not be a primary determinant for the antiapoptotic function associated with L-745,870.
We also showed that L-745,870 attenuated ischemia-induced CA1 neuronal cell death with concomitant increase in the NAIP expression in rescued CA1 neurons. Indeed, selective upregulation of NAIP might mediate a broad range of protection against oxidative stress-induced cell death. For example, a previous study has shown that the small molecule alkaloid, K252a, which is structurally quite different from L-745,870, upregulated NAIP levels and exerted a significant protective effect against ischemic damage in hippocampal CA1 neurons (Xu et al, 1997).
In conclusion, the dopamine D4 receptor antagonist, L-745,870, exerts a potent neuroprotective effect against ischemia-induced cell death via increases in NAIP. Since L-745,870 is clinically well tolerated (Bristow et al, 1997), this compound may represent an effective therapeutic strategy for the clinical prevention of neuronal cell death after ischemia. Future studies not only on the molecular mechanism by which L-745,870 induces the NAIP expression but also on the effectiveness of this compound in various ischemic conditions will clarify therapeutic potentials of L-745,870 in the treatment of several types of acute as well as chronic neurodegenerative diseases caused by oxidative stress. Further, our NAIP-ELISA-based drug screening may facilitate the discovery of novel neuroprotective compounds.
Footnotes
Acknowledgements
The authors thank Dr Kenji Yamamoto and all the members of our laboratory for their scholarly input.