
Abstract
Transgenic mice overexpressing endothelin-1 (ET-1) in astrocytes (GET-1) displayed more severe brain edema and neurologic dysfunction after experimental ischemic stroke. However, it was not clear whether astrocytic ET-1 contributed to cytotoxic or vasogenic edema associated with stroke. In this study, the role of astrocytic ET-1 in cytotoxic edema and brain injury was investigated. Upon acute water intoxication, the GET-1 mice had a lower survival rate and more severe neurologic deficits. Such an exacerbated condition in the GET-1 mice may be a result of a significant increase in cerebral water content and increased expression of the water channel protein, aquaporin 4 (AQP-4). The GET-1 mice treated with OPC-31260, a nonpeptide arginine vasopressin V2 receptor antagonist, were alleviated from the cerebral water accumulation and neurologic deficit during the early time period after water intoxication. In addition, a significant reduction of AQP-4 expression was observed in astrocytic end-feet AQP-4 in the hippocampus of the GET-1 mice treated with OPC-31260. Therefore, ET-1-induced AQP-4 expression and cerebral water accumulation are the key factors in brain edema associated with acute water intoxication. The V2 receptor antagonist, OPC-31260, may be one of the effective drugs for the early treatment of ET-1-induced cytotoxic edema and brain injury.
Keywords
Introduction
Cytotoxic edema occurs in a number of pathologic conditions, including pediatric diabetic ketoacidosis, liver failure, and in other situations, such as in the early stage of a severe burn, heat stroke, as well as traumatic brain injury (Blei, 2008; Li et al, 2001; Liu et al, 1999; Wolfsdorf et al, 2007). Brain edema is also one of the complications occurring after stroke and is one of the leading causes of death. During the early phase of ischemic stroke, the dominant effect of hypoxia leads primarily to cytotoxic edema, reaching maximum by 24 h (Rosenberg, 1999). Previously, it was shown that transgenic mice (GET-1 mice) overexpressing endothelin-1 (ET-1) in the astrocytes using the glial fibrillary acidic protein promoter showed more severe neurologic deficit, increased infarct volume, hemispheric swelling, and cerebral water content after transient focal ischemia induced by middle cerebral artery occlusion (MCAO) and reperfusion (Lo et al, 2005). The increase in water accumulation and edema in the brains of the GET-1 mice correlated with an increased expression of the water channel protein, aquaporin 4 (AQP-4) in the astrocytic end-feet in blood vessels. In addition, the GET-1 brains also showed more Evans Blue extravasation and decreased endothelial occludin expression after transient MCAO, indicating the breakdown of the blood–brain barrier (BBB) and increased vasogenic edema. However, it is still unclear whether astrocytic ET-1 has a major role in cytotoxic or vasogenic edema formation.
Early after ischemia, depletion of high-energy phosphates due to the abrogation of oxidative phosphorylation upsets the energy-dependent ion transporters and disturbs intracellular ion distribution, which leads to the swelling of astrocyte end-feet within several minutes. Astrocyte swelling lasts for 24 h after cerebral ischemia and is followed by necrosis (Garcia et al, 1994). As astrocytes are the most abundant in the central nervous system, cellular swelling in astrocytes due to pathologic diseases, such as stroke, is most severely affected. During ischemic stroke, cytotoxic edema is usually formed earlier than vasogenic edema, which is caused by the BBB breakdown and movement of fluid containing plasma protein from the intravascular to the extravascular space. Vasogenic edema formation poses a risk of hemorrhage in the damaged vessel and contributes to a net increase in brain volume and pressure (Marmarou, 2007).
Despite the frequent occurrence of cerebral edema after stroke and the undesirable consequences, there is still no effective therapy to prevent or slow cerebral edema. Several agents have been used to treat edema, including mannitol and glycerol; however, prolonged administration of these agents has adverse effects or a gradual loss of effectiveness (Yildizdas et al, 2006). Controversial results have been shown regarding the effectiveness of corticosteroids in treating cellular edema as well (Gomes et al, 2005). Other antiedemic agents are, therefore, important in the treatment of stroke patients. Arginine vasopressin (AVP), a neuropeptide hormone, has been reported to regulate astroglial cell volume in vitro, and responsible for both vasogenic and cytotoxic brain edema formations (Del Bigio and Fedoroff, 1990). The predominant type of AVP receptors in the central nervous system is the V1a subtype, which is expressed in different regions of the brain, including the cerebral cortex, blood vessels, bed nucleus of stria terminalis, accumbens nucleus, central nucleus of amygdale, stigmoid hypothalamic nucleus, suprachiasmatic nucleus, hippocampus, choroids plexus, and pituitary (Gerstberger and Fahrenholz, 1989). Hosli et al (1991) showed that cortical astrocytes expressed vasopressin receptors, which have characteristics similar to those of the V1 subtype. However, another study suggested the presence of the V2 receptor in the hypothalamic supraoptic nuclei in the brain (Wotjak et al, 1994). Treatment with V1 antagonists has been shown to prevent ischemia-induced cerebral edema development (Shuaib et al, 2002), suggesting that the V1 vasopressin receptor is important in water regulation in brain cells (Rosenberg et al, 1990). Another study also indicated that the vasopressin receptor V1, but not the V2 subtype, is involved in the pathogenesis of secondary brain damage after focal cerebral ischemia (Rosenberg et al, 1990). Previous study has shown that OPC-31260, a non-peptide arginine vasopressin V2 receptor antagonist, is effective in treating water retention diseases, such as hyponatremia caused by inappropriate antidiuretic hormone secretion, congestive heart failure, and liver cirrhosis (Saito et al, 1997). Laszlo et al (1999) also showed that OPC-31260 at doses of 10 to 30 mg/kg produced a dose-dependent inhibition of subarachnoid hemorrhage-induced cerebral edema formation, accompanied by an increase in urinary volume and decrease in urine osmolarity without a significant alteration of urine electrolytes. Therefore, the role of the vasopressin receptor V1 versus V2 subtype in cerebral edema formation after ischemic stroke remains controversial.
As the GET-1 mice exhibited severe brain edema after transient MCAO, we determined whether astrocytic ET-1 has an important role in cytotoxic edema formation by using water-intoxicated mice as a model for cytotoxic brain edema. We also investigated whether astrocytic ET-1 in brain edema in the GET-1 mice is mediated by the V2 vasopressin receptor by treatment with OPC-31260.
Materials and methods
Water Intoxication and Assessment
An acute water intoxication model was used to induce cytotoxic edema in the brain, in which injected water was absorbed through mesenterial vessels producing hypo-osmotic hyponatremia (Manley et al, 2000). Transgenic and nontransgenic mice were littermates or subsequent generation siblings and were therefore strain matched. Age-matched Ntg and GET-1 mice received intraperitoneal injection of distilled water equal to 20% of body weight with DDAVP (1-desamino-8-
Water Content Measurement
At 90 mins after water intoxication when the neurologic score peaked in both Ntg and GET-1 mice, the brain tissue was removed for brain water content analysis (Lo et al, 2005). The brain was weighed immediately to obtain a wet weight and was then dried in an oven at 105°C for 48 h and weighed again to obtain a dry weight. The percentage of water content was calculated as ((wet weight−dry weight)/wet weight) × 100%.
Immunocytochemical Analysis
At 90 mins after water intoxication, when the neurologic score peaked in both Ntg and GET-1 mice, the brain tissue was removed and fixed in 1% formalin, then processed for immunocytochemical analysis. Coronal brain slices −2.0 mm from the bregma were used for immunocytochemical study. Brain sections were incubated with antibodies against glial fibrillary acidic protein (GFAP; 1:500) and AQP-4 (1:400). Signals were visualized using the Vectastain ABC Kit (Vector Laboratories, Burlingame, CA, USA) with 3,3′-diaminobenzidine tetrahydrochloride (Zymed, South San Francisco, CA, USA). All the conditions were followed according to our previous study (Lo et al, 2005).
Western Blot Analysis
A separate water intoxication experiment was conducted to collect protein samples from the Ntg and GET-1 brains. The brains were divided into left and right hemispheres. The cortex and hippocampal regions were isolated in both the hemispheres. The cortex together with the hippocampus of the left hemisphere was homogenized in lysis buffer (50 mmol/L Tris-HCL, pH 6.8, 150 mmol/L NaCl, 5 mmol/L EDTA (ethylenediaminetetraacetic acid), 0.5% sodium deoxycholate, 0.5% NP-40 (Nonidet P-40), plus proteinase inhibitor cocktail). The homogenate was centrifuged (3,000 g for 5 mins at 4°C), and the supernatant was used for the western blot analysis of GFAP expression. To obtain the tissue membrane fractions for AQP-4 analysis, the membranous protein of the cortex and hippocampus from the right hemisphere was extracted using the ReadyPrep Protein Extraction Kit (Bio Rad, CA, USA, Catalog no. 163-2088). All the procedures were followed according to the manufacturer's instruction manual. Blots were incubated with antibodies against GFAP (1:1,000, Z0334, Dako, Demark), AQP-4 C terminus (1:750, AB3594, Chemicon, Temecula, CA, USA), and α-tubulin (1:5,000, sc-5286, Santa Cruz, CA, USA). Signals were visualized by ECL (Amersham, Piscataway, NJ, USA) and quantitated using PhotoImager (Molecular Dynamics, Uppsala, Sweden). Values for protein levels were expressed as percentage of the Ntg nonoperated group after normalization with individual α-tubulin levels for equal loading, while in AQP-4 western analysis Ponceau-S red staining was used to ensure equivalent sample applications.
Drug Treatment
OPC-31260 (5-dimethylamino-1-[4-(2-methylbenzoylamino)benzoyl]-2,3,4,5-tetrahydro-1H-benzazepine hydrochloride) was dissolved in water at a concentration of 30 mg/kg and administrated to the Ntg and GET-1 mice in a single dosage through oral gavage 30 mins before water intoxication (Laszlo et al, 1999). The aquaretic effect of OPC-31260 was reported to be 6 to 8 h (Laszlo et al, 1999). OPC-31260 was generously provided by Otsuka Pharmaceutical (Tokyo, Japan) as a gift.
Statistical Analysis
The statistical tests were conducted using the GraphPad Prism software (San Diego, CA, USA) as specified, and the data are presented as mean±s.e.m. The neurologic score data comparison between the Ntg and GET-1 mice (with or without OPC-31260 treatment) were analyzed using the Mann–Whitney test. For all other measurements, one-way ANOVA (analysis of variance) followed by Bonferroni's posttest was used. P<0.05 was considered statistically significant.
Results
GET-1 Mice Had a Higher Death Rate Than Ntg Mice After Water Intoxication
Both cytotoxic and vasogenic edema coexist in ischemic stroke. To address the role of astrocytic ET-1 in cytotoxic or vasogenic edema formation, water intoxication was performed in both Ntg and GET-1 mice. After water intoxication induction, both groups of mice showed a reduction in locomotive activity within 20 mins and became comatose. In the Ntg mice, no deaths were observed from 0 to 90 mins, and the survival rate started to decrease only after 90 mins, from 100% to 92%. Thereafter, no additional dead mice were observed until the end of the experiment. However, the GET-1 mice died much earlier and faster than did the Ntg mice; the survival rate of the GET-1 mice started to decrease at 45 mins after water intoxication and continued to decrease quickly within 45 mins, from 100% to 82%, and became stable thereafter until the end of the experiment (Figure 1A). All surviving mice in both groups appeared normal after 24 h.
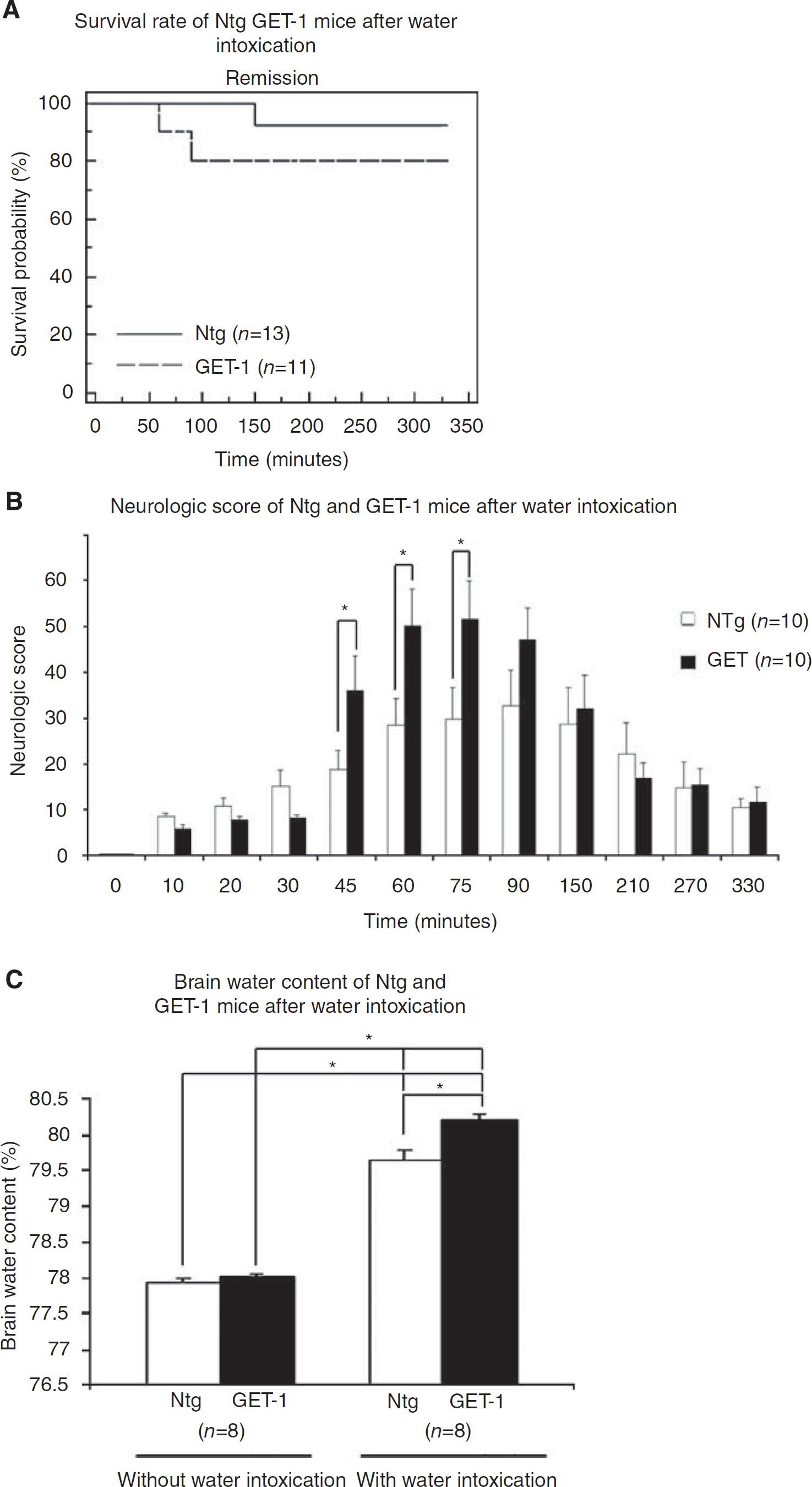
Effect of water intoxication on cerebral edema formation. Ntg and GET-1 mice were intraperitoneally administrated with water. (
GET-1 Mice Exhibited More Severe Neurological Deficits Compared With Ntg Mice After Water Intoxication
After water intoxication, most of the Ntg mice behaved normally at the first 30 mins but with slight motor deficits, as their movements became stiff. At 45 or 60 mins onwards, some of the Ntg mice showed a decrease in consciousness and body coordination ability. As indicated in the neurologic deficit score, the Ntg mice exhibited gradual neurologic dysfunction at 30 mins (15.0±3.5) that peaked at 90 mins (32.7±7.9) (Figure 1B). After 150 mins, the neurologic score gradually decreased and returned to normal at 330 mins, suggesting a full recovery of neurological function. The GET-1 mice receiving the same treatment showed significantly worse neurological deficit compared with the Ntg mice, as indicated by a dramatic increase in the neurological deficit score after 45 mins (36.0±7.6, ∗P<0.05 by Mann–Whitney test) to 60 mins and peaked at 75 mins (51.6±8.3, ∗P<0.05 by Mann–Whitney test). During that period, the GET-1 mice showed more severe deficits of consciousness and respiration, an increase in cranial nerve reflex, and body coordination. A substantial decrease in the survival rate in the GET-1 mice at 45 to 90 mins after water intoxication may be due to the sudden worsening of the neurologic score during this time period. After 150 mins, there was no difference in neurologic scores between the GET-1 and Ntg mice (Figure 1B).
GET-1 Mice Accumulated More Water in Their Brains Than Ntg Mice After Water Intoxication
To further understand the role of astrocytic ET-1 in water intoxication-induced neurological deficit in GET-1 mice, the cerebral water content in both Ntg and GET-1 mice was measured (Figure 1C). Before water intoxication, the cerebral water content in the Ntg and GET-1 mice was approximately the same (Ntg: 77.92%±0.06%; GET-1: 78.02%±0.03%). At 90 mins after water intoxication, a significant increase in cerebral water content was observed in the Ntg mice (from 77.92%±0.06% to 79.63%±0.149%; P<0.0001) and in the GET-1 mice (from 78.02%±0.03% to 80.02%±0.087%; P<0.0001). In addition, when the water-intoxicated Ntg mice were compared with the GET-1 mice, the cerebral water content in the GET-1 mice (80.2±0.087; P<0.01; n=8) was significantly higher than that of the Ntg mice (79.6±0.15; n=8), suggesting that the GET-1 mice were more susceptible to water intoxication-induced cytotoxic edema.
Increased Expression of GFAP in the GET-1 Brain After Water Intoxication
To further understand the role of overexpressing astrocytic ET-1 in cytotoxic edema formation and glial reactivity, the level of GFAP expression was determined by immunocytochemical analysis. Glial fibrillary acidic protein, an intermediate filament cytoskeletal protein, is increased in the expression of reactive astrocytes and is used as an indicator of brain injury (Liao et al, 2008). Under normal conditions, immunocytochemical analysis showed that both Ntg and GET-1 mice had GFAP signals sparely expressed over the hippocampal region (Figures 2Ai and 2Bi). At higher magnification, GFAP-labeled astrocytes in the control Ntg and GET-1 mice appeared to be stallate shaped (Figures 2Aiii and 2Biii). When the Ntg mice were subjected to water intoxication, the level of GFAP expression and the number of GFAP-positive astrocytes were induced significantly in the hippocampal regions (Figures 2Ai and 2Aii). At higher magnification, the reactive astrocytes with long processes in the Ntg mice were stained strongly with GFAP (Figure 2Aiv). In the GET-1 mice before water intoxication, similar situations were observed as those in the Ntg mice (Figure 2Biii). However, after water intoxication, a significantly higher increase in GFAP staining was observed in the hippocampus of the GET-1 mice (Figure 2Biv). The expression of GFAP in astrocytes of the GET-1 mice was even more intense with more number of astrocytes over the hippocampus than those of the Ntg mice (Figures 2Aiv and 2Biv). These reactive astrocytes appeared to have more and longer astrocytic processes after water intoxication than those from the Ntg mice (Figure 2Biv). The expression of GFAP was also determined by western blot analysis and quantitated (Figures 2Av and 2Bv). The results were in line with those obtained from immunocytochemical analysis. Significant upregulation of GFAP was observed in both Ntg and GET-1 mice after water intoxication. However, the GFAP expression of GET-1 mice was 46% higher than that of the Ntg mice. Other regions, such as the putamen, also showed an increase in the GFAP signal after water intoxication in both Ntg and GET-1 mice, but a large difference was not observed between the two groups of mice (data not shown), suggesting that the GET-1 mice were more prone to water intoxication-induced response in the hippocampal regions.
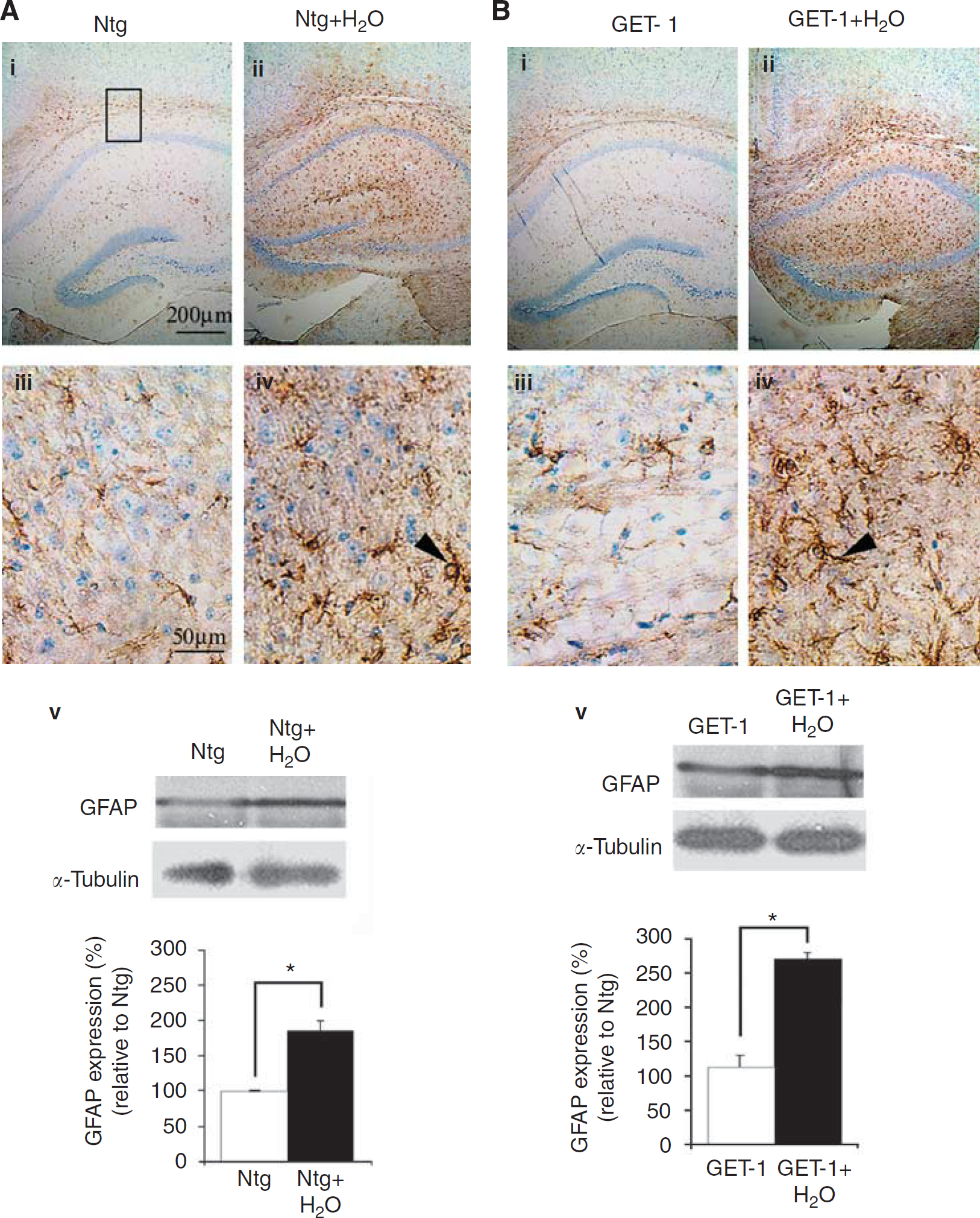
Immunocytochemical analysis of GFAP expression after water intoxication. Mice brains were collected at 90 mins after water intoxication when the neurological score peaked in both Ntg and GET-1 mice. Representative micrograph shows the localization and expression of GFAP in the astrocytes of the hippocampus of the Ntg and GET-1 brains at (
Increased Expression of Aquaporin 4 in Astrocytic End-Feet After Water Intoxication
The expression level of water channel protein, AQP-4, was also determined after water intoxication by immunocytochemical analysis. Under normal conditions, the expression of AQP-4 was observed in the hippocampal regions of the Ntg and GET-1 brains (Figures 3Ai and 3Bi). At higher magnification, AQP-4 expression was localized at the astrocytic end-feet in contact with the cerebral vessels forming perivascular sheath and allowing easy identification of cerebral blood vessels (Figures 3Aiii and 3Biii) as described in previous reports (Nicchia et al, 2000). After water intoxication, the expression of AQP-4 in the Ntg mice was significantly upregulated with more widespread and diffused staining over the hippocampal region (Figure 3Aii). The AQP-4 staining surrounding the cerebral vessels became more scattered and patchy as well as less polarized. Therefore, upregulation of the AQP-4 expression made the vessels appear much longer and thicker (Figure 3Aiv). However, more intense staining of AQP-4 was observed in the hippocampal regions of the GET-1 mice compared with that of the Ntg mice after water intoxication (Figure 3Bii). At higher magnification, the staining in the GET-1 brain was increased compared with that of the Ntg mice and longer immunostaining was observed along the perivascular sheath (Figure 3Biv). The level of AQP-4 expression was quantitated by Western blot (Figures 3Av and 3Bv). The results were in line with those of the immunocytochemical analysis. A significant upregulation of AQP-4 was observed in both Ntg and GET-1 mice after water intoxication. However, the GET-1 mice showed 16% higher AQP-4 expression than did the Ntg mice, suggesting that AQP-4 was increased in the GET-1 mice after water intoxication.
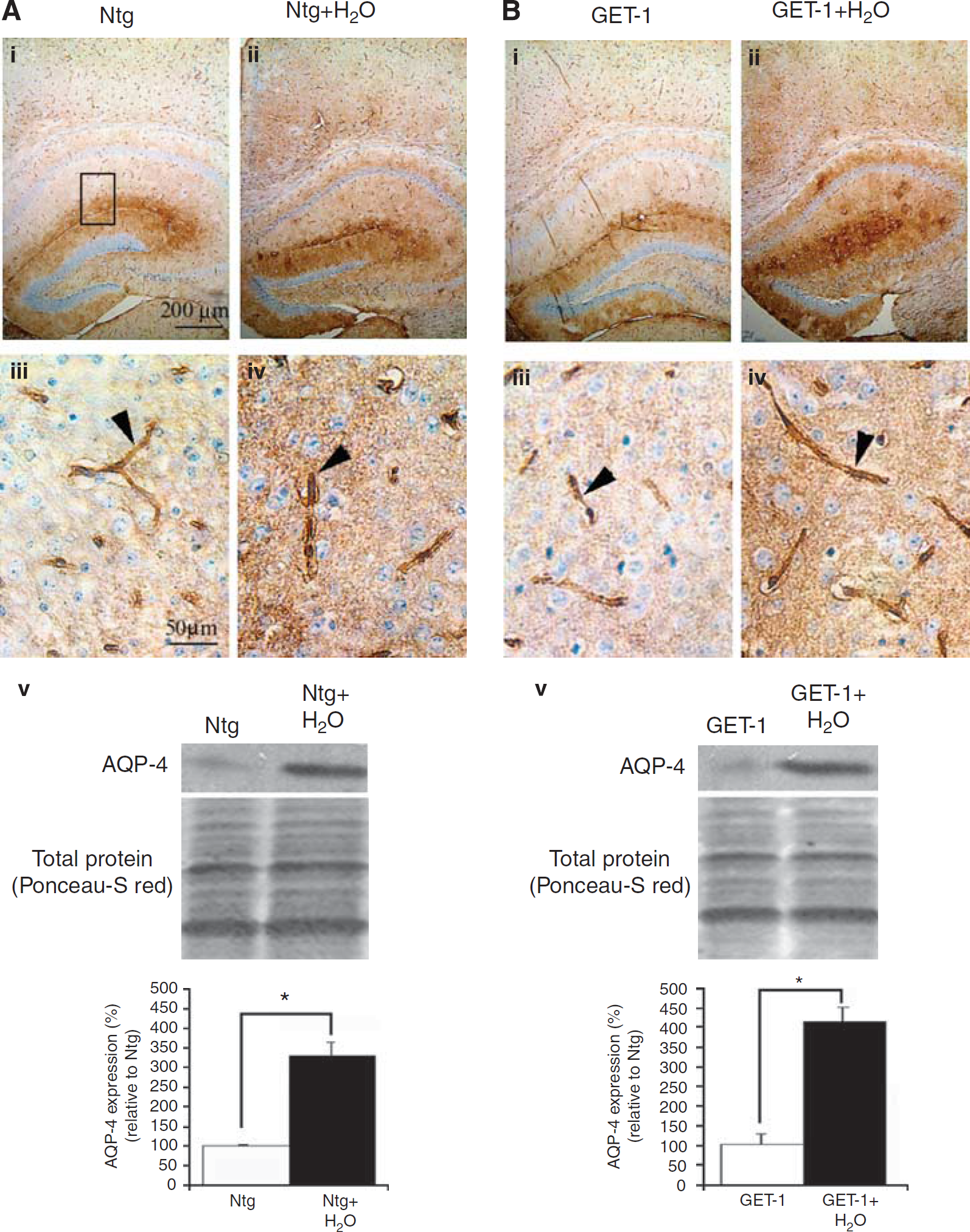
Immunocytochemical analysis of AQP-4 expression after water intoxication. Representative micrograph shows the localization and expression of AQP-4 in the astrocytes of the hippocampus of the Ntg (
Treatment of OPC-31260 Ameliorates Cerebral Neurological Deficit in GET-1 Mice After Water Intoxication
To address whether astrocytic ET-1-induced cytotoxic edema formation was also mediated by the V2 vasopressin receptor, the GET-1 mice were treated with OPC-31260, a V2 vasopressin receptor antagonist, before water intoxication. As shown in Figure 4A, for the GET-1 mice without drug treatment, the neurological deficit quickly worsened at 45 mins (36.0±7.6) and peaked at 75 mins (51.6±8.3) and then recovered gradually after 210 mins (16.8±3.5). After OPC-31260 treatment, significant ameliorations of neurological deficit were observed within the first 20 mins (Ntg: 7.7±0.8, GET-1: 3.9±2.0, ∗P<0.05 by Mann–Whitney test), and a trend in improvement was also observed at 45 to 90 mins, but slightly worsened at later time points (90 to 200 mins). The neurological deficit in the GET-1 mice after OPC-31260 treatment peaked at 90 mins (45.7±8.6), which was later than that of the Ntg mice. After that, the OPC-31260-treated GET-1 mice gradually recovered after 210 mins (23.1±8.0). The right shift in the neurologic deficits of the OPC-31260-treated GET-1 mice suggested that OPC-31260 delayed the occurrence of deficits. Throughout the whole experiment, similar mortality rates were observed in both GET-1 mice with or without drug treatment (data not shown).
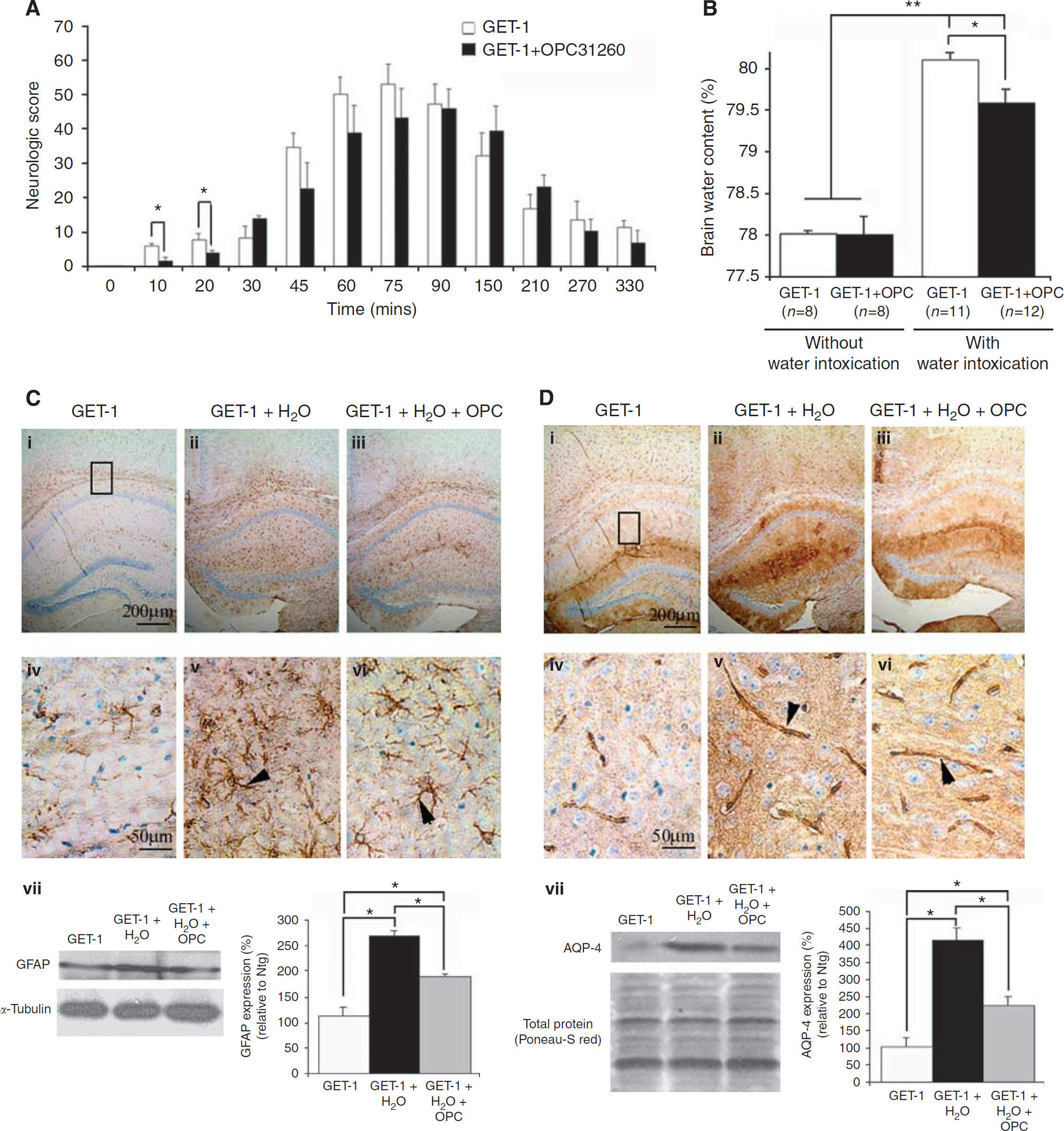
Effect of OPC-31260 (30 mg/kg) treatment on cerebral edema formation in GET-1 mice after water intoxication. The GET-1 mice were administrated with OPC-31260 treatment 30 mins before water intoxication. (
Treatment of OPC-31260 Significantly Abolished Water Accumulation in GET-1 Mice After Water Intoxication
After water intoxication, the GET-1 mice displayed a significant increase in cerebral water accumulation (80.2±0.1, n=11). To determine whether OPC-31260 could diminish cerebral edema caused by water intoxication, water content was quantitated. OPC-31260 treatment significantly diminished water accumulation in the brain (79.6±0.2, n=12) (Figure 4B). The significant reduction in cerebral water accumulation correlated with the improvement in neurologic deficits observed in the OPC-31260-treated GET-1 mice, suggesting that astrocytic ET-1-induced cytotoxic edema formation may be mediated by the V2 vasopressin receptor.
Treatment of OPC-31260 After Water Intoxication Decreased GFAP in the Astrocytes
Immunocytochemical and Western blot analyses were also carried out to investigate the effect of OPC-31260 administration after water intoxication on the GFAP expression level in the GET-1 brains. OPC-31260 treatment significantly reduced ~30% the level of GFAP staining in the hippocampal regions of the GET-1 brain (P=0.045) (Figures 4Ciii and 4Cvii). At higher magnification, the OPC-31260-treated GET-1 mice were shown to have shorter processes, which was similar to the GET-1 controls without water intoxication (Figure 4Cvi). In addition, both the staining intensity and the number of activated astrocytes were much lower than those of the GET-1 brain without drug treatment (arrowheads in Figures 4Civ and 4Cvi). The OPC-31260-treated GET-1 mice showed a significant reduction of GFAP expression determined by Western blot analysis when compared with the nondrug-treated group, suggesting that OPC-31260 partially blocked ET-1-induced glial reactivation (Figure 4Cvii).
Treatment of OPC-31260 After Water Intoxication Aquaporin 4 in Astrocytic End-Feet
The GET-1 mice showed significant increases in AQP-4 expression in the hippocampal regions after water intoxication. OPC-31260 treatment significantly downregulated AQP-4 expression level in the GET-1 brain, as the intensity of AQP-4 staining was almost comparable with that of the controls without water intoxication (Figures 4Di and 4Diii). At higher magnification, the intensity was much reduced and less diffused although immunostaining along the perivascular sheath was observed (Figures 4Dv and 4Dvi). Western blot analysis also showed a significant downregulation of AQP-4 expression (∼46%; P=0.045) in the OPC-31260-treated group when compared with that in the nontreated group (Figure 4Dvii), suggesting that OPC-31260 could effectively reduce AQP-4-mediated water accumulation in the brains of GET-1 mice.
Discussion
Endothelin-1 is a potent vasoconstrictor normally produced by endothelial cells in the brain. Marked induction of ET-1 level has been reported in ischemic stroke conditions and has been suggested to have an important role in the pathogenesis of ischemia-induced brain lesions (Barone et al, 1994). The induction of ET-1 under ischemic conditions is localized to astrocytes in addition to endothelial cells in the ischemic brain, suggesting the role of astrocytic ET-1 in ischemic injury (Tsang et al, 2001). Recently, transgenic mice overexpressing astrocytic ET-1 (GET-1) were shown to be more susceptible to ischemic brain damage with an increased water accumulation, increased brain edema (cytotoxic edema), and impaired integrity of the BBB (vasogenic edema) (Lo et al, 2005). Thus, the contribution of astrocytic ET-1 in cytotoxic versus vasogenic edema cannot be distinguished. In this study, the water intoxication model was used to further investigate the contribution of astrocytic ET-1 to cytotoxic edema formation.
After water intoxication, GET-1 mice showed a lower survival rate, significantly more severe neurological deficit, and increased cerebral water accumulation compared with the Ntg mice. The higher expression of AQP-4 may account for the significant increase in cerebral water content after water intoxication in the GET-1 brain, suggesting that astrocytic ET-1 contributes to cytotoxic edema formation. Such finding is in agreement with a previous report that increased AQP-4 transcripts were found to be associated with cerebral edema formation (Saito et al, 1997). Aquaporin 4 is most abundant in the perivascular glial processes, indicating that these cells are actively involved in water transport and water homeostasis in the brain (Nielsen et al, 1997). Previous reports have shown the important role of AQP-4 in cerebral edema development by making use of transgenic mice with AQP-4 overexpression in astrocytes or AQP-4-deleted mice (Manley et al, 2000; Yang et al, 2008). However, these reports did not address the role of astrocytic ET-1 in cytotoxic edema. A previous study has shown that the overexpression of ET-1 leads to more water accumulation in the brain after transient MCAO (Lo et al, 2005). At present, it is still not clear whether astrocytic ET-1 has direct roles in the transcription or translocation of astrocytic AQP-4 leading to cytotoxic edema. The increased level of ET-1 synthesis in astrocytes and vascular endothelial cells associated with ischemic brain injury has been reported (Tsang et al, 2001). The blocking of ETA receptor, but not of ETB, reduced swelling in the ipsilateral hemisphere of the brain of GET-1 mice after transient MCAO, suggesting that the exacerbated effects of astrocytic ET-1 in cerebral edema is mediated through the ETA receptor in the brain (Lo et al, 2005). Matsuo et al (2001) also showed that MCAO-induced brain edema is mediated by the ETA receptor. In addition, ETA receptor-mediated neurohypophysial AVP secretion has been reported in the explanted culture of the hypothalamo-neurohypophysial complex (Rossi, 2004). Therefore, it is possible that the overexpression of astrocytic ET-1 in the GET-1 brains after water intoxication activates the central AVP action through the ETA receptor leading to the upregulation of astrocytic AQP-4 and higher cerebral water accumulation than in the Ntg brain.
In this study, the GET-1 brain also showed an increased reactive astrocytes, which are characterized by the enhanced expression of GFAP and the increased number of astrocytes, after water intoxication. Previously, similar observations were made in the GET-1 brain after transient MCAO (Lo et al, 2005). Astrocytes' response to brain damages results in changes in gene expression, cellular hypertrophy, and cell proliferation leading to more number of astrocytes. However, it is still largely unknown whether the reactive astrocytes contribute to beneficial or detrimental effects after brain insults (Chen and Swanson, 2003). Recently, an in vivo study, making use of transgenic mice with conditional knockout of GFAP ablated of reactive gliosis after cerebral ischemia, showed that reactive astrocytes exert a neuroprotective effect after brain injury (Li et al, 2008). Kimelberg (1991) also suggested that astrocytes might exert a protective effect and counteract cytotoxic edema in stroke by releasing osmotically active molecules, such as taurine. Nonetheless, the increased level of GFAP expression and number of GFAP-positive cells have been linked to the level of brain injury (Lo et al, 2005).
The secreted AVP acts on the V2 receptor and regulates water homeostasis. The action of vasopressin through the V2 receptor has been implicated in several pathologic conditions associated with abnormal water retention, such as hyponatremia, syndrome of inappropriate antidiuretic hormone secretion, and endolymphatic hydrops in Ménière's disease (Lemmens-Gruber and Kamyar, 2006). The vasopressin V2 receptor antagonist is an important therapeutic agent in the treatment of these water retention disorders through the renal system. The V2 receptor antagonist blocks AVP-mediated water reabsorption in the kidney medullary collecting duct through the inhibition of adenylate cyclase, thus reducing the renal cAMP level that is involved in AQP-2 expression and translocation in the medulla. In the brain, however, V2 receptors are believed to be restricted to the cerebellum of adult brains (Kato et al, 1995) whereas V1 receptors are widely distributed in the central nervous system and highly expressed in the hypothalamus area (Hernando et al, 2001). Therefore, one might suggest that the wide distribution of the vasopressin V1 receptor in the brain is the basis for effective treatment of cerebral edema with V1 receptor antagonists over V2 receptor antagonists (Shuaib et al, 2002). As the GET-1 mice displayed more severe cytotoxic edema than the Ntg mice, the effect of V2 receptor antagonists, OPC-31260, on cytotoxic edema formation was determined as the predominant type of edema appears during the early phase of ischemic stroke (Rabinstein, 2006). Water intoxication induced cytotoxic edema with a significant increase in cerebral water content in both Ntg and GET-1 mice. Immunocytochemical and Western blot analyses also showed that GFAP expression is upregulated in both Ntg and GET-1 mice after water intoxication, indicating that the reactive astrocytes undergo gliosis and change in morphology in response to brain injury (Ridet et al, 1997). With OPC-31260 treatment, GET-1 mice showed a significant reduction in neurological deficits at the early time points as well as cerebral water accumulation and AQP-4 expression after being induced by cytotoxic edema. Immunocytochemical and Western blot studies also showed that OPC-31260-treated GET-1 mice showed a significant reduction of GFAP expression when compared with the nondrug-treated group, suggesting that OPC-31260 partially blocks astrocytic ET-1-induced glial reactivation associated with brain injury. The effectiveness of the vasopressin V2 receptors antagonist, OPC-31260, in reducing water accumulation and cerebral edema formation in the GET-1 mice suggested that V2 receptors are involved in astrocytic ET-1-induced cytotoxic edema formation after transient cerebral ischemia. The present data are also in agreement with the findings of Molnar et al (2008) suggesting that the vasopressin V2 receptor antagonist, OPC-31260, inhibits cerebral edema formation induced by global cerebral ischemia. In addition, OPC-31260 has been shown to effectively prevent subarachnoid hemorrhage-induced edema (Laszlo et al, 1999) that is predominantly of the vasogenic type. However, the V1 AVP receptor, but not the V2 receptor, is shown to be involved in water regulation and in the pathophysiology of secondary brain damage after focal cerebral ischemia (Rosenberg et al, 1990), although the role of the V1 AVP receptor in cytotoxic or vasogenic edema was not distinguished. In a previous negative report on the V2 receptor antagonist for treatment of edema, SK&F 101926 or [adamantaneacetyl(1), O-Et-D-Tyr(2), Val(4), Abu(6), Arg(8,9)]-vasopressin), in which its specificity has been questioned, was used. In this study, OPC-31260 was used to treat cytotoxic edema. OPC-31260 was widely used as the V2 receptor antagonist and has been used in cerebral edemic studies (Laszlo et al, 1999). A previous finding showed that the antagonistic activity of OPC-31260 on the V2 receptor was more than 80 times more potent than those for V1a receptors in rodents (Nakamura et al, 2006). An in vitro study of a competitive displacement assay also showed that OPC-31260 was bound 100 times more for binding to the V2 receptor than to the V1 receptor (Yamamura et al, 1992). Therefore, the non-specific effects of OPC-31260 on the V2 receptor would be minimized.
Astrocytes in the brain are swollen during cytotoxic edema. An electrophysiological study suggested that V1-like vasopressin receptors were present in astrocytes and involved in vasopressin-mediated cell volume regulation. The AVP V1 receptor antagonist, but not the V2 receptor antagonist, was shown to significantly inhibit AVP-mediated astrocytic volume increase (Sarfaraz and Fraser, 1999). The protective effects of the V1 receptor antagonist against brain edema formation are suggested to be due to the inhibition of AQP-4 activity or expression by blocking the V1a receptors (Kleindienst et al, 2006). In this study, AQP-4 expression was also significantly reduced in GET-1 mice with the V2 receptor antagonist, OPC-31260, treatment after water intoxication, posing the possibility that the V2 receptor may also contribute to the vasopressin-dependent AQP-4-mediated cerebral edema formation in ischemic stroke. However, OPC-31260 could significantly delay and reduce astrocytic ET-1-induced cytotoxic edema formation. Astrocytic ET-1 in GET-1 mice does not appear to cross the BBB as the mice have no sign of hypertension as expected if there was increased ET-1 in the plasma due to BBB breakdown (Lo et al, 2005; Leung et al, 2004). In addition, the GET-1 mice showed no major effect on kidney function as their urine-concentrating ability also appeared to be normal (Lo et al, 2005), suggesting that astrocytic ET-1-induced brain edema is possibly mediated by the central V2 receptor. The GET-1 mouse model would serve as an invaluable model to further study the role of astrocytic ET-1-induced edema mediated through the V2 receptor in the central nervous system.
In summary, overexpression of ET-1 in astrocytes contributed to cytotoxic edema formation after water intoxication. The V2 receptor antagonist, OPC-31260, significantly prevented cerebral edema formation contributed by astrocytic ET-1 after water intoxication, suggesting the central effects of V2 receptor on cerebral edema formation. Water intoxication-induced lethality due to cytotoxic edema has been reported previously (Gardner, 2002). Overhydration due to sudden and excessive intake of water would cause fluid imbalances and hyponatremia, which are common among people undergoing athletic training, such as for marathons and strenuous work or in patients with kidney problems. The altered ET-1 system has been previously reported in disease conditions, such as stroke, hypertension, and chronic heart disease. Thus, this study suggested that those patients may have a higher risk of developing cytotoxic edema when they intake water in excess within a short period of time. Treating patients with water intoxication with OPC-31260 at the early critical stage could help to delay neurological deficits and reduce lethality. The current study will provide a better understanding of the therapeutic use of vasopressin receptor antagonist for treatment of cytotoxic brain edema in stroke patients.
Footnotes
The authors declare no conflict of interest.