
Abstract
Nuclear factor-kappa B (NF-κB) is activated by oxidative stress such as that induced by transient focal cerebral ischemia (tFCI). Whether NF-κB has a role in cell survival or death in stroke is a matter of debate. We proposed that the status of oxidative stress may determine its role in cell death or survival after focal ischemia. To characterize the coordinated expression of genes in NF-κB signaling after mild cerebral ischemia, we investigated the temporal profile of a NF-κB-pathway-focused DNA array after 30 mins of tFCI in wild-type (WT) mice and human copper/zinc-superoxide dismutase transgenic (SOD1 Tg) mice that had a significantly reduced level of superoxide. Differentially expressed genes among 96 NF-κB-related genes were further confirmed and compared in the WT and SOD1 Tg mice using quantitative polymerase chain reaction, Western blotting, and immunohistochemistry. Persistent upregulation of NF-κB seen at 7 days in the WT mice was decreased in the SOD1 Tg mice. Lymphocytotrophic cytokine genes such as interleukin-2, interleukin-12, and interferon-α1 were increased in the SOD1 Tg mice compared with the WT mice after tFCI. In addition, antiapoptosis factors bcl-2 and tumor necrosis factor receptor-associated factor 1 rapidly increased in the SOD1 Tg mice compared with the WT mice. This study indicates that reduced oxidative stress by SOD1 overexpression increased NF-κB-related rapid defenses, such as immune response and antiapoptosis factors, and prevented brain damage after tFCI-induced oxidative stress.
Keywords
Introduction
Nuclear factor-kappa B (NF-κB) is induced by different stimuli, such as reactive oxygen radicals, ultraviolet irradiation, proinflammatory cytokines, lymphocytokines, B- or T-cell activation, and growth factors (Hazan et al, 1990; Li and Verma, 2002; Osborn et al, 1989; Schreck et al, 1991). Active NF-κB targets the response element of the promoters of NF-κB target genes (Pahl, 1999) and controls transcription of hundreds of target genes. Nuclear factor-kappa B plays a critical role in regulating cell survival by suppressing apoptosis (Barkett and Gilmore, 1999). Studies have suggested that NF-κB promotes cell death in focal ischemia (Schneider et al, 1999; Zhang et al, 2005). We have reported that oxidative stress increases NF-κB DNA binding activity and causes a transient loss of the IκB kinase (IKK) complex (IKKα, β, and γ), the upstream component of NF-κB signaling, after 30 or 60 mins of middle cerebral artery (MCA) occlusion and reperfusion (Huang et al, 2001; Song et al, 2005). However, other investigators have shown a decrease in NF-κB activity after an initial increase at 3 h (Irving et al, 2000). The loss of NF-κB activity could play a part in the development of tumor necrosis factor (TNF)-induced cytotoxicity in cerebral ischemia, providing further evidence for a neuroprotective role for NF-κB (Botchkina et al, 1999). The difference in these findings may result from the nature of the ischemic injuries (permanent versus transient, duration and severity of ischemia and reperfusion) and from the interaction of other signaling pathways with oxidative stress. The antiapoptotic function of NF-κB in the nervous system has been extensively studied by Mattson et al (Barger and Mattson, 1996; Yu et al, 1999; Mattson et al, 2000), and these investigators have reported that the NF-κB subunit plays a role in regulating neuronal survival in neurodegenerative diseases, probably by targeting genes such as manganese-superoxide dismutase, bcl-2, and bcl-x.
Many genes associated with the dynamic process of cerebral ischemia have been identified; some are protective against ischemic insult and others contribute to delayed cell death. We wanted to determine which genes are involved simultaneously in oxidative stress and the NF-κB signaling pathway after cerebral ischemia in wild-type (WT) mice and human copper/zinc-superoxide dismutase transgenic (SOD1 Tg) mice. Among the 96 NF-κB signaling genes, there was a significant change in genes related to the NF-κB family, to cytokines, their receptors, antiapoptosis factors, and proinflammatory complement genes. Overexpression of SOD1 induced upregulation of lymphocytotrophic cytokines and antiapoptotic genes at early time points after transient focal cerebral ischemia (tFCI).
Although the immune system was once thought to be independent of cerebral neural influence, recent studies have demonstrated the elaborate interactions between the immune and nervous systems (Gendron et al, 2002; Steinman, 2004). Interleukin (IL)-2 is implicated in the generation of effector phenotypes from antigen-stimulated T cells (Wagner et al, 1980). The T-cell growth factor IL-2 is clearly an important lymphocytotrophic cytokine and acts through IL-2 receptors (IL-2R) in the brain; IL-2R and the IL-2 proteins are present in both the normal brain and the brain undergoing inflammatory processes (Merrill, 1990). No difference has been reported in IL-2-gene expression between control and ischemic brains (Zhai et al, 1997), and there is little role for cell-mediated immunoreaction by IL-2 in the pathogenesis of cerebral vasospasm (Nagata et al, 1993). However, the role of IL-2 in the ischemic brain is not fully elucidated. Our present results show that upregulation of IL-2 and IL-2Rα mRNA after tFCI was significantly higher in SOD1 Tg mice than in WT mice at early time points after reperfusion. Interleukin-2 played a major role in immune cell proliferation as well as in immune cell protection against cell death by induction of antiapoptotic mechanisms during an immune reaction (Ellery and Nicholls, 2002).
The purpose of the present series of experiments was to characterize and compare the temporal gene profile between WT and SOD1 Tg mice after 30 mins of tFCI. We used an NF-κB pathway-focused gene array containing 96 genes related to NF-κB-mediated signal transduction (Ghosh et al, 1998). Many genes, including the NF-κB family, cytokines and their receptors, adaptor proteins, and signal transduction kinases, were differentially expressed after 30 mins. After we confirmed gene changes using quantitative real-time polymerase chain reaction (QPCR), we further focused on several genes that showed similar profiles in both a complementary DNA (cDNA) array and in QPCR experiments. Selected genes were analyzed and compared between the WT and SOD1 Tg mice by Western blot analysis and immunohistochemistry. Our results show that lymphocytotrophic cytokines and antiapoptotic factors were also highly expressed in SOD1 Tg mice at early time points after tFCI.
Materials and methods
Focal Cerebral Ischemia
Experiments were performed in accordance with National Institutes of Health guidelines and were approved by Stanford University's Administrative Panel on Laboratory Animal Care. Tg mice of the TgHS/SF-218 strain, which carries the SOD1 gene with a CD-1 background, were derived from the founder stock (Epstein et al, 1987). There were no observable phenotypic differences between the Tg mice and their WT normal littermates. Tg mice (3-month-old males, 35 to 40 g) with a three-fold overexpression of SOD1 activity in brain cells (Epstein et al, 1987), and WT mice were subjected to tFCI and reperfusion in a randomized, blind manner. CD-1 mice (40 to 45 g), used in a study with χ-phenyl-n-tert-butyl-nitrone, were purchased from Charles River Laboratories (Wilmington, MA, USA). Transient focal cerebral ischemia was induced by intraluminal MCA occlusion with a nylon monofilament suture as described previously (Yang et al, 1994). The mice were anesthetized with 2.0% isoflurane in 30% oxygen and 70% nitrous oxide using a face mask. Rectal temperature was controlled at 37°C with a homeothermic blanket. The left femoral artery was cannulated for measurement of blood pressure and arterial blood gases, with samples for analysis being taken immediately after cannulation, 10 mins after occlusion, and 10 mins after reperfusion. The left common carotid artery was exposed and the left external carotid artery and its branches were electrocoagulated. An 11.0-mm 5-0 surgical monofilament nylon suture, blunted at the tip, was introduced into the left internal carotid artery through the external carotid artery stump. After 30 mins of proximal MCA occlusion, blood flow was restored by removal of the suture. To allow for reperfusion, the occluding filament was gently withdrawn. The animals were then allowed to recover from anesthesia. In sham-operated mice, the same surgical procedure was performed, except that the filament was not advanced to occlude the MCA. Physiologic parameters were monitored throughout the studies and values were the same as previously reported (Fujimura et al, 1999).
RNA Preparation
The animals were deeply anesthetized with isoflurane 1, 6, and 24 h and 7 days after restoration of cerebral blood flow (n = 4). The sham-operated animals underwent exposure of carotid arteries without insertion of the nylon suture (n = 4). After decapitation, the brains were quickly removed and the perfused MCA territory was obtained. It was immediately frozen in powdered dry ice and kept at −80°C until use. Extraction of total RNA was performed using TRIzol reagent (15596-026; Invitrogen, Carlsbad, CA, USA) in accordance with the instructions of the manufacturer.
Complementary DNA Array
Single-strand cDNA was synthesized by reverse transcription of the RNA (3 μg) at 42°C for 90 mins with the use of biotin-16-dUTP (1093070; Roche, Mannheim, Germany), M-MLV reverse transcriptase (M1701; Promega, Madison, WI, USA), and primer mix from the NF-κB Signaling Pathway Genearray kit (MM-016N; SuperArray, Bethesda, MD, USA). Labeling, hybridization, and washing of the membrane were performed according to the manufacturer's instructions. Subsequently, the membranes were treated with CDP-star (MS050R; Applied Biosystems, Bedford, MA, USA) and exposed on X-ray film. The film was scanned and densitometries were performed with the use of Multi-Analyst software (ST32151N; Bio-Rad, Hercules, CA, USA). The spot density of the internal control housekeeping genes (i.e., β-actin and RPL13A) on the membrane was also determined and the ratio of each gene to the housekeeping gene was calculated. This ratio was obtained from each animal and the data from the ischemic brains at each time point were compared with those from the sham-operated brains.
Quantitative Real-Time Polymerase Chain Reaction
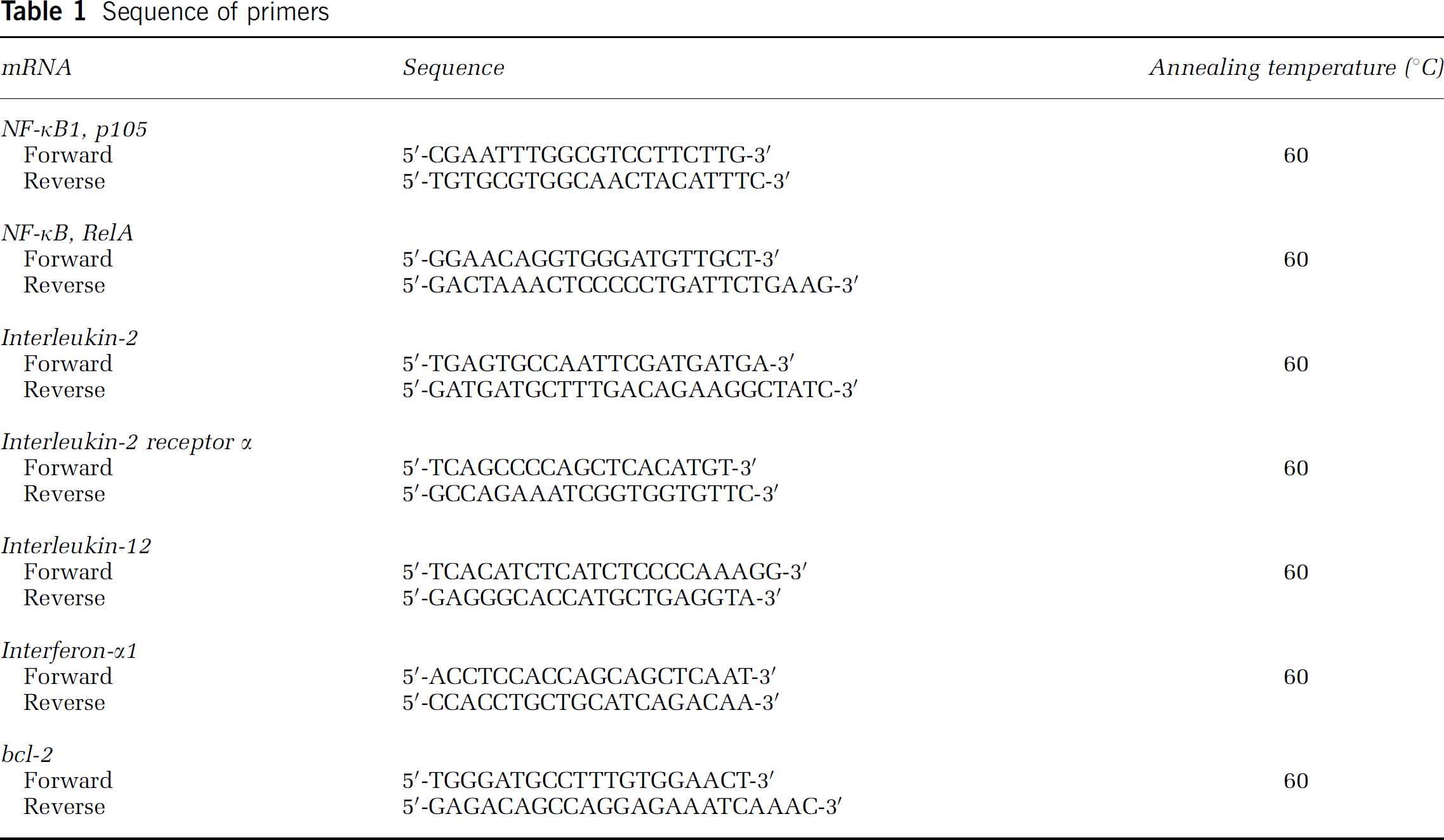
All QPCRs were performed using the Mx3000 PCR System (Stratagene, La Jolla, CA, USA). All QPCR amplifications were performed using the recommended buffer supplied by the manufacturer. The QPCR mixture consisted of 2 μL of each template (n = 4), 5 pmol of each primer (Table 1), and QuantiTect SYBR Green (204143; Qiagen, Valencia, CA, USA) including ROX as an internal control. Triplicate reactions were performed for each template amount. QPCR cycling conditions were as follows: initial denaturation at 95°C for 2 mins, followed by 40 cycles at 95°C for 15 secs, 1 min annealing at 60°C, and a 30-sec extension at 72°C. Data analysis was performed using Mx3000 software (Stratagene). CTs were determined using the signal/noise ratio set to standard deviation above background-subtracted mean fluorescence values.
Sequence of primers
Western Blotting
The animals were decapitated 1, 6, and 24 h and 7 days after reperfusion under deep anesthesia with isoflurane (n = 4). Samples from the sham-operated animals were also obtained (n = 4). Samples were obtained from the MCA territory brain tissue on the ischemic sides, including the striatum and cortex, and were quickly frozen in powdered dry ice and kept at −80°C until use. For protein extraction, the tissue was homogenized by gently douncing 35 times in a glass tissue grinder (Wheaton, Millville, NJ, USA) in 7 volumes of cold suspension buffer (20 mmol/L HEPES potassium hydroxide (pH 7.5), 250 mmol/L sucrose, 10 mmol/L potassium chloride, 1.5 mmol/L magnesium chloride, 1 mmol/L edetic acid, 1 mmol/L ethyleneglycotetraacetic acid, and 0.7% protease inhibitor cocktail (P8340; Sigma-Aldrich, St Louis, MO, USA)). The homogenate was centrifuged at 10,000 g for 15 mins at 4°C, and the supernatant was used for this study. The cytosolic and nuclear fractions were prepared from the ischemic brain using ProteoExtract™ (539790; Calbiochem, La Jolla, CA, USA). Assays to determine the protein concentration were performed by comparison with a known concentration of bovine serum albumin using a kit (23227; Pierce, Rockford, IL, USA). A lysate equivalent to 10 μg of protein from each fraction was run on a sodium dodecyl sulfate-polyacrylamide gel for 120 mins at 100 V, together with a size marker (RPN800; GE Healthcare, Piscataway, NJ, USA). The protein on the gel was subsequently transferred onto a polyvinylidene fluoride transfer membrane (LC2002; Invitrogen) in a buffer containing methanol, glycine, Tris base, and sodium dodecyl sulfate, after which the membrane was placed in 5% powdered milk in phosphate-buffered saline with 0.1% Tween-20 (Bio-Rad) for 1 h to block nonspecific binding. It was then incubated with primary antibodies for 24 h at 4°C. The primary antibodies used were 1:200 dilution of rabbit polyclonal antibodies against p50 (ab7971; Abcam, Cambridgeshire, UK), p65 (3034; Cell Signaling Technology, Cambridge, MA, USA), and the X-chromosome-linked inhibitor of apoptosis protein (XIAP) (2042; Cell Signaling Technology), 1:200 dilution of mouse monoclonal antibody against bcl-2 (610538; BD Biosciences, San Diego, CA, USA) and TNF receptor-associated factor (TRAF) 1 (AP1002; Oncogene Research Products, San Diego, CA USA), 1:200 dilution of rat monoclonal antibody against IL-2Rα (CBL1334; Chemicon International, Temecula, CA, USA), 1:400 dilution of rabbit polyclonal antibody against TFIID (SC-204; Santa Cruz Biotechnology, Santa Cruz, CA, USA), 1:3,000 dilution of α-tubulin monoclonal antibody (T5168; Sigma-Aldrich), or 1:5000 dilution of β-actin monoclonal antibody (A5411; Sigma-Aldrich). After washing, the membrane was incubated with horseradish peroxidase-conjugated anti-mouse immunoglobulin G (Amersham International, Buckinghamshire, UK) or horseradish peroxidase-conjugated anti-rabbit immunoglobulin G at 1:5,000 dilution for 60 mins. The signal was then detected with a chemiluminescent kit (Pierce). Multi-Analyst 1.0.2 software (Bio-Rad) was used for data analysis.
Statistical Analysis
The data are expressed as mean ± s.d. Comparisons among multiple groups were performed by one-way ANOVA with appropriate Bonferroni or Dunnet tests (GraphPad Prism; Oberlin, San Diego, CA, USA). P-values less than 0.05 were considered statistically significant.
Results
Comparison of mRNA Expression Between Wild-Type and Copper/Zinc-Superoxide Dismutase Transgenic Mouse Brains after 30 mins of Transient Focal Cerebral Ischemia
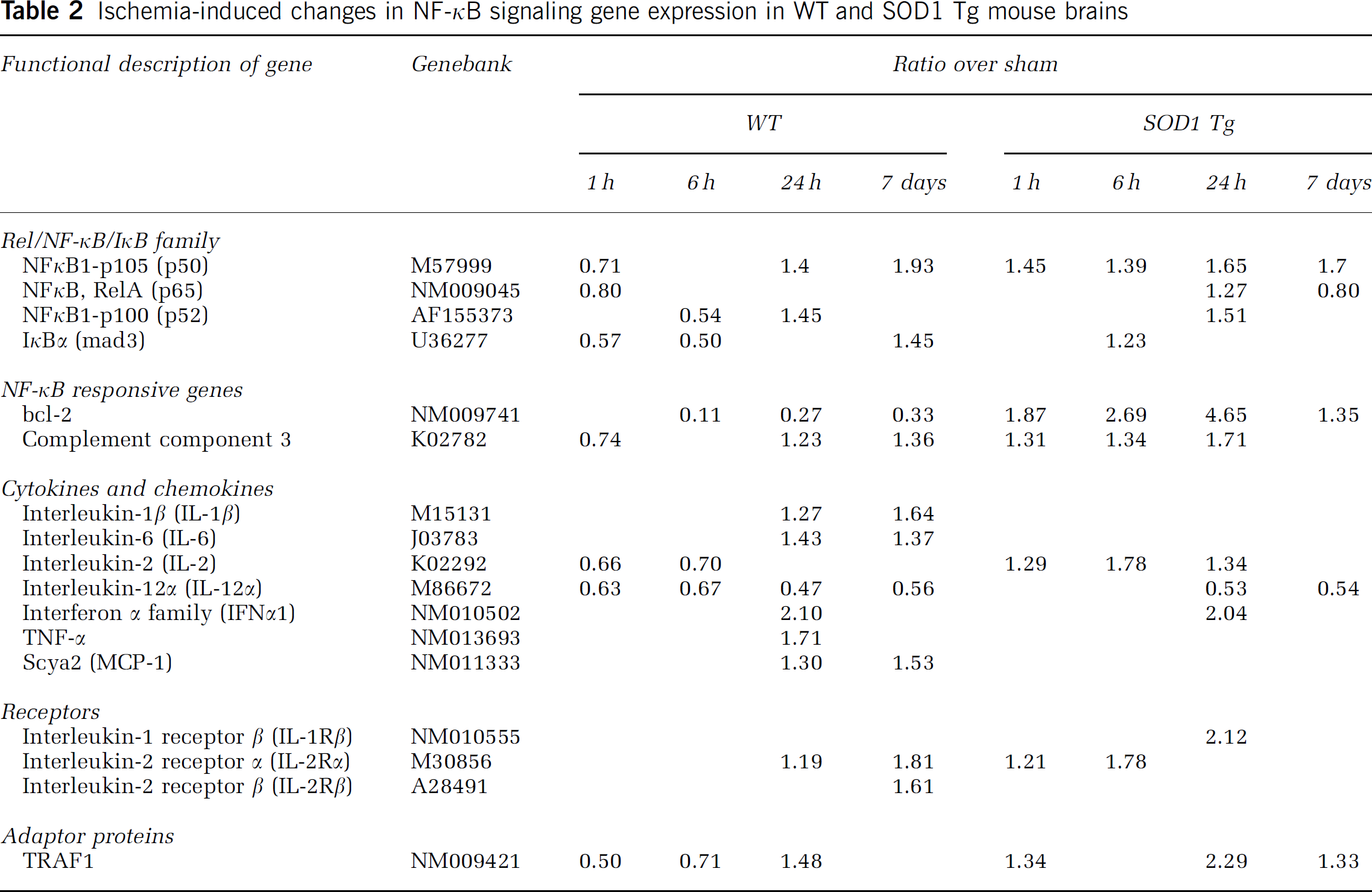
Oxidative stress is a major source of injury from cerebral ischemia and reperfusion (Bowler et al, 2002). When the MCA territory of the WT and SOD1 Tg mouse brains was measured after tFCI using mitochondrial dehydrogenase staining with triphenyltetrazolium chloride, the infarct volumes after 30 mins of occlusion were 24.3 and 16.42 mm3 in the WT mice and the SOD-overexpressing animals, respectively (data not shown). The SOD1 Tg mice had an infarct volume that was 32% less than that in the WT mice after 30 mins of MCA occlusion and reperfusion. To analyze differential expression profiles between the WT and SOD1 Tg mice after tFCI, we used mRNA extracted from the MCA territory for the NF-κB-pathway-focused cDNA array, where 96 cDNAs were spotted on nylon membranes. Table 2 summarizes ratio changes over the sham-operated mice, with a temporal profile of the WT and SOD1 Tg mice after cerebral ischemia. After 30 mins of ischemia in the WT mice, some genes of NF-κB signaling, the NF-κB family, cytokines, receptors, and adaptor proteins were down-regulated at the early times of 1 and 6 h, while the majority of the other genes showed increased expression at 24 h and 7 days. Among the NF-κB-family genes, NF-κB1 p105 showed increased expression throughout the 7 days after an early decrease at 1 h. However, in the SOD1 Tg mice, NF-κB1 p105 mRNA maintained a similar level after tFCI. In the WT mice, marked upregulations of inflammatory cytokines and chemokines, IL-1β, IL-6, TNFα, and monocyte chemoattractant protein-1 (MCP-1), were observed 24 h and 7 days after 30 mins of tFCI. In contrast to the WT mice, IL-1β, IL-6, TNFα, and MCP-1 did not increase in the SOD1 Tg mice. Compared with the WT mice, IL-2, the T-lymphocyte stimulator, increased from 1 to 24 h in the SOD1 Tg mice. The striking upregulation was measured with the spots containing the cDNA for IL-1Rβ at 24 h in the SOD1 Tg mice. IL-1Rβ functioned as a negative or ‘decoy' receptor anti-inflammatory (Colotta et al, 1993, 1994). With regard to adaptor proteins, TRAF1 was down-regulated 1 and 6 h after tFCI and upregulated at 24 h. The mRNA of TRAF1, an antiapoptotic factor (Tsitsikov et al, 2001), especially showed a rapid decrease in the WT mice. In contrast, the SOD1 Tg mice showed an increase after tFCI. Proinflammatory complement component 3 increased at 24 h and 7 days in the WT mice. Persistent upregulation of complement component 3 mRNA in the WT mice was reduced in the SOD1 Tg mice at 7 days. Cerebral ischemia in the WT mice caused an increase in proinflammatory cytokines, but a decrease in antiapoptotic and lymphocytotrophic genes. However, reduced oxidative stress caused by overexpression of SOD1 increased expression of antiapoptotic and lymphocytotrophic genes. Several distinctive features were noted in the WT animals compared with the SOD1 Tg mice, that is, that SOD1 caused a decrease in expression of harmful genes and SOD1 increased survival genes induced by ischemia. Selected genes were further confirmed and compared between the WT and SOD1 Tg mice using QPCR, Western blotting, and immunohistochemistry.
Ischemia-induced changes in NF-κB signaling gene expression in WT and SOD1 Tg mouse brains
Copper/Zinc-Superoxide Dismutase Reduced the Persistent Upregulation of Nuclear Factor-Kappa B p105 and RelA mRNA after Transient Focal Cerebral Ischemia
The mRNA upregulation of NF-κB p105 (for NF-κB subunits, p50) and RelA (for NF-κB subunits, p65) was measured by QPCR throughout the 7 days after tFCI in the WT mice (Figures 1A and 1B). Nuclear factor-kappa B p105 and RelA, particularly, were increased by 2.7- and 2.3-fold, respectively, in the WT mice at 7 days. However, in the SOD1 Tg mice, the levels of NF-κB p105 and RelA mRNA that were increased at 6 and 24 h returned to the basal level at 7 days (Figures 1A and 1B). The protein levels of p50 and p65 in the cytosol and nucleus were analyzed by Western blotting. In accordance with the QPCR data, we found an increase in the nuclear p50 and p65 proteins in the ischemia-damaged brains of the WT mice throughout the 7 days (Figures 2A and 2B). The nuclear p50 level was significantly increased by 4.3-fold 7 days after tFCI in the WT mice and the nuclear p65 level was slightly increased. Whereas, in the SOD1 Tg mice, NF-κB and the p50 and p65 proteins did not show significant increases in either the nucleus or cytosol (Figures 2A and 2B).
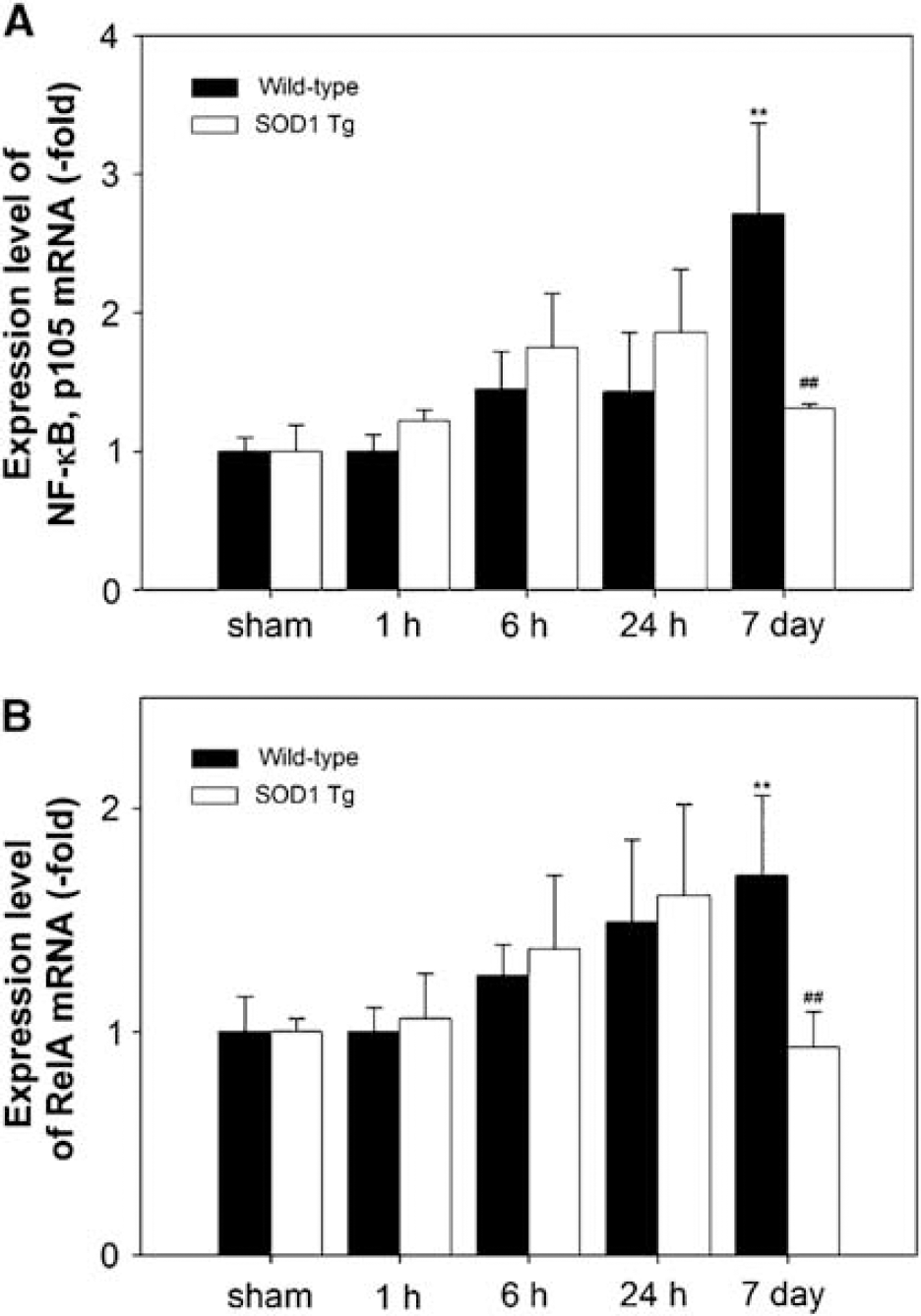
Quantitative real-time polymerase chain reaction analysis of temporal expression of NF-κB p105 (for NF-κB subunits, p50) and RelA (for NF-κB subunits, p65) after 30 mins of tFCI in WT and SOD1 Tg mice. QPCR was performed using total RNA extracted from the ischemic brains of the WT and SOD1 Tg mice. mRNA expression was detected using primer sets listed in Table 1. Time points include sham, and 1, 6, and 24 h, and 7 days after tFCI. (
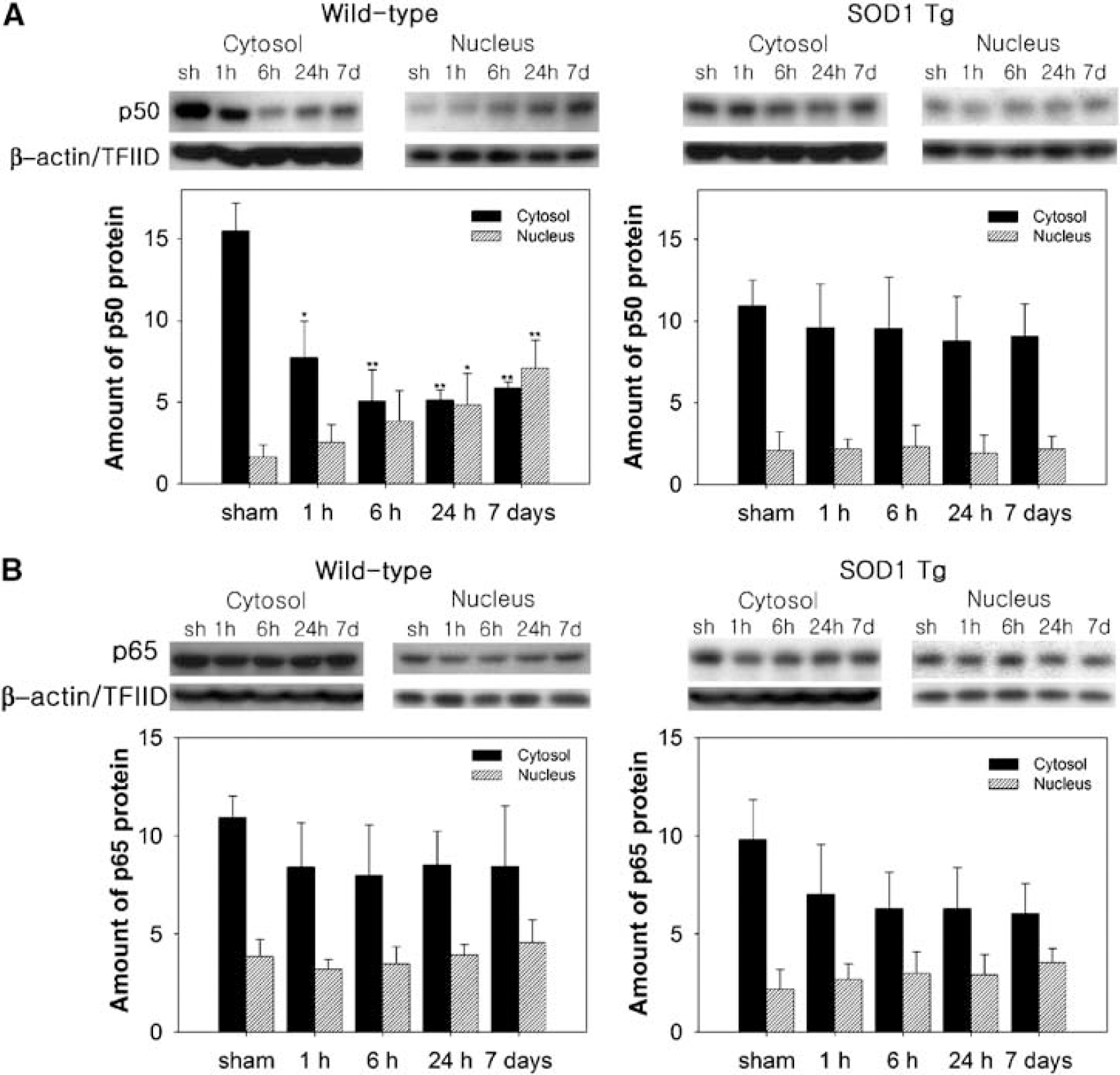
Nuclear translocation of NF-κB subunits p50 and p65 after 30 mins of tFCI in WT and SOD1 Tg mice. Nuclear and cytosolic extracts at different time points after tFCI were prepared and separated by sodium dodecyl sulfate gel electrophoresis. Western blot analysis revealed that p50 and p65 immunoreactivity was evident as a single band of molecular mass of 50 and 65 kDa, respectively. β-Actin and TFIID were used as internal controls for the cytosol and nucleus, respectively. (
Overexpression of Copper/Zinc-Superoxide Dismutase Induced Transient Upregulation of IL-2, IL-2Rα, IL-12, and Interferon-α1 after Transient Focal Cerebral Ischemia
The transcription factor NF-κB is implicated in various aspects of T-cell development and function, and IKK-induced NF-κB activation is essential for the generation and survival of mature T-cells (Schmidt-Supprian et al, 2003). T-cell growth factor IL-2 mRNA was upregulated in the WT mice 24 h and 7 days after tFCI (Figure 3A). Whereas, in the SOD1 Tg mice, IL-2 mRNA increased rapidly 1 h after tFCI, remained at a high level by 24 h, and decreased at 7 days. Overexpression of SOD1 induced rapid upregulation of IL-2 mRNA, however, in the WT mice, IL-2 increased gradually. IL-2 increases the expression of IL-2R to achieve its biologic effects, which leads to the generation of the high affinity receptors (Harel-Bellan et al, 1986). The immediate upregulation of IL-2Rα mRNA was measured in the WT and SOD1 Tg mice 1 h after tFCI (Figure 3B). Similar to the induction of IL-2 mRNA in the SOD1 Tg mice, these mice also had a higher level of IL-2Rα mRNA than the WT mice from 1 to 24 h (Figure 3B). Upregulation of the IL-2Rα protein was further confirmed by Western blot. In the SOD1 Tg mice, transient protein upregulation of IL-2Rα at 1 h was higher than in the WT mice after tFCI (Figure 3C). Expression of the mRNA of other lymphocytotrophic cytokines, such as IL-12 and interferon (IFN)-α, was increased after tFCI in the WT and SOD1 Tg mice (Figures 3D and 3E). Increases in IL-12 and IFN-α1 mRNA were 2.3- and 5.0-fold higher, respectively, at 6 h in the SOD1 Tg mice compared with the WT mice.
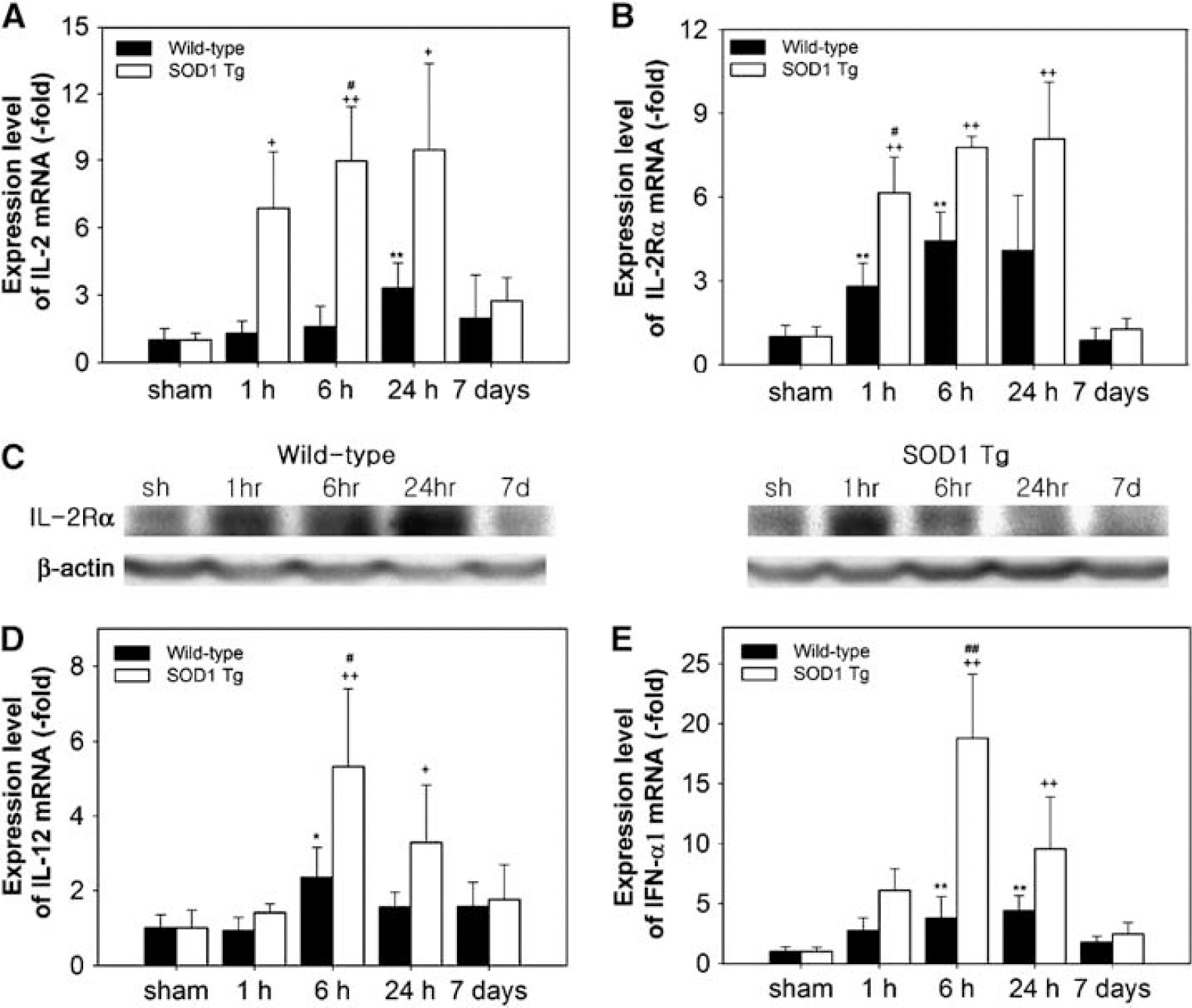
Changes in expression of IL-2, IL-2Rα, IL-12, and IFN-α1 after 30 mins of tFCI in WT and SOD1 Tg mice. (
Copper/Zinc-Superoxide Dismutase Increased Antiapoptotic Signals
Once NF-κB is activated via various types of stimulation, free NF-κB translocates to the nucleus, where it binds to specific response elements of target genes including antiapoptotic factors as well as cytokines. Cerebral ischemia induced 1.8- and 1.6-fold increases in bcl-2 mRNA levels at 1 and 6 h, respectively, in the ipsilateral brain of the WT mice (Figure 4A). In the SOD1 Tg mice, bcl-2 mRNA showed a 3.0-fold upregulation at 1 h, which was higher than in the WT mice (Figure 4A). Upregulation of bcl-2 mRNA has been reported in sublethal forebrain ischemia (Wu et al, 2003). We next examined whether the bcl-2 protein level is consistent with the mRNA level in the ischemic brains after tFCI. However, Western blots showed decreased immunoreactivity of the bcl-2 protein in the ischemic striatum, which was sensitive to injury at all time points in the WT mice after tFCI (Figures 4B and 4C). The increase in bcl-2 mRNA levels was not consistent with the decrease in protein levels in the WT mice after tFCI. In contrast to the WT mice, the bcl-2 proteins in the SOD1 Tg mice significantly increased in the striatum after tFCI (Figures 4B and 4C). Nuclear factor-kappa B can regulate genes that function at the earliest checkpoint to suppress TNF-α-mediated apoptosis (Wang et al, 1998). mRNA of the TNFα-mediated antiapoptotic component, TRAF1, was decreased 1 h after tFCI in the WT mice (Table 2). The cytosolic TRAF1 protein also rapidly decreased after tFCI in the WT mice (Figures 4B and 4C). However, in the SOD1 Tg mice, mRNA and the cytosolic protein level of TRAF1 increased after 30 mins of tFCI (Table 2 and Figure 4B). In response to TNF, recruitment of XIAP to the receptor complex through interaction with TRAF1 is required to inhibit apoptosis. Therefore, we measured the levels of the XIAP protein in the WT and SOD1 Tg mice after tFCI. XIAP protein levels were also decreased from 1 to 24 h in the striatum of the WT mice after tFCI (Figures 4B and 4C). However, overexpression of SOD1 increased expression of the XIAP protein in the striatum after tFCI (Figures 4B and 4C). Cells positive for terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling (TUNEL) were observed in the ischemic caudate of the WT mice (data not shown), bcl-2 was expressed in the ischemic caudate of the SOD1 Tg mice. An overlapped image of bcl-2 and TUNEL demonstrated that most bcl-2-immunopositive cells were not colocalized with TUNEL-positive cells (data not shown). Free radical species play an important role in apoptosis and in the pathogenesis of infarction after focal cerebral ischemia. After 30 mins of tFCI, infarct damage and apoptotic cells were significantly decreased in the SOD1 Tg mouse brains compared with the WT brains (data not shown), which was in accordance with the reduced infarction and apoptosis in SOD1 Tg mice after 1 or 3 h of tFCI (Kamii et al, 1996; Kondo et al, 1997; Yang et al, 1994). These results suggest that overexpression of SOD1 prevents apoptosis via a marked upregulation of antiapoptotic components such as bcl-2, TRAF1, and XIAP.
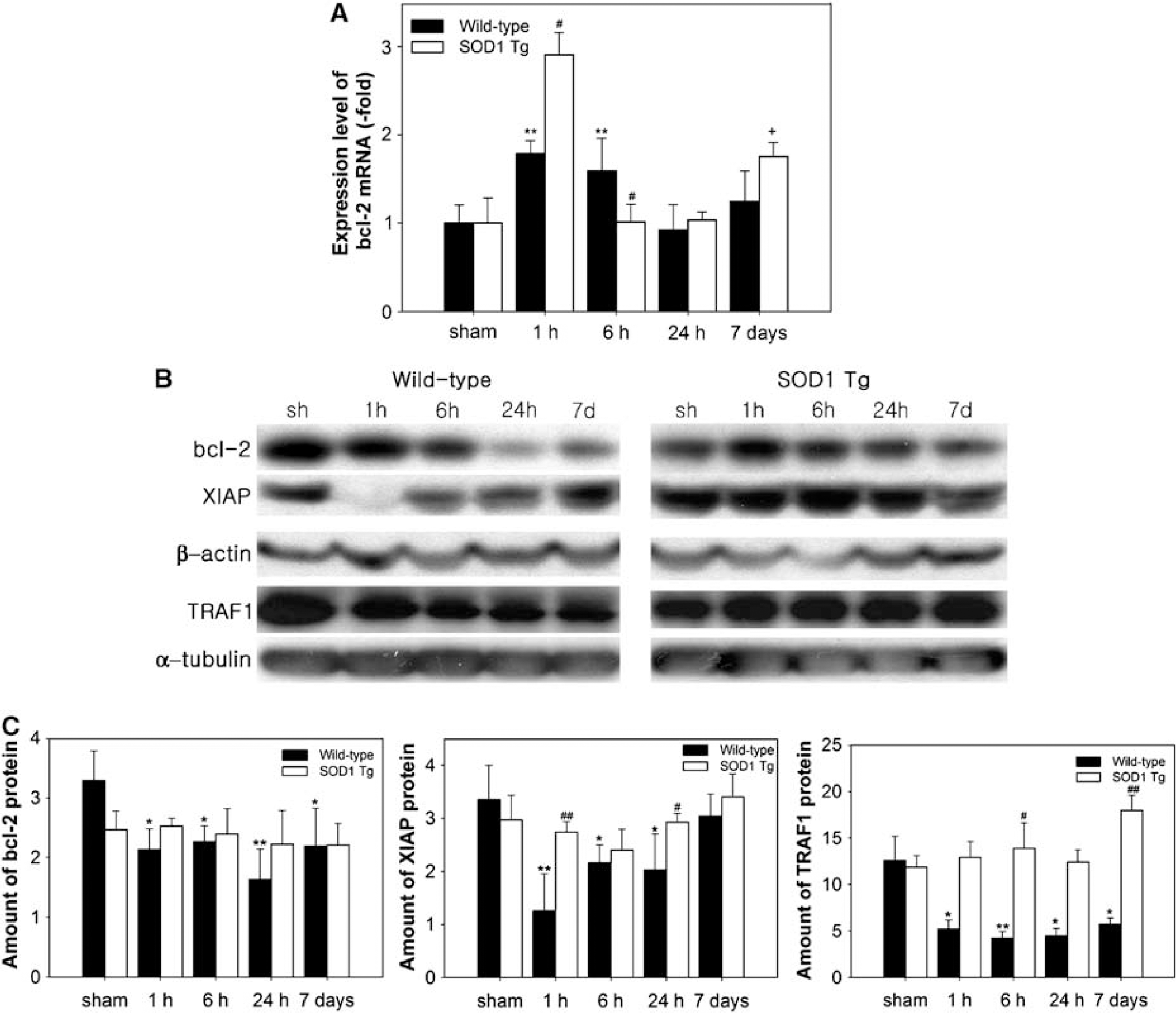
Expression levels of antiapoptotic factors in ischemic WT and SOD1 Tg mouse brains after 30 mins of tFCI. (
Discussion
In focal cerebral ischemia, the oxidative responsive transcription factor, NF-κB, can trigger different intracellular programs leading to either neuronal survival or neuronal apoptosis, depending on the nature and temporal course of the insult. One of the main purposes of our study was to find changes in genes of the NF-κB signaling pathway after cerebral ischemia and reperfusion. Nuclear factor-kappa B is one of the central mediators of neuronal cell survival and death because a wide variety of inducers and target genes are related to it. Our in vivo results showed that cerebral ischemia induced persistent upregulation of NF-κB, p105, and RelA mRNA throughout 7 days in the WT mice. In contrast to the WT mice, overexpression of SOD1 showed transient increases in p105 and RelA mRNA after tFCI. Clemens (2000) suggested that NF-κB is only transiently activated in neurons that survive, but is persistently activated in neurons that are destined to die after global cerebral ischemia. A decrease in persistent NF-κB activation caused by antioxidants was consistent with downregulation of NF-κB subunits after tFCI caused by SOD1 overexpression at 7 days (Stephenson et al, 2000). Regulation of NF-κB at a post-translational level has been suggested by Brand et al (1996). Transcriptional upregulation of NF-κB p105 mRNA after 6 mins of ischemia was recently reported (Kawahara et al, 2004), and is in accordance with our results, that is, upregulation of p105 mRNA after 30 mins of cerebral ischemia. This reflects the transcriptional regulation of NF-κB, p105, and RelA after cerebral ischemia. Moreover, we demonstrated that the level of the nuclear p50 protein was highly increased throughout the 7 days after cerebral ischemia in the WT mice. Qiu et al (2001) showed that in response to hypoxia, there was an enhanced elevation of the nuclear p50 protein at 24 and 72 h in the rat hippocampus. A cell death-promoting role for p50 was reported showing that p50 knockout significantly reduced ischemic damage in an MCA occlusion reperfusion model (Schneider et al, 1999). From these results, we can speculate that SOD1 overexpression prevented persistent NF-κB upregulation at a late time point, 7 days, and that it might protect against neuronal cell death that occurs over a period of days in transient cerebral ischemia. Our results have shown that overexpression of SOD1 reduced infarct size in the ischemic brain (Kamii et al, 1996; Yang et al, 1994).
Although a few studies have been put forth to explain the immune response in the brain, stroke has been shown to be associated with altered immune function, especially in the brain (Becker et al, 2003; Kato et al, 1996). The local cerebral immune and inflammatory responses to stroke involve upregulation of several inflammatory mediators, including cytokines, chemokines, and intercellular adhesion molecules that are controlled by NF-κB activity (Arvin et al, 1996; Hill et al, 1999; Pahl, 1999). The T-cell growth factor, IL-2, is an important stimulus for immune responses and activation of T lymphocytes (Abbas, 2003). Our results showed that IL-2 and IL-2Rα transcripts in the reduced oxidative stress caused by SOD1 overexpression or χ-phenyl-n-tert-butyl-nitrone administration were dramatically upregulated from 1 h after tFCI. However, induction of IL-2 and IL-2Rα was gradually increased in the WT or vehicle-treated mice. Our results support the hypothesis that reduced oxidative stress induced a rapid defense immune mechanism at the initiation of cerebral ischemia by oxidative stress. It has been reported that antioxidants such as N-acetyl-
In the present study, reduced oxidative stress showed several distinctive features. Persistent upregulation of NF-κB-family genes, NF-κB p105, and RelA in the WT mice was blocked by overexpression of SOD1. Lymphocytokines such as IL-2, IL-12, and IFN-α1, relevant for immune response, were rapidly induced in the SOD1 Tg mice after cerebral ischemia. In addition, antiapoptotic factors were increased with less oxidative stress. The neuroprotective expression profile of NF-κB-related genes promoted by reduced oxidative stress will elucidate the putative role of each molecule in stroke and offer a potential therapeutic target for treatment.