
Abstract
Adequate hydration is essential for normal brain function and dehydration induces cognitive deterioration. In addition, dehydration has emerged as a stroke risk factor. However, it is unknown whether alterations in cerebrovascular regulation are responsible for these effects. To address this issue, C57BI/6 mice were water deprived for 24 or 48 hours and somatosensory cortex blood flow was assessed by laser-Doppler flowmetry in a cranial window. Dehydration increased plasma osmolality and vasopressin levels, and suppressed the increase in blood flow induced by neural activity, by the endothelium-dependent vasodilator acetylcholine and the smooth muscle relaxant adenosine. The cerebrovascular dysfunction was associated with oxidative stress and cognitive deficits, assessed using the Y maze. The vasopressin la receptor antagonist SR49059 improved the dehydration-induced oxidative stress and vasomotor dysfunction. Dehydration upregulated endothelin-1 in cerebral blood vessels, an effect blocked by SR49059. Furthermore, the endothelin A receptor antagonist BQ123 ameliorated cerebrovascular function. These findings show for the first time that dehydration alters critical mechanisms regulating the cerebral circulation through vasopressin and oxidative stress. The ensuing cerebrovascular dysregulation may alter cognitive function and increase the brain's susceptibility to cerebral ischemia.
INTRODUCTION
Water is essential for cellular homeostasis and life. Without water, humans can survive only for days, and severe dehydration leads to cognitive dysfunction, delirium, and coma.1,2 During sporting events, dehydration is associated with decrements in the athletes' physical performance. 3 Mild levels of dehydration alter mood and cognitive function and reduce concentration, alertness, and short-term memory in children 4 and young adults.5–7 In the elderly, dehydration is well known to compromise cognitive function.8,9 Aging blunts thirst and drinking responses, making older people more vulnerable to body fluid imbalance, 10 and increasing their susceptibility to cognitive decline. 11 Indeed, dehydration is a predisposing factor for confusion in long-term care residents. 12 Furthermore, plasma hypertonicity, a marker of dehydration, increases the risk of ischemic stroke in hospitalized patients 13 and may precipitate cerebral ischemic events in susceptible elderly individuals. 14
The mechanisms responsible for these effects have not been clearly elucidated. Dehydration leads to reductions in brain volume and changes in neural activity in brain areas involved in fluid homeostasis, 15 but little is known about the factors responsible for the alteration in cognition. 16 One possibility is that dehydration disrupts critical cerebrovascular homeostatic mechanisms, such as the increases in cerebral blood flow (CBF) induced by neural activity or by endothelial cells, that assure that the brain receives a supply of oxygen and glucose well matched to its energy needs. 17 In support of this possibility, alterations in cerebrovascular regulation are often associated with cognitive dysfunction. 18 Furthermore, vasopressin (AVP), the plasma level of which increases with dehydration, is involved in the cerebrovascular dysfunction induced by administration of slow pressor doses of angiotensin II (ANGII). 19 The effect is due to AVP-mediated expression of the potent vasoactive peptide endothelin-1 (ET-1) in cerebral blood vessels, which, in turn, is responsible for the cerebrovascular alterations.19,20
Therefore, in this study, we sought to determine whether dehydration alters the mechanisms regulating the cerebral microcirculation. We found that dehydration induces a profound disruption of the ability of neural activity, endothelial cells, and smooth muscle cells to regulate CBF, which is associated with cognitive dysfunction assessed using the Y-maze. The effect depends on AVP-mediated oxidative stress and induction of ET-1 in cerebral blood vessels. The findings provide the first evidence to date that dehydration alters critical regulatory mechanisms of the cerebral circulation, which may reduce vascular reserves and contribute to the associated cognitive dysfunction and increased stroke risk.
MATERIALS AND METHODS
Mice
All the methods used in this study have been described in detail in previous publications from this laboratory,19–21 and are briefly summarized. All experimental procedures were approved by the IACUC (Institutional Animal Care and Use Committee) of Weill Cornell Medical College and were performed according to the IACUC, NIH, and ARRIVE guidelines (http://www.nc3rs.org/ARRIVE).
Water Deprivation Experiments
For water deprivation (WD) experiments, adult male (3 months old) wild-type C57BL/6J mice (25 to 30 g; Jackson Laboratory, Bar Harbor, ME, USA) were deprived of access to water, but not to food, for 24 or 48 hours. Control mice were given free access to food and water. Body weights were recorded before and after WD. Taking into account the differences in weight and water turnover between mice and humans, 22 the reduction in body weight and the increase in plasma osmolality observed in the present study indicate moderate to severe dehydration. After the WD period, mice were either used for neocortical CBF measurements or killed for biochemical measurements in blood and brain.
General Surgical Procedures for Neocortical Cerebral Blood Flow Studies
Mice were anesthetized with isoflurane in a mixture of N2 and O2 (Induction, 5%; Maintenance, 2%). The trachea was intubated and mice were artificially ventilated with an oxygen-nitrogen mixture. The O2 concentration in the mixture was adjusted to provide an arterial pO2 (Pao2) of 120 to 130 mm Hg.19–21 One of the femoral arteries was cannulated for recording mean arterial pressure (MAP) and collecting blood samples. Rectal temperature was maintained at 37°C using a rectal probe connected to a thermostatically controlled heating pad. End-tidal CO2, monitored by a CO2 analyzer (Capstar-100; CWE Inc., Ardmore, PA, USA), was maintained at 2.6% to 2.7% to provide a pCO2 of 30 to 35 mm Hg.19–21 After surgery, isoflurane was discontinued and anesthesia was maintained with urethane (750 mg/kg, intraperitoneally) and chloralose (50 mg/kg, intraperitoneally). Throughout the experiment, the level of anesthesia was monitored by testing corneal reflexes and motor responses to tail pinch.
Monitoring Cerebral Blood Flow
A small craniotomy (2 × 2 mm) was performed to expose the parietal cortex, the dura was removed, and the site was superfused with Ringer's solution (37°C; pH 7.3 to 7.4; composition in mmol/L: 137 NaCl, 5 KCl, 1 MgCl2, 1.95 Na2HPO3, 15 NaHCO3, 2 CaCl2). Neocortical CBF was continuously monitored at the site of superfusion with a laser-Doppler probe (Perimed, Kings Park, CA, USA) positioned stereotaxically on the cortical surface. The outputs of the flowmeter and blood pressure transducer were connected to a data acquisition system (PowerLab, AD Instruments, Colorado Springs, CO, USA) and saved on a computer for offline analysis. Cerebral blood flow values were expressed as percentage increases relative to the resting level. Zero values for CBF were obtained after the heart was stopped by an overdose of isoflurane at the end of the experiment. Although laser-Doppler flowmetry is not quantitative, it monitors relative changes in CBF quite accurately. 23
Experimental Protocol for Cerebral Blood Flow Experiments
After mean arterial pressure and blood gases were stable (pCO2: 30 to 35 mm Hg; pO2: 120 to 130 mm Hg; pH: 7.3 to 7.4) (Supplementary Table 1), the cranial window was superfused with Ringer's solution (vehicle), and CBF responses were recorded. To minimize the confounding effects of anesthesia on vascular reactivity, the time interval between the administration of urethane-chloralose and the testing of CBF responses was kept consistent among the different groups of mice studied. The whisker-barrel cortex was activated for 60 seconds by stroking the contralateral vibrissae, and the evoked changes in CBF were recorded. The endothelium-dependent vasodilator acetylcholine (ACh; 10 μmol/L; Sigma, St Louis, MO, USA) or the smooth muscle relaxant adenosine (400 μmol/L; Sigma) was superfused on the exposed neocortex for 5 minutes. In some studies, CBF responses were tested with vehicle superfusion or after 30 minutes of superfusion with the reactive oxygen species (ROS) scavenger Mn(lll)tetrakis(4-benzoic acid) porphyrin chloride (MnTBAP, 100 μmol/L; Calbiochem, Billerica, MA, USA), the Via receptor (V1aR) antagonist SR49059 (1 μmol/L; Tocris, Bristol, UK), the ETA receptor antagonist BQ123 (1 μmol/L; Tocris) or the ANG 1A receptor antagonist losartan (5 μM; Sigma). The specificity and selectivity of these pharmacological agents was tested in previous studies.19,24
Electrocorticogram and Field Potentials
Mice were anesthetized and surgically prepared as described in section
Reactive Oxygen Species Detection
Reactive oxygen species production in the somatosensory cortex was assessed by hydroethidine (DHE) microfluorography as previously described.19–21 The DHE (2 μmol/L; Invitrogen, Grand Island, NY, USA) was superfused on the somatosensory cortex for a total of 60 minutes. The brain was removed, frozen, and coronal sections (thickness, 20 μm) were cut through cortex underlying the cranial window using a cryostat. Sections were analyzed using a Nikon Eclipse E800 fluorescence microscope (Melville, NY, USA) equipped with a custom filter set for detection of DHE oxidation products.19–21 Images were acquired with a digital camera (Coolsnap, Roper Scientific, Tucson, AZ, USA) and analyzed in a blinded manner using the IPLab software (Scanalytics, Fairfax, VA, USA), as described previously. 26 Fluorescent intensities of all sections (20/animal) were added, divided by the total number of pixels analyzed, and expressed as relative fluorescence units.
Immunohistochemistry
For confocal microscopy, mice (
Vasopressin and Endothelin-1 Measurement
Plasma AVP was measured using a competitive EIA kit (Cayman Chemicals, Ann Arbor, MI, USA) after purification over a phenyl matrix (Bond elut-PH; Varian, Walnut Creek, CA, USA) according to the manufacturer's instructions. Endothelin-1 was measured in cerebrovascular preparations using the commercially available ELISA kit (Enzo Life Sciences, Farmingdale, NY, USA). Homogenates, including large, medium size, and small cerebral blood vessels, were prepared and purified as previously described.19,21
Behavioral Assessment by the Y Maze
Cognitive function was assessed using the Y maze as previously described. 27 The Y maze test does not involve learning new rules and takes advantage of the natural tendency of rodents to explore new environments. Briefly, the Y maze consisted of three identical arms made of transparent plastic joined in the middle to form a ‘Y’ (20 cm high, 10 cm wide, and 30 cm long). The mice were handled daily and allowed to acclimate to the apparatus for a week before testing. Mice were placed into one of the arms of the maze (start arm) and allowed to explore only two of the arms for 5 minutes (training trial). The third arm, which remained closed, was randomly chosen in each trial. The closed arm was opened in the test trial, serving as the novel arm. After a 30-minute intertrial interval, the mice were returned to the same start arm and were allowed to explore all three arms for 5 minutes (test trial). Sessions were video recorded and replayed for determination of the parameters of interest by a blind observer. The number of visits and time spent in each arm were recorded in a blinded manner. To avoid confounding effects of changes in the motor activity, the number of visits to the novel arm was expressed as a percentage of the total number of visits to all three arms. 27
Data Analysis
Sample size was determined according to power analysis based on previous published works published by our laboratory on CBF regulation. No animal was excluded, animals were randomly assigned to treatment and control groups and analysis were performed in a blinded manner. Data are expressed as mean ± s.e.m. Intergroup differences were analyzed using the analysis of variance with Tukey's
RESULTS
Water Deprivation Increases Plasma Osmolality and Plasma Vasopressin
Water deprivation for 24 and 48 hours resulted in a reduction in body weight and mean arterial pressure (
Water Deprivation Impairs Cerebrovascular Responses Induced by Different Mechanisms
Water deprivation attenuated the increase in CBF produced by neural activity (whisker stimulation) both at 24 and at 48 hours (Figure 2A). Furthermore, responses to the endothelium-dependent vasodilator ACh and to the smooth muscle relaxant adenosine were also reduced (Figures 2B and 2C). To rule out that the global attenuation in cerebrovascular responses was secondary to suppression of neural activity, we examined the effect of dehydration on the electrocorticogram and on the field potentials induced in the somatosensory cortex by whisker stimulation. As illustrated in Figures 3A to 3D, dehydration did not affect the amplitude of the field potentials or the frequency distribution of the electrocorticogram. In contrast, the anesthetic isoflurane attenuated the field potentials and the electrocorticogram at all frequencies (Figures 3A to 3C), providing a positive control for the sensitivity of the monitoring system. Therefore, the cerebrovascular dysfunction induced by dehydration is not associated with alterations in spontaneous or evoked neural activity in the neocortex in which CBF was measured.
Water Deprivation Impairs Cognitive Function Assessed by the Y Maze Test
Cerebrovascular dysfunction is often associated with cognitive deficits. 27 Therefore, we used a two-trial spatial-memory task in a Y maze 27 to determine whether also the cerebrovascular dysfunction induced by dehydration was associated with cognitive alterations. As shown in Figure 2D, dehydration was associated with a reduction in the number of visits to the novel arm. Furthermore, water-deprived mice spent less time in the novel arm compared with sham animals (Figure 2E). Therefore, the cerebrovascular dysfunction induced by dehydration is associated with cognitive impairment at the Y maze.
Oxidative Stress Is Involved in the Cerebrovascular Dysfunction Induced by Water Deprivation
Oxidative stress is responsible for cerebrovascular dysfunction in a number of disease models, including ANGII-induced hypertension, chronic intermittent hypoxia, or Alzheimer's disease.19,21,26,27 Therefore, we investigated whether WD increases ROS production in the somatosensory cortex in which CBF is recorded. Water deprivation induced a time-dependent increase in ROS assessed by DHE microfluorography (Figure 4A). To determine whether the increased ROS production was responsible for the cerebrovascular dysfunction, we investigated the effect of neocortical superfusion with the ROS scavenger MnTBAP. We found that this agent ameliorated the attenuation in the CBF response to whisker stimulation, ACh and adenosine (Figures 4B to 4D), implicating oxidative stress in the cerebrovascular dysfunction induced by WD.
The Vasopressin Receptor Antagonist SR49059 Prevents the Cerebrovascular Alterations and Cognitive Dysfunction Induced by Water Deprivation
The systemic effects of dehydration are initiated by the release of AVP from hypothalamic nuclei into the systemic circulation. 28 Since AVP has powerful effects on the cerebral circulation by activating V1aR,29–31 we tested whether this peptide is involved the cerebrovascular dysfunction induced by WD. The V1aR antagonist SR49059 (2 mg/kg per day in 0.1 mL of saline) was administered intraperitoneally during the WD period. SR49059 did not affect the increase in osmolality induced by dehydration (Figure 5A), the weight reduction (Figure 5B), or baseline cerebrovascular responses (Figures 5C to 5E), but attenuated the increase in ROS (Figure 5F). SR49050 did not affect the number of AVP+-positive cells in the PVN or the AVP+ cells expressing c-Fos (Figures 1H and 1I), indicating that this agent did not suppress PVN activity. Furthermore, SR49059 ameliorated the CBF responses, to ACh and adenosine more than whisker stimulation (Figures 5C to 5E). SR49059 also improved the dehydration-induced cognitive deficits assessed by the Y maze test, especially at 48 hours (Figures 5G and 5H). To determine whether sustained activation of V1aR on cerebral blood vessels by AVP was driving the neurovascular dysfunction, we examined the effect of topical application of SR49059 to the cerebral cortex of water-deprived mice. In contrast to systemic administration, topical application of SR49059, at a concentration effective in counteracting the cerebrovascular actions of AVP, 24 had no effect on the attenuation of CBF responses induced by dehydration (Figures 6A to 6C). This observation suggests that AVP does not mediate the dysfunction by directly activating cerebrovascular V1aR.
Neocortical Superfusion with the ETAR Antagonist BQ123 Ameliorates the Cerebrovascular Alterations Induced by Water Deprivation
In a model of ANGII hypertension, AVP induces cerebrovascular dysfunction by upregulating the vasoactive peptide ET-1 in cerebral microvessels. 2 To test whether a similar mechanism is involved in the cerebrovascular effects of dehydration, we examined whether dehydration increased ET-1 in cerebral blood vessels and, if so, whether ET-1 is involved in the cerebrovascular dysfunction. Dehydration increased ET-1 in neocortical blood vessels (Figure 7A). Furthermore, neocortical superfusion of the endothelin type A receptor (ETAR) antagonist BQ123 improved all cerebrovascular responses (Figures 7B to 7D). The increase in ET-1 levels was counteracted by systemic administration of SR49059, supporting the hypothesis that AVP is responsible for the ET-1 upregulation in cerebral blood vessels (Figure 7A).
Dehydration is well known to increase plasma levels of ANGII, 33 a peptide that causes cerebrovascular dysfunction through ANG 1A receptor. 19 Neocortical superfusion with losartan, at a concentration effective in counteracting the cerebrovascular effects of ANGII (5 μmol/L), 19 did not rescue the dysfunction, although it tended to improve the CBF response to whisker stimulation and ACh at 24 hours (Figures 6D and 6E). The CBF response to adenosine was not improved (Figure 6F).
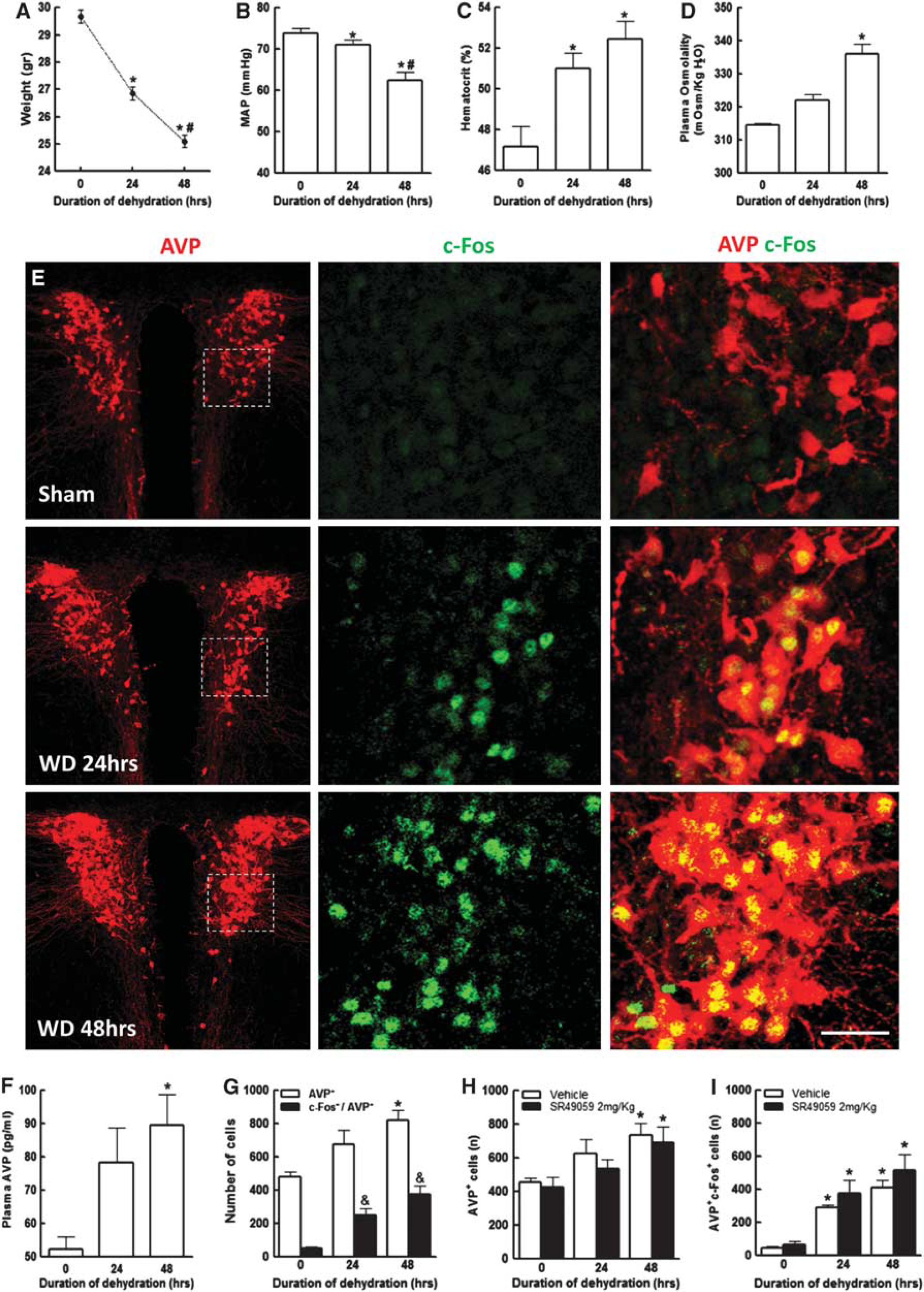
Body weight and mean arterial pressure (MAP) are reduced in dehydrated mice in a time-dependent manner (
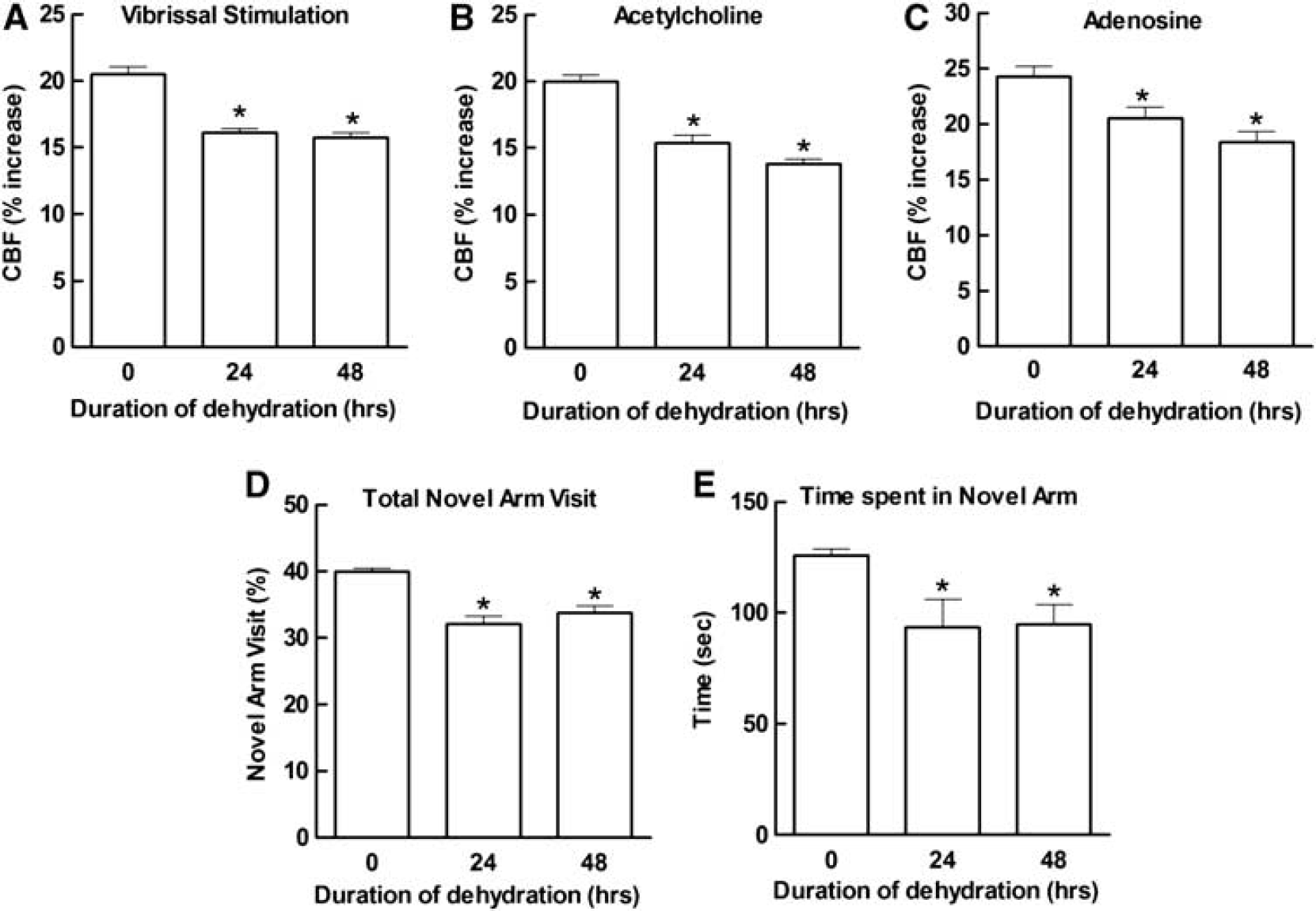
Dehydration impairs cerebral blood flow (CBF) responses induced by neural activity, and by the endothelium-dependent vasodilator acetylcholine (ACh) or the smooth muscle relaxant adenosine (
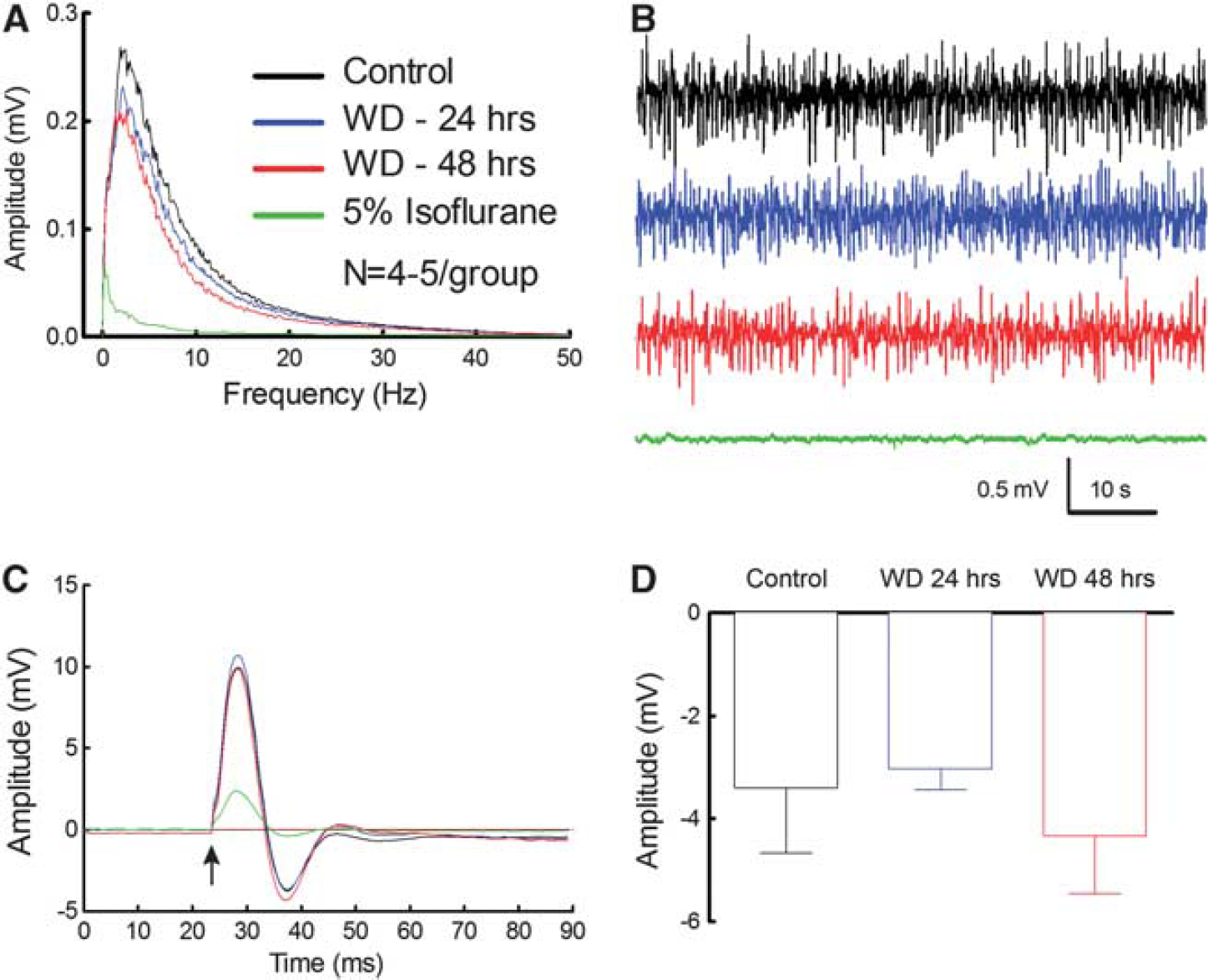
Spontaneous and evoked neural activity is not altered by dehydration. Dehydration does not affect the amplitude
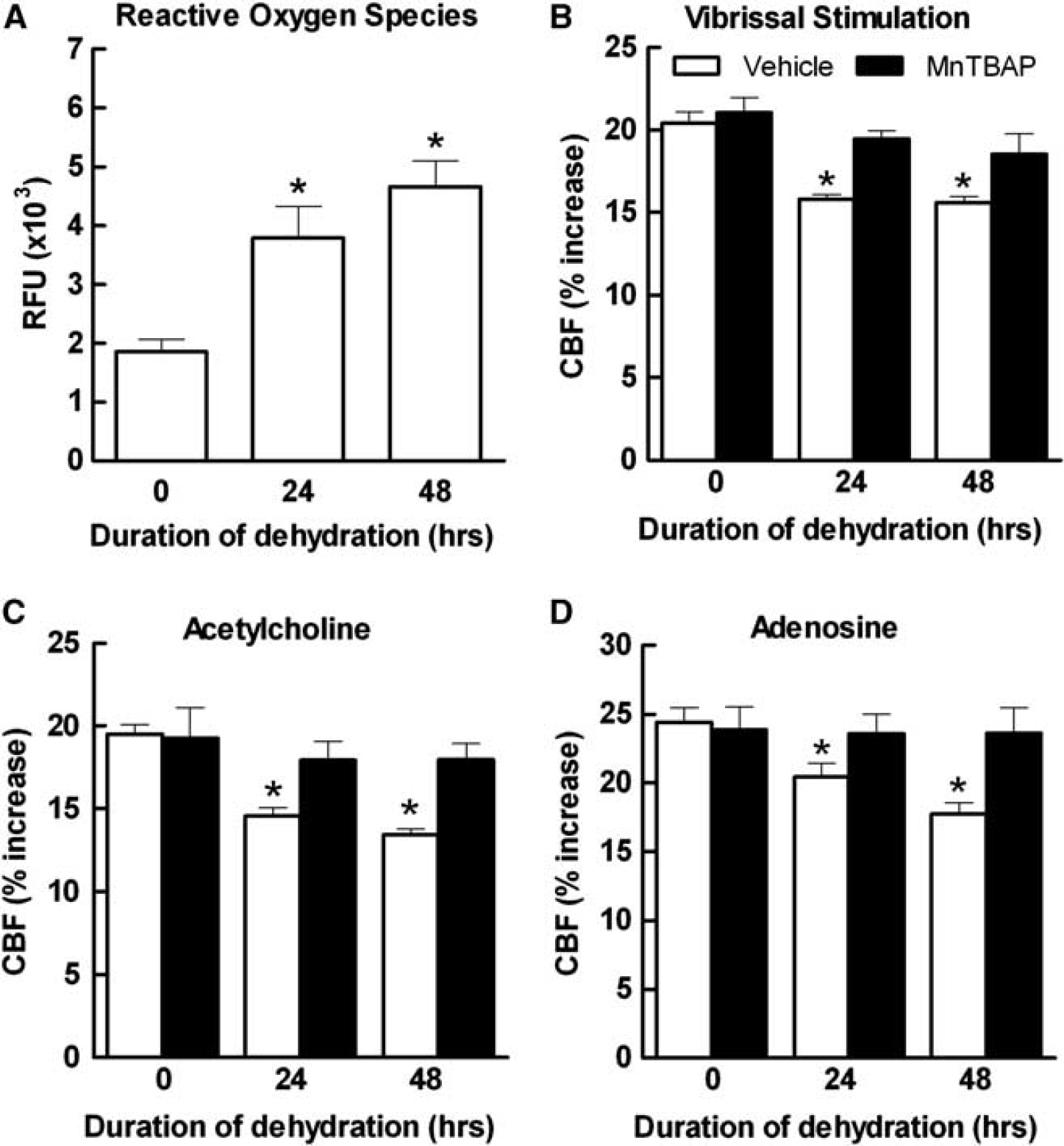
Dehydration increases oxidative stress in the somatosensory cortex assessed by hydroethidine (DHE),
DISCUSSION
We found that WD induces oxidative stress in the brain and disrupts major cerebrovascular regulatory mechanisms involving neurons, endothelial cells, and smooth muscle cells. The cerebrovascular dysfunction is associated with reduced cognitive performance, assessed by the Y maze. Oxidative stress, cerebrovascular dysfunction, and cognitive deficits are ameliorated by systemic administration of the V1aR antagonist SR49059, implicating the dehydration-induced increase in AVP in the mechanisms of the dysfunction. Dehydration upregulates ET-1 in cerebral blood vessels in an AVP-dependent manner, and mediates the cerebrovascular dysfunction through ETAR. A lesser contribution of ANG 1A receptor was also observed, but only for the response to whisker stimulation and ACh. These novel observations, collectively, show for the first time that dehydration-induced cognitive dysfunction is associated with deleterious effects on cerebrovascular function, which are related to the increase in AVP induced by dehydration.
The findings of this study cannot be attributed to changes arterial pressure or blood gases because these variables were controlled and did not differ among groups. Similarly, the cerebrovascular and cognitive changes cannot be attributed to the weight loss or increase in plasma osmolality induced by dehydration because treatment with SR49059 rescued the cerebrovascular and cognitive changes without affecting these parameters. Furthermore, it is unlikely that a global suppression in neural activity is responsible for the cerebrovascular and cognitive alterations because dehydration did not affect spontaneous or evoked cortical neural activity. The increase in AVP plasma levels is unlikely to results entirely from hemoconcentration, because AVP immunoreactivity in the PVN was increased, indicating upregulation of hypothalamic AVP synthesis. During WD rodents reduce food intake, a compensatory response known as ‘dehydration anorexia’, which increases with the duration of dehydration. 22 However, changes in systemic metabolism are unlikely to mediate the cerebrovascular effects of dehydration. This is because in mice WD in excess of 24 hours produces only minimal additional dehydration, but induces substantial weight loss. 22 Since the neurovascular dysfunction was already well developed at 24 hours of dehydration, we doubt that metabolic effects of the reduced food intake had a major role in the cerebrovascular alterations.
We found that WD increases ROS production in the somatosensory cortex and that the ROS scavenger MnTBAP abrogates the cerebrovascular dysfunction. Therefore, it is likely that the cerebrovascular dysfunction is related to oxidative stress. Since AVP, whose levels are increased after WD, stimulates vascular superoxide production through activation of V1aR, 34 we tested whether this mechanism was also involved in the cerebrovascular and cognitive effects of WD. We found that SR49059 attenuated the ROS production and ameliorated the cerebrovascular and cognitive dysfunction, implicating V1aR in its mechanisms. Since a residual ROS increase was observed after SR49059 treatment V1aR-independent pathways are also likely to play a role.
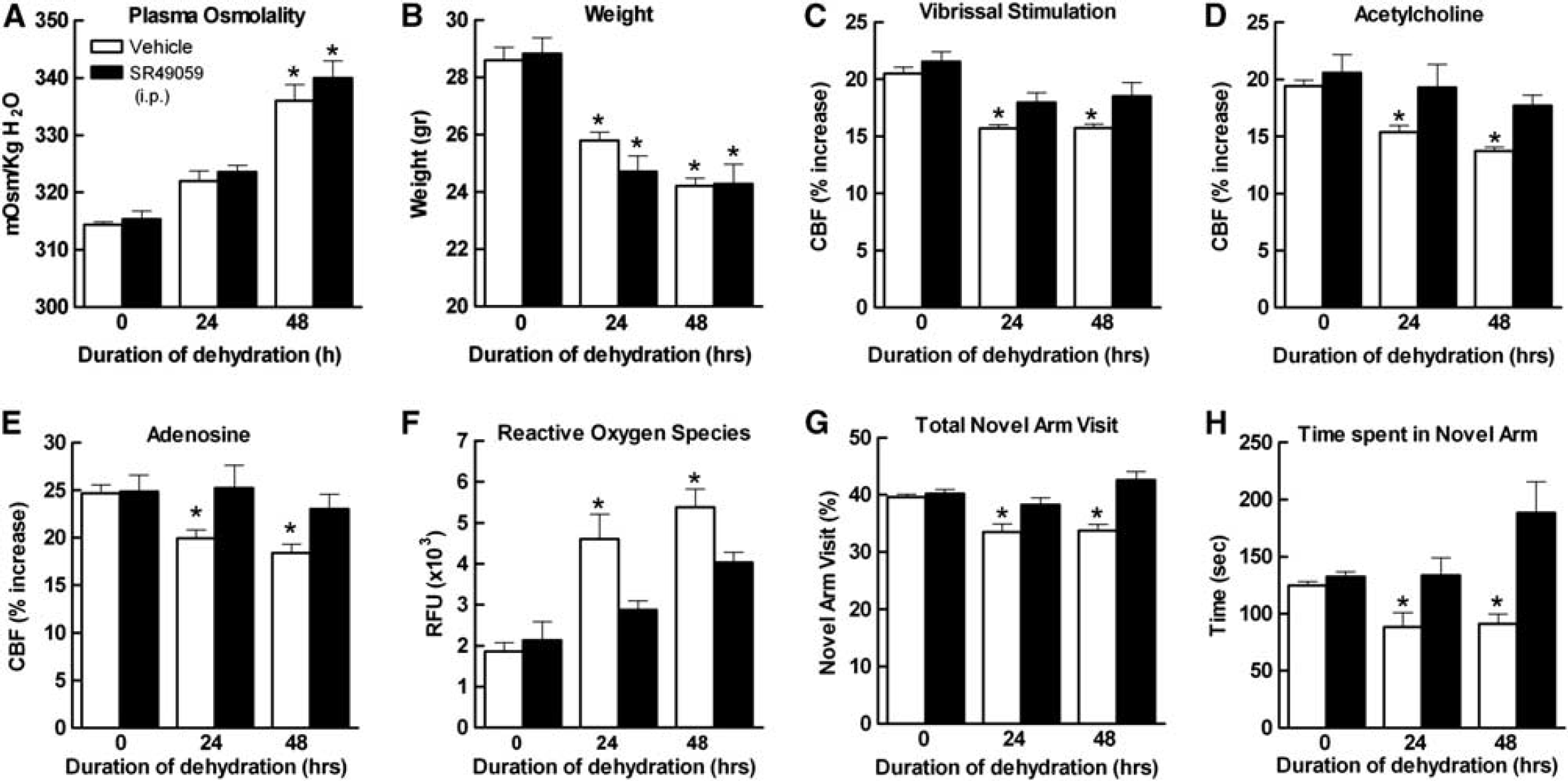
Systemic administration of SR49059 (2 mg/kg; intraperitoneal) has no effect on the reduction in body weight and the increase in plasma osmolality induced by dehydration (
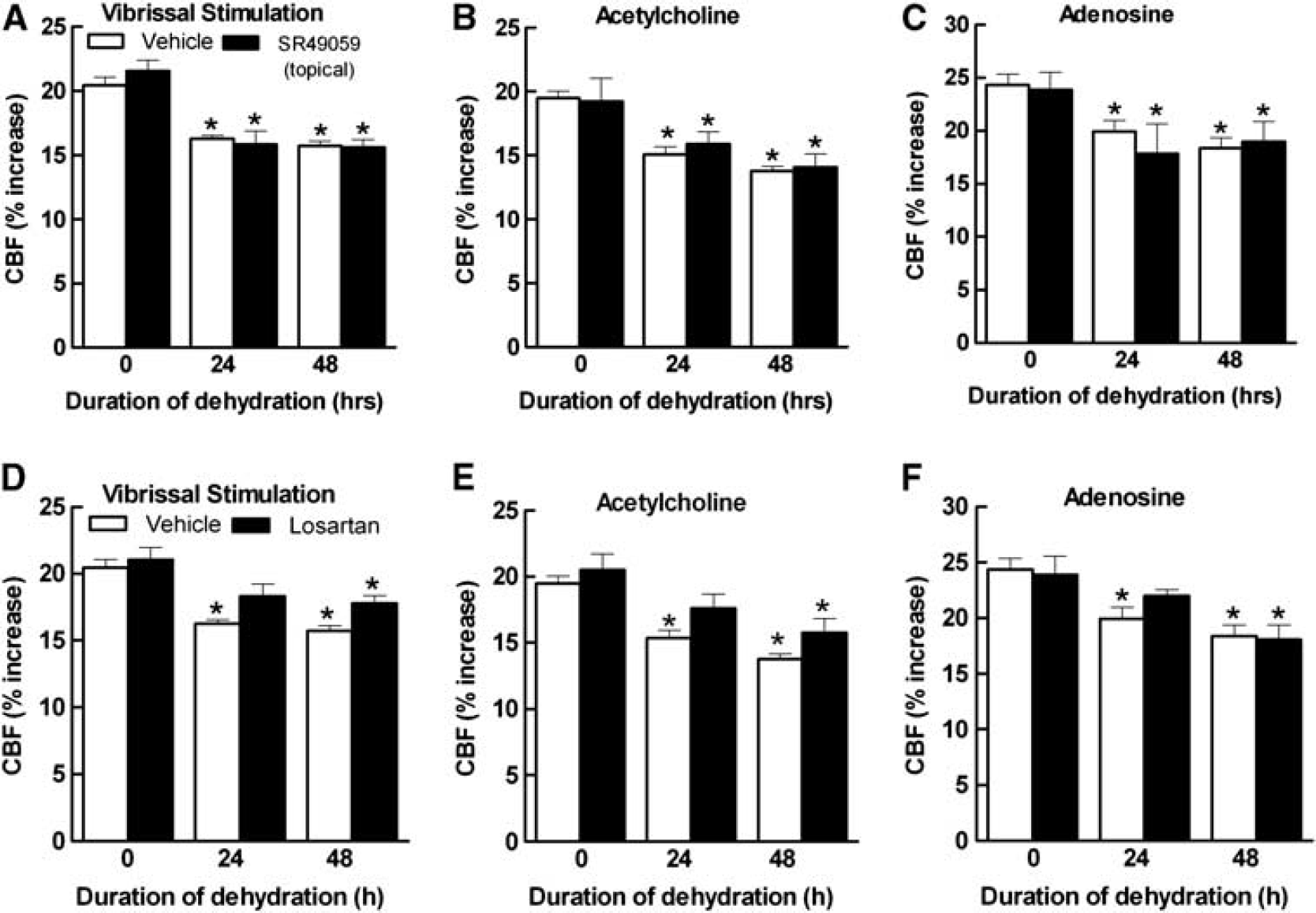
Neocortical application of SR49059 (1 mol/L) has no effect on the dehydration-induced suppression of the cerebral blood flow (CBF) response to whisker stimulation
Vasopressin has profound vascular effects and could be directly responsible for the cerebrovascular dysfunction by acting on cerebrovascular V1aR. 35 However, this possibility is not supported by our data since acute neocortical application of a V1aR antagonist did not reverse the cerebrovascular dysfunction. Another possibility is that vasopressin induces the expression of an intermediate mediator, which in turn is responsible for the cerebrovascular alterations. Indeed, in ANGII hypertension AVP induces vascular ET-1 expression, which mediates vascular dysfunction by activating ETAR.19,20 To determine whether this was the case also in WD, we measured ET-1 levels in cerebral vessels and we found that dehydration increases ET-1. Furthermore, the ETAR antagonist BQ123 reversed the cerebrovascular dysfunction induced by WD. These data, collectively, are consistent with the hypothesis that AVP released during WD induces the expression of ET-1 in cerebral blood vessels resulting in cerebrovascular dysfunction. In addition, ANG 1A receptor inhibition tended to improve the CBF responses to whisker stimulation and ACh suggesting a potential contribution of ANGII to the dysfunction, mainly at 24 hours. However, the alteration in smooth muscle reactivity to adenosine can be entirely attributed to ETI, since it was not improved by losartan.
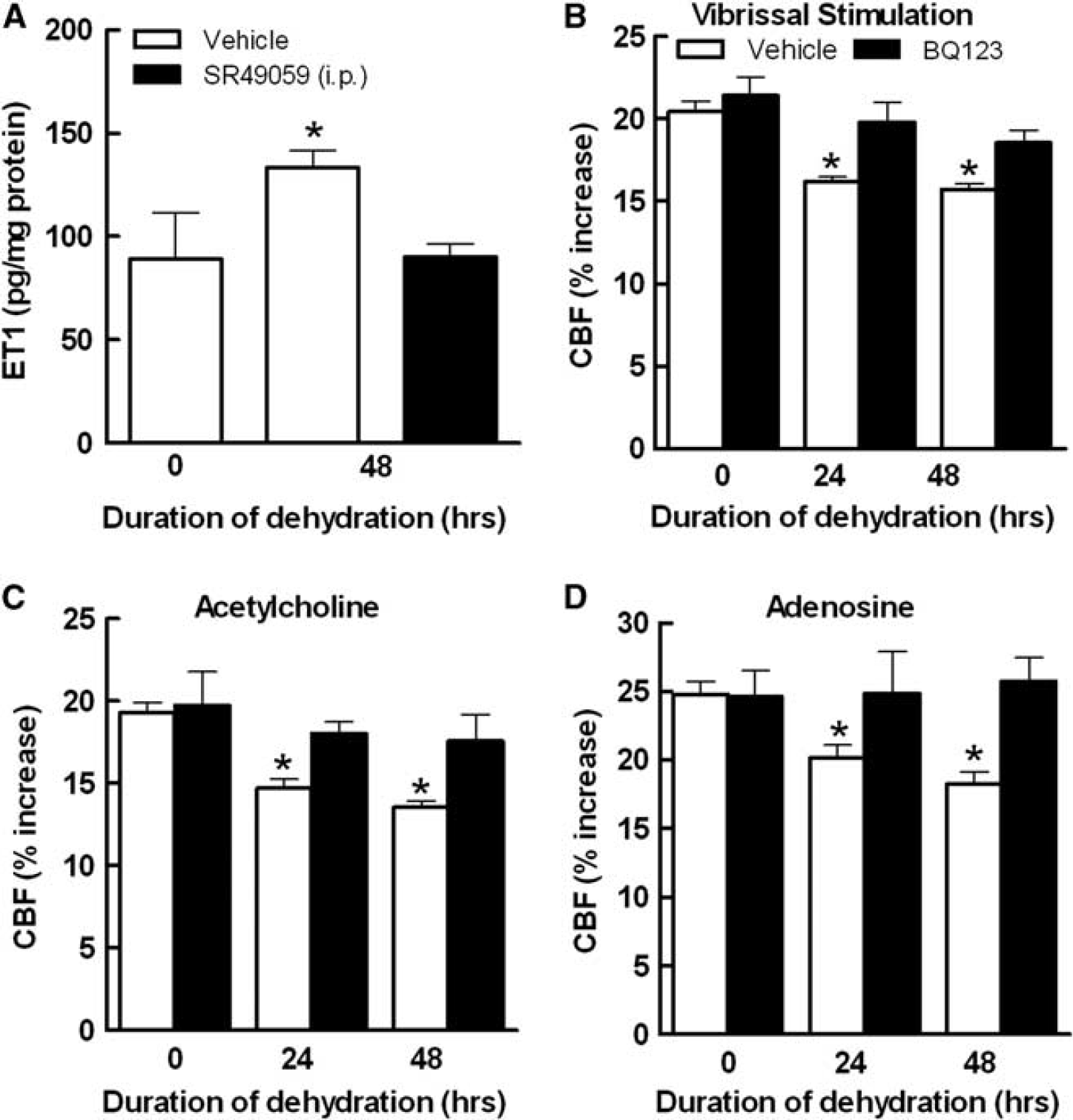
Dehydration increases endothelin-1 (ET-1) levels in cerebral blood vessels, and effect blocked by SR49059
A remarkable aspect of the cerebrovascular dysfunction of dehydration is that it also involves smooth muscle relaxation. The involvement of smooth muscle cells has not been observed in other models of cerebrovascular dysfunction associated with oxidative stress, which usually impairs functional hyperemia and endothelium-dependent responses.21,27,36 Responses to smooth muscle relaxants are attenuated in the presence of overt damage to smooth muscle cells, for example, in aging and amyloid angiopathy.37–39 However, since the attenuated response to adenosine is rescued by SR49059 or topical application of MnTBAP and BQ123, structural damage to smooth muscle cells by dehydration is unlikely. Rather, dehydration must engage distinct signaling pathways that result in a reversible smooth muscle cell dysfunction. The impairment of smooth muscle cell function could also be responsible for the attenuation of the CBF responses to whisker stimulation and ACh, because the vasomotor apparatus is dysfunctional. However, it remains to be established whether the upstream vasoactive signals generated by neural activity and endothelial cells are suppressed by dehydration, which could also contribute to the dysfunction.
In conclusion, we have showed that WD alters key regulatory mechanisms of the cerebral circulation and induces cognitive dysfunction. The cerebrovascular alterations are characterized by attenuation of functional hyperemia and endothelium-dependent CBF responses, as well as disruption of smooth muscle relaxation. The mechanisms of the cerebrovascular dysfunction involve release of AVP, and activation of V1aR, which, in turn, leads to oxidative stress and upregulation of ET-1 in cerebral arterioles. However, ANGII could also have a relatively minor role in the attenuation of responses initiated by neural activity and endothelial cells, especially at 24 hours. The data suggest that the cognitive dysfunction induced by dehydration may involve alterations in critical homeostatic mechanisms regulating the cerebral circulation and leading to vascular insufficiency. The findings provide novel insights into the mechanistic bases of dehydration-induced cognitive deficits and for the increased susceptibility to ischemic brain injury associated with hyperosmolar states.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.