
Abstract
Treatment efficacy for ischemic stroke represents a major challenge. Despite fundamental advances in the understanding of stroke etiology, therapeutic options to improve functional recovery remain limited. However, growing knowledge in the field of epigenetics has dramatically changed our understanding of gene regulation in the last few decades. According to the knowledge gained from animal models, the manipulation of epigenetic players emerges as a highly promising possibility to target diverse neurologic pathologies, including ischemia. By altering transcriptional regulation, epigenetic modifiers can exert influence on all known pathways involved in the complex course of ischemic disease development. Beneficial transcriptional effects range from attenuation of cell death, suppression of inflammatory processes, and enhanced blood flow, to the stimulation of repair mechanisms and increased plasticity. Most striking are the results obtained from pharmacological inhibition of histone deacetylation in animal models of stroke. Multiple studies suggest high remedial qualities even upon late administration of histone deacetylase inhibitors (HDACi). In this review, the role of epigenetic mechanisms, including histone modifications as well as DNA methylation, is discussed in the context of known ischemic pathways of damage, protection, and regeneration.
CEREBRAL ISCHEMIA AND ENDOGENOUS TOLERANCE
According to World Health Organization statistics, stroke is the second leading cause of death and long-term disability in developed countries (http://www.who.int/mediacentre/factsheets/fs310/en/index.html). In spite of major advances in the understanding of the disease etiology, only few and limited treatment options within a very narrow time range are as yet applicable. 1 An impressive amount of neuroprotective agents have been identified in animal models, however, none of them proved efficient in clinical trials until now. 2 This might be partly due to inappropriate preclinical modeling of the disorder and stroke care as well as insufficient preclinical testing. 3 Growing knowledge of the intricate disease mechanisms suggests that an effective treatment requires the ability to interact with not one, but multiple and diverse pathophysiological cascades induced by the insult.
As a reduction of blood flow occurs and leaves brain tissue unsupplied with oxygen and glucose, a highly complex series of spatial and temporal events begins to evolve. Cells in the ischemic core, the zone most affected by restricted energy supply, die within minutes. Energy failure prevents the maintenance of the fine-balanced membrane potential, crucial for proper functioning and survival. As a result, the neurotransmitter glutamate is excessively released, which, in turn, results in an overload of calcium, causing excitotoxic cell death. 4 However, cells in the penumbra, where energy metabolism is partially preserved, are predominantly confronted with propagating excitotoxicity, oxidative and nitric stress, and secondary phenomena like inflammation, spreading depolarization, and delayed cell death. 5 All these processes (Figure 2A) go hand in hand with great changes in gene expression that affect hundreds of genes.
Different players regulate the genomic response, concurrently inducing both, beneficial and harmful pathways.6,7 A crucial modulator is the transcription factor, hypoxia-inducible factor (HIF), activated upon hypoxia.8,9
Because of the slowly progressing nature of the mechanisms observed in the penumbra, where neurons may survive several hours or even days, 10 a window for pharmacological intervention is provided. 4 Identified neuroprotective agents target the whole range of the different pathways involved: some inhibit excitotoxic cell death, others interfere with oxidative stress or promote cell survival signaling, still others are able to enhance blood flow, stimulate neurogenesis and plasticity, or display anti-inflammatory properties. 11
The fact that epigenetic molecules—being the key regulators of transcription—may simultaneously act on all these different levels of ischemic brain injury makes them promising candidates for clinical use.
THE EPIGENETIC MACHINERY
DNA methylation occurs at CpG islands and confers repression to the gene localized in the surrounding (Figure 1). The methylation step is carried out by DNA methyltransferases (DNMTs) and the subsequent repressive effect is mediated by the recruitment of chromatin modifiers that bind to the methylated site. 12 Other epigenetic marks, particularly histone acetylation and phosphorylation, directly influence the structure of the chromatin. Acetylation at histone lysine residues neutralizes their positive charge and thus reduces the association to the negatively charged DNA. Consequently, this open form of chromatin allows transcription factors and transcriptional co-activators to access specific gene promoters and induce active transcription. 13 The lack of acetyl groups generates condensed heterochromatin, usually associated with gene repression.14,15 Acetylation and deacetylation occur in a dynamic process. While the acetylation of lysine residues (of histones as well as non-histone proteins) is carried out by histone acetyl transferases (HATs) using an acetyl group stemming from Acetyl-Co-A, the removal of acetyl groups is performed by histone deacetylases (HDACs) (Figure 1). The family of HDACs consists of four classes based on sequence similarity. Class I (HDAC 1 to 3 and 8), class II (HDAC 4 to 7, 9, and 10), and class IV (HDAC 11) HDACs are zinc dependent, while the so-called ‘Sirtuins’ of class III act in a nicotinamide adenine dinucleotide-dependent manner.16,17 DNA methylation and histone acetylation have been more extensively studied than other post-transcriptional modifications such as histone methylation, phosphorylation, sumoylation, or ubiquitinilation. However, knowledge on histone methylation and its fundamental role in transcriptional regulation is accumulating. In a dynamic process, lysine and arginine residues can be methylated by histone methyltransferases and demethylated by histone demethylases. The methylation marks can act as both, activating or repressive marks depending on the specific position of the residue. 18
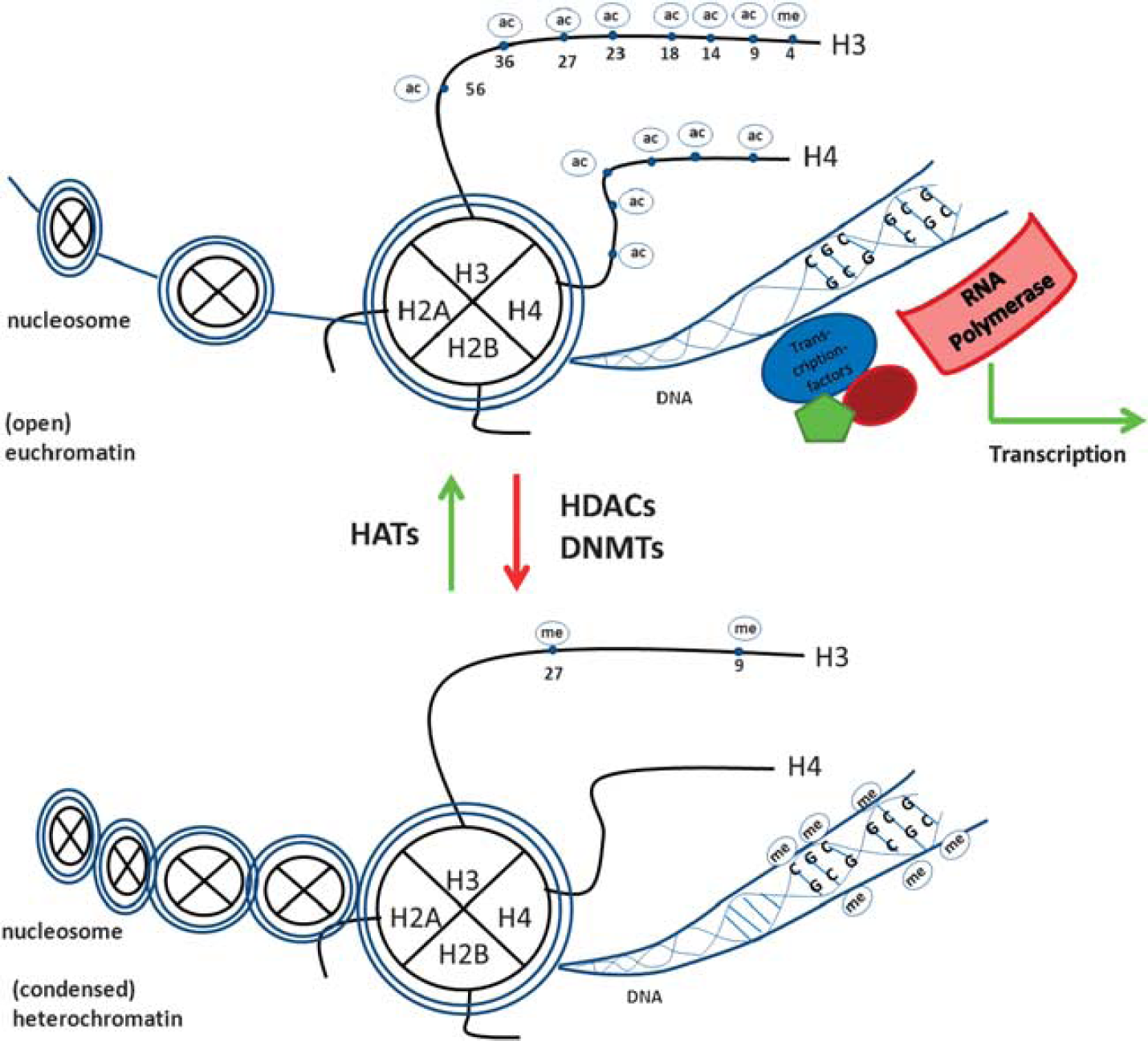
Epigenetic regulation of transcription. Transcription is possible within open (eu-) chromatic regions, where typical epigenetic marks, such as histone 3 and 4 acetylation (ac), as well as histone 3 lysine 4 trimethylation (me) appear together with the lack of DNA methylation (me). However, genes in more condensed (hetero-) chromatic regions are repressed. Characteristic epigenetic marks are DNA methylation together with histone 3 lysine 9 di-/trimethylation, as well as lysine 27 trimethylation.
Ultimately, all these marks are known to act together and form a complex histone code, responsible for the orchestration of gene expression, the regulation of access to the chromatin, and finally, the recruitment of interacting proteins such as transcription factors and chromatin modifiers.18,19
EPIGENETICS AND THE NERVOUS SYSTEM
An orchestrated expression of genes is essential for cell differentiation as well as cell survival. It implies the control of homeostasis, but also requires dynamic modulation in response to environmental stimuli, including stress or injury. Postmitotic cells, such as neurons, are exceptional in this context, as they are not subject to continuous renewal and their life span usually coincides with that of the whole organism. This fact renders their preservation crucial and makes an adaptation to stress as well as a potential for recovery necessary, not only to their own survival. 20
A body of evidence is accumulating that epigenetic modifications regulate a wide range of neuro-physiologic as well as neuro-pathologic processes. In the brain, the epigenetic machinery covers basic phenomena such as differentiation, the preservation of tissue specific transcription profiles, as well as complex processes from synaptic plasticity to learning and memory. 21 Mutations and maladaptations of the epigenetic system on the level of DNA methylation, histone acetylation, and methylation are implied in neuro-developmental22,23 and neurodegenerative24,25 contexts, as well as neuropsychiatric diseases26,27 and drug addiction28,29 (for a review, see Graff et al. 30 ). More specifically, the role of HDACs as regulators of transcription has been identified to be fundamental, and perturbation of acetylation homeostasis is now acknowledged to be a central event in neurologic pathologies. Currently, the research focus lies on HDAC inhibition. There are multiple ongoing clinical trials using HDAC-inhibiting drugs in the context of diverse cancer pathologies. Suberoylanilide hydroxamic acid (SAHA), a class I and II HDACi, has already been approved as a treatment for cutaneous T-cell lymphoma. 31 In cancer cells, HDACi function as potent inducers of cell cycle arrest, differentiation, and apoptosis. 32 Paradoxically, strong evidence from rodent models is accumulating that HDAC inhibition shows significant neuroprotective properties and attenuates cell death in most diverse neuropathological conditions such as Huntington’s disease, 33 spinal muscular atrophy, 34 status epilepticus, 35 and experimental autoimmune encephalitis 36 (for reviews, see Shein and Shohami 37 and Abel and Zukin 38 ). Further, targeted manipulation of aberrant histone acetylation shows promising results in the treatment of ischemic damage.
In this review, the current knowledge on various epigenetic mechanisms, such as histone acetylation, methylation, phosphorylation, and DNA methylation and their interaction, will be discussed in the context of known pathways in cerebral ischemia. Given the current state of research, the main focus lies on the effects of HDAC inhibition (Figure 2B). The role of micro RNAs will not be considered. For comprehensive reviews see Saugstad 39 and Rink and Khanna 40 ).
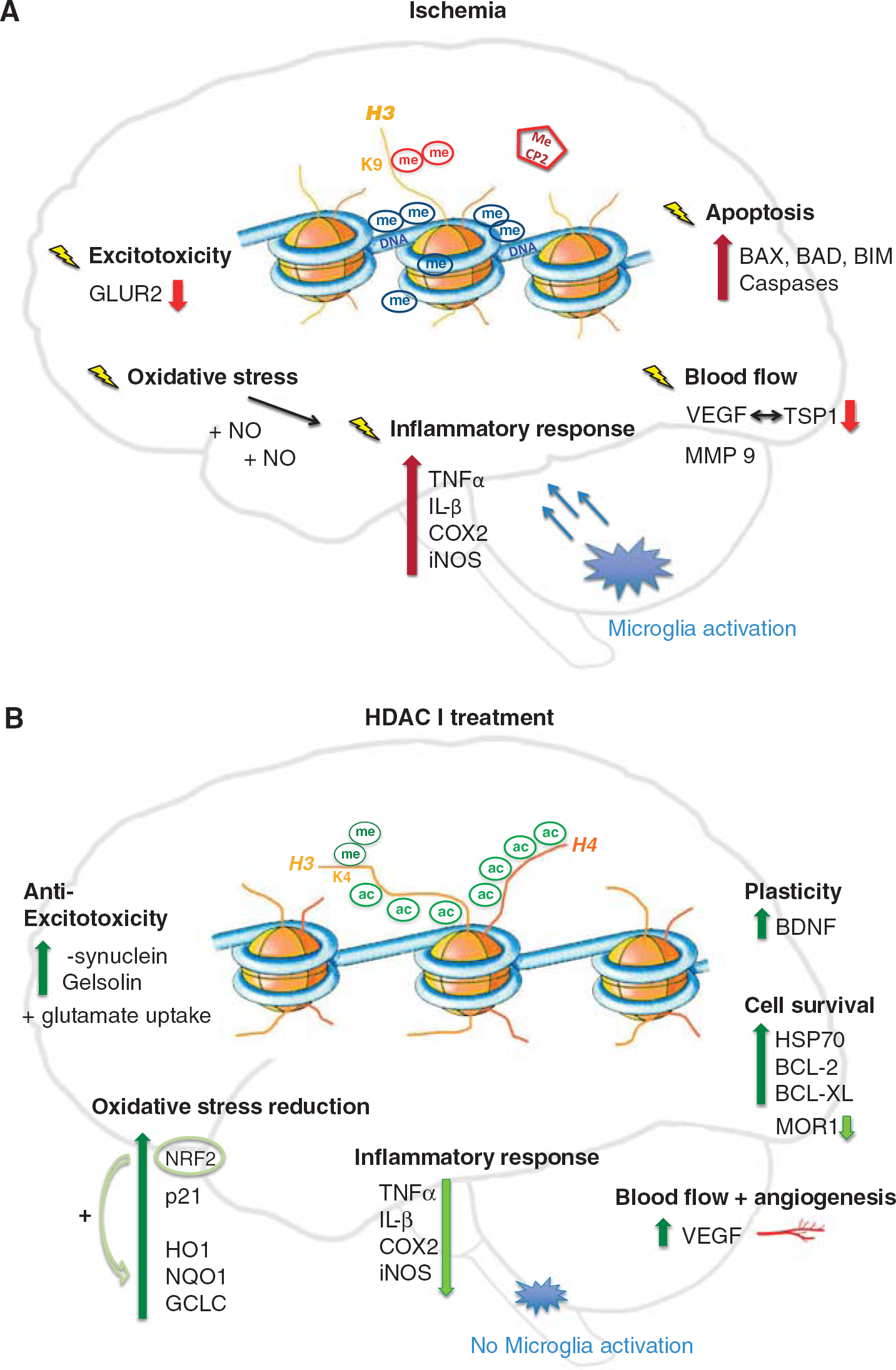
Ischemic pathways of damage (
GLOBAL EPIGENETIC CHANGES IN RESPONSE TO ISCHEMIA
DNA Methylation
Ischemia leads to great alterations in gene expression.6,41,42 Generally, increased transcriptional repression can be observed together with the corresponding silencing epigenetic marks. The global amount of DNA methylation rises after an ischemic insult and this increase correlates with augmented brain injury.43,44 Interestingly, the manipulation of transcription on the epigenetic level can yield a protective state. It has, for example, been demonstrated that pharmacological inhibition of DNA methylation ameliorates neurologic outcome in a rodent model of ischemia. 43 Further, mice expressing reduced levels of DNA methyltransferase1 (DNMT1) in postmitotic neurons are protected from ischemic brain injury. A complete absence of DNMT1, however, does not lead to functional recovery. 44 A possible explanation for the observed phenomenon might be that reduced levels of DNMT1 influence chromatin structure and allow transcription factor binding, such as HIF-1, and thereby upregulate neuroprotective genes. This exemplifies that, in spite of being visible on a global scale—such as a global change in DNA methylation—it is more likely that changes occur on a gene- and promoter-specific level rather than operating over large genomic regions 45 ).
Histone De-/acetylation
Global transcriptional repression upon ischemia is similarly reflected on the histone acetylation level. In and around the ischemic core, a general decrease of histone H3 acetylation46–50 as well as of H4 acetylation levels51–54 was identified in multiple studies. Vast deacetylation processes occur quickly upon an ischemic insult and induce a fatal course of events. However, because of the dynamic nature of epigenetic modification, it is possible to reverse this detrimental state of repression. Promising therapeutic agents that have been subject to extensive analysis are the diverse group of HDACi. A number of different classes exist and the development of new derivatives is intensively investigated aimed at designing inhibitors that exhibit decreased toxicity, high potency, blood–brain barrier permeability, and improved selectivity.55–58
An overview of HDACi highlighting mechanisms of action, blood–brain barrier permeability, clinical assessment, and safety information can be found in Table 1.
HDAC inhibitors
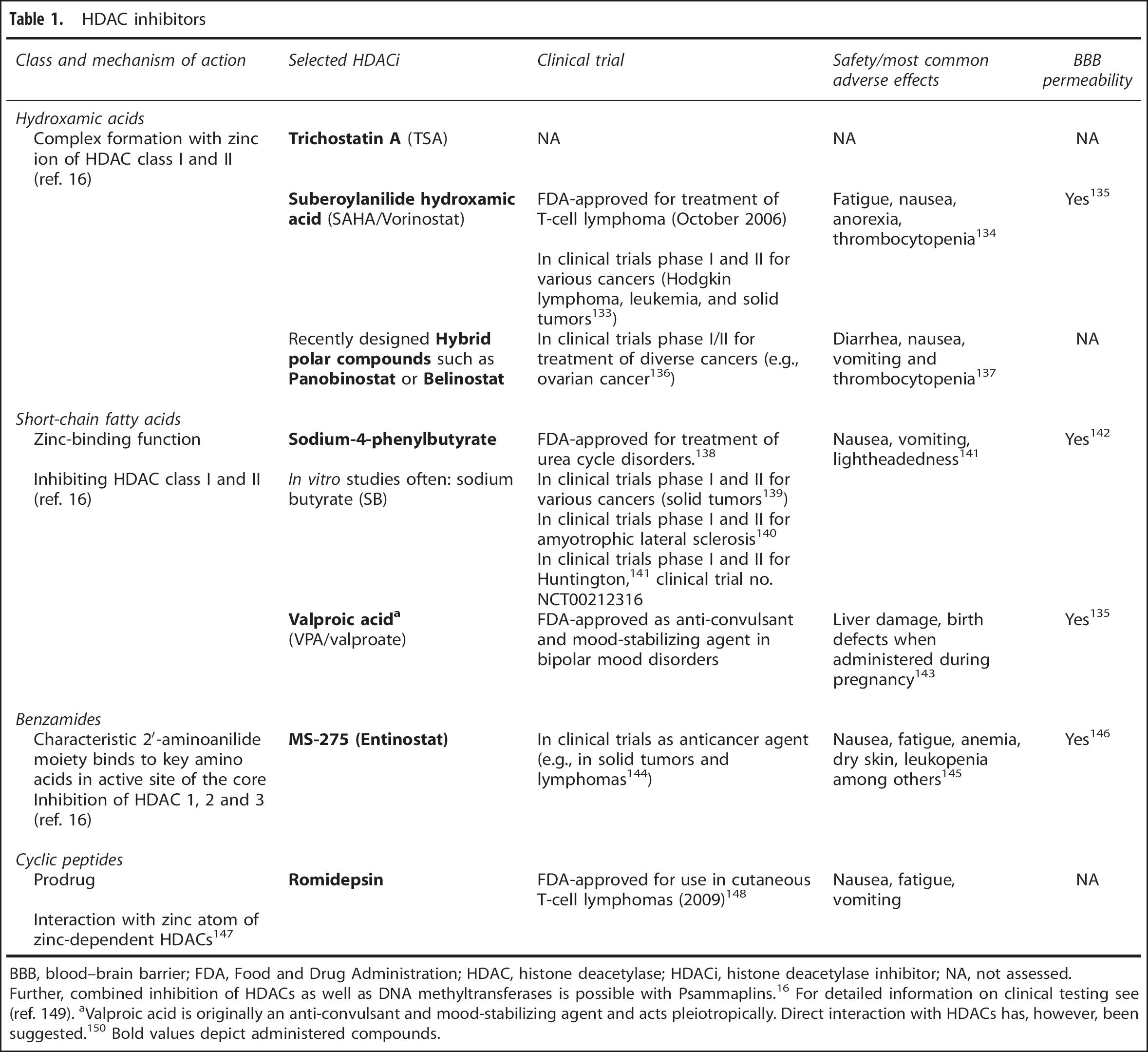
BBB, blood–brain barrier; FDA, Food and Drug Administration; HDAC, histone deacetylase; HDACi, histone deacetylase inhibitor; NA, not assessed.
Further, combined inhibition of HDACs as well as DNA methyltransferases is possible with Psammaplins. 16 For detailed information on clinical testing see (ref. 149). aValproic acid is originally an anti-convulsant and mood-stabilizing agent and acts pleiotropically. Direct interaction with HDACs has, however, been suggested. 150 Bold values depict administered compounds.
In stroke, treatment with valproic acid (VPA), trichostatin A (TSA), sodium butyrate (SB), SAHA, or MS-275 at appropriate doses leads to maintenance of histone 3 and 4 acetylation levels during ischemia, or their re-establishment after late administration. In spite of pan-HDACi being selective for only ~2% to 5% of all genes,59,60 they can influence multiple and most diverse pathways involved in the course of ischemic pathology and interestingly do not only induce gene activation, but global transcriptional changes including the downregulation of some genes (Figure 2B). Thus, HDACi are thought to restore the transcriptional balance in response to ischemic injury. As a consequence, pan-HDAC inhibition, applied in rodent models, both as pre- and post-treatment, significantly reduces infarct volume, attenuates brain damage, and promotes functional recovery in a number of studies.46,47,49,54,61,62 A beneficial effect can be monitored upon HDACi administration up to 7 hours poststroke. 48 For a meta-analysis of outcome regarding the beneficial effects of different HDACi in stroke see Gibson and Murphy. 63 Improved outcome parameters will not further be considered in the following chapters as the focus lies on the pathways involved in damage, protection, and regeneration. However, a concise list of reviewed papers with details on administration as well as information on reduced infarct volume and neurologic deficits is available in Table 2.
Publications using HDAC inhibition in ischemic models
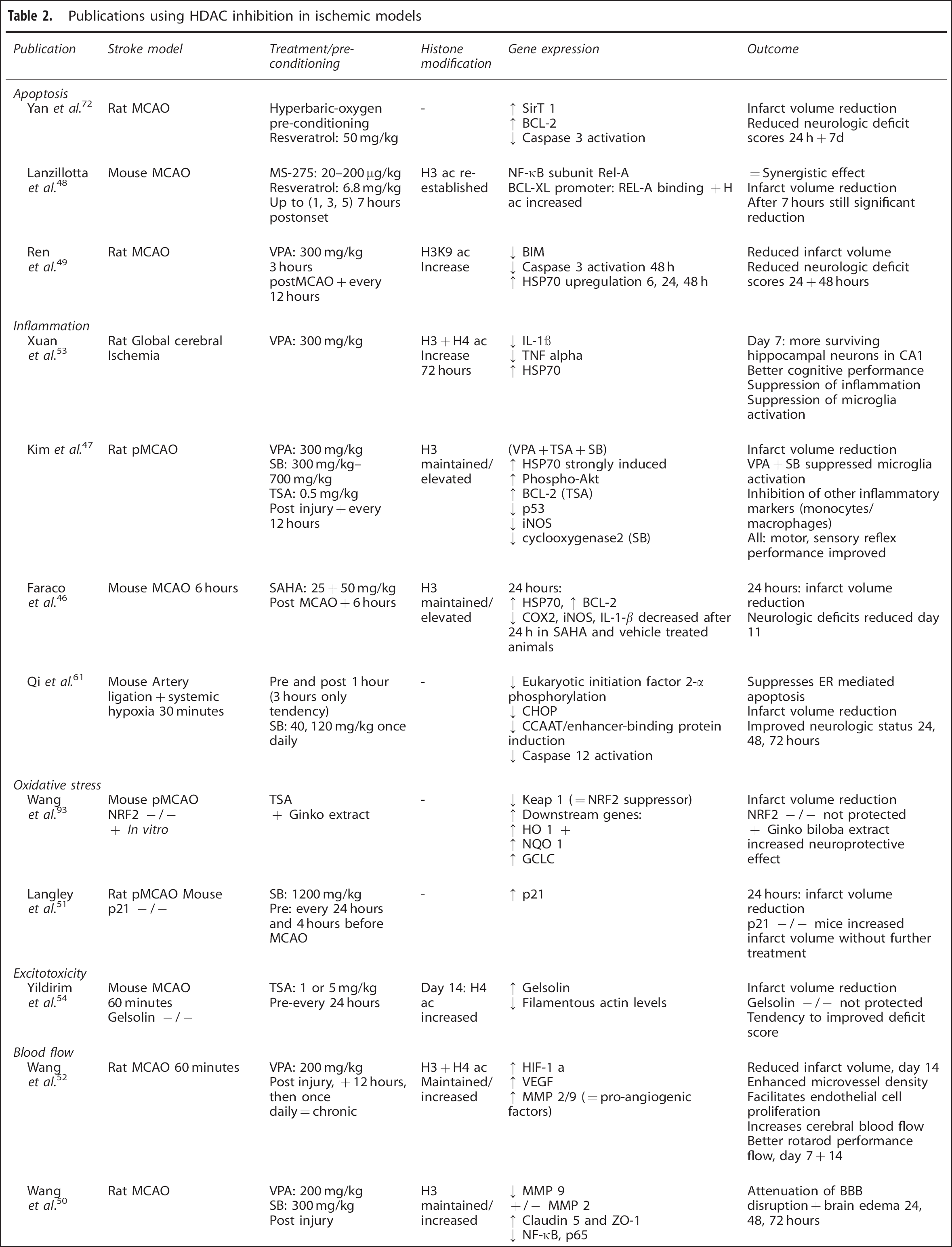
BDNF, brain-derived neurotrophic factor; CHOP, C/EBP homology protein; ER, estrogen receptor; GCLC, glutamate cystein ligase; GFAP, Glial fibrillary acidic protein; HDAC, histone deacetylase; MCAO, middle cerebral artery occlusion; MMP, matrix metalloprotease; pMCAO, permanent middle cerebral artery occlusion; SAHA, suberoylanilide hydroxamic acid; SB, sodium butyrate; TNF, tumor necrosis factor; TSA, trichostatin A; VEGF, vascular endothelial growth factor; VPA, valproic acid. ↑ = increased expression/levels after treatment; ↓ = downregulation after treatment; + / − = unaffected.
Histone Acetylases
Hypoxic conditions compel cells to rely on anaerobic glycolysis for energy production. With the reduction of aerobic metabolism, acetyl-Co-A levels drop. Histone acetylases require acetyl-CoA as a cofactor to perform the histone acetylation step. Furthermore, HATs are activated upon phosphorylation. With ATP being scarce, hypoxia might lead to an inhibition of HAT activation. 64 Suppression of HAT activation might well contribute to the observed decrease in histone acetylation levels upon stroke. However, HAT activity does not seem to be altered by cerebral ischemia. 46 It adds to the complexity that the HATs CBP/p300 and SRC-1 are known to be involved in hypoxia-mediated transactivation of HIF-1.64,65 Hypoxia-inducible factor activation is regarded as a major protective event in the endogenous defense against hypoxic damage. The involvement of HATs in this process would mark them as protective players. However, it was shown that genetic inhibition of the HAT CBP/p300 yields neuroprotective effects after experimental global cerebral ischemia. 66
Histone Deacetylases
Knowledge on HDACs and their role in the ischemic as well as in the healthy brain of rodent models is accumulating. Brain maps with expression analyses of HDACs are now available for mice, 67 rats, 68 and also humans (Website: ©2012 Allen Institute for Brain Science. Allen Human Brain Atlas (Internet). Available from: http://human.brain-map.org/). However, no comprehensive work exists that compares expression patterns among species and would allow for conclusions on similarities between rodent models and human patients. Analyzing ‘normal’ adult mouse brains, for example, revealed that HDAC isoforms 1 to 11 are all expressed throughout the brain showing a cell- and region-specific pattern.67,69 In the cerebral cortex, the distribution of HDAC 1 to 3 is found to be particularly widespread and prominent. 67 Interestingly, HDAC 2 is predominantly localized in the nucleus, indicating its active role in chromatin remodeling, whereas HDAC 1 (ref. 67) and HDAC 6 (ref. 70) are rather situated in the cytoplasm, pointing to a possible involvement with non-histone targets.
Ischemia, induced by middle cerebral artery occlusion (MCAO), leads to alterations of HDAC expression. While, in mice, no expression can be found in the ischemic core 1 week after the insult, substantial upregulation of HDAC 3, 6, and 11 occurs within hours after stroke in the cortex and was shown to last during the early phases. 69 Later, after 1 week, HDAC 1, 2, and 3 are still upregulated in the peri-infarct area. 67
Treatment with the neuroprotective pan-HDACi SAHA reduces ischemia-induced alterations in HDAC expression. 67 Not only pan-HDACi, but the manipulation of single HDACs as well, can induce protective states. In an in vitro model of stroke, oxygen glucose deprivation (OGD), specific knockdown of HDAC 3 and 6 promotes the survival of cortical neurons. 69 The important role of HDAC 6 could also be confirmed in an oxidative stress model, where selective inhibition of HDAC 6 alone leads to an attenuation of cell damage comparable to that of pan-HDAC inhibition. 70 Currently, selective HDAC 6 inhibitors are being developed and optimized. 71 Summing up, in approaches to pin down the role of protection to single HDAC isoforms in rodent models, HDAC 3 and HDAC 6 were identified as potential mediators in neurotoxicity. However, it remains to be elucidated in how far the protection is mediated via epigenetic changes in transcriptional activity in neurons and glial cells, or whether non-transcriptional actions in cytoplasmic compartments may significantly modulate the response to injury. Cytoplasmic targets might also be relevant in the explanation of a very recent finding that—on the first glance—stands in contrast to the widely observed neuroprotection upon HDACi. The upregulation of the HDAC SIRT1 has been described to confer protection in stroke. 72 However, histone acetylation levels are not considered in this study and SIRT1 is known to have a wide range of non-histone targets 73 that constitute potential mediators of the protective effect.
Interestingly, in a genome-wide association study for ischemic stroke, carried out in humans by the International Stroke Genetics Consortium and the Welcome Trust Case Control Consortium2, a new association has been identified between HDAC 9 and large vessel stroke, one of the brain infarct subtypes. According to the study, carriers of the A-allele (about 9% of the study population) as well as homozygous carriers (1% of the population) have an increased risk for stroke. However, nothing is known about the underlying molecular pathways.74,75
These findings highlight the complexity that has to be faced—as different epigenetic candidates might be involved in different subtypes of stroke. Moreover, given these circumstances, it might be necessary to re-evaluate our concepts of infarct maturation resulting from destructive and protective mechanisms as a more sophisticated phenomenon, orchestrated by individual genetic as well as epigenetic factors.
Epigenetic Crosstalk Upon Ischemia
Repression is not only monitored on the level of DNA methylation and histone acetylation, but further evidence for crosstalk and tight orchestration of the epigenetic response also exists.
A relatively recent finding demonstrates that treatment with HDACi not only increases histone acetylation levels, but also affects histone methylation levels in neurons as well as astrocytes.76,77 Pan-HDACi, as well as Class I HDACi, increase histone 3 lysine 4 (H3K4) di- and trimethylation (me2/me3), which constitute typical marks of transcriptional activation. This may be accompanied by a decrease in repressive H3K9me2 levels.76,77 Moreover, HDACi treatment not only influences histone methylation, but is also described to modulate microRNA pathways poststroke. 78 These findings suggest that the whole epigenetic machinery works in concert in physiologic as well as pathologic conditions and adaptive responses in the brain.
Regulation of global gene silencing after stroke is mediated at least in part by the transcription factor repressor element silencing 1 transcription factor (REST), a potential candidate for orchestrating epigenetic modifications in postischemic neurons.79–81 REST is activated after neuronal damage and its derepression leads to the repression of many neuronal genes involved in synaptic plasticity and remodeling. REST silences target genes via association with distinct corepressor elements, which in turn recruit HDACs 1 and 2, but also associate with the transcriptional repressor methyl-CpG binding protein 2 (MeCP2), as well as the repressive histone methyltransferase G9a. 80 The depletion of REST prevents repressive epigenetic modifications after ischemia, restores gene expression, and rescues hippocampal neurons. 81 However, the complex etiology suggests more than one pathway involved in the regulation and response to ischemia. Repressive epigenetic marks predominate in ischemic damage while a protected state is associated with open chromatin and globally enhanced gene activation.
THE EPIGENETIC MACHINERY IN PATHWAYS OF ISCHEMIC BRAIN DAMAGE AND PROTECTION
Excitotoxicity
The major excitatory neurotransmitter in the brain is glutamate. After an ischemic insult, glutamate uptake is impaired, excessive amounts accumulate, and lead to an increased calcium influx into neurons, which in turn sets diverse degenerating cascades into motion. Diverse proteins have been identified that are involved in the prevention of excitotoxic injury, when it comes to HDAC inhibition. They tackle the problem of glutamate accumulation either by modulating its uptake or by dealing with secondary phenomena such as the developing calcium overload. In an in vitro model of excitotoxicity, it was demonstrated that valproate pre treatment prevents glutamate-induced excitotoxicity in cortical neurons 82 and cerebellar granule cells83,84 highlighting the role of α-synuclein as a mediator of protection against excitotoxicity. 84 HDACi application also preserved white matter functioning and stimulated astrocytic glutamate uptake in an OGD model. 85 Getting rid of excessive amounts of glutamate allows normal synaptic activity and can save the neurons. The gene of glutamate receptor 2 (GluR2) involved in synaptic functioning for example is repressed upon ischemia. 79 This goes hand in hand with H3K9 deacetylation and enrichment of H3K9 dimethylation over the promoter 81 in hippocampal CA1 neurons. With the help of HDAC inhibition as well as knockdown of the transcription factor REST, which assembles epigenetic repressors like HDAC 1 and 2 and G9a, gene silencing can be attenuated and hippocampal neurons are rescued. Furthermore, a beneficent effect could be observed with TSA pretreatment in mice undergoing MCAO. In this context, an upregulation of the protein gelsolin was assessed.54,86 Gelsolin is a protein involved in actin remodeling and the stabilization of calcium channels and hence has a role in excitotoxic cell death.87,88 In treated wild-type mice that underwent experimental stroke, low calcium levels coincide with gelsolin upregulation. As HDACi pretreatment does not ameliorate the state of gelsolin-deficient mice in ischemic models, gelsolin is identified as an important mediator in ischemic cell death. 54
Oxidative Stress
In the course of ischemia, a high amount of reactive oxygen species (ROS), particularly nitric oxide (NO), is generated. 89 ROS accumulation is a fatal event, as it contributes to the destruction of cellular macromolecules such as DNA. To promote survival, cells have developed several strategies for the detection of DNA damage and epigenetic modulators are recognized as key molecules in repair processes of oxidative damage. 90 Histone 2AX is phosphorylated (γ-H2AX) upon oxidative stress and accumulates with progressing injury.91,92 Oxidative stress constitutes an important element that contributes to subsequent injury in ischemia. Both, in models of pure oxidative stress, 70 as well as ischemic models, HDACi are able to attenuate neuronal death. A key regulator of antioxidant-responsive genes is the transcription factor nuclear factor erythroid 2-related factor 2 (NRF2). Upon pre treatment with diverse HDACi, the repression of NRF2 suppressor Keap1 is monitored in an in vitro model of stroke. 93 As a result, NRF2 translocates to the nucleus and binds to gene promoters with antioxidant response elements to coordinate the expression of anti-oxidative enzymes. After TSA administration, downstream gene transcripts with antioxidant response elements, such as heme oxygenase1 (HO1), NQO1, and glutamate cystein ligase are boosted. In line with this, in vivo experiments with NRF2 knockout mice show no protection in spite of HDACi treatment, whereas animals, where NRF2 pathways have been activated display an enlarged protective effect. 93 These findings identify NRF2 as a key element in the antioxidant response, the regulation of which is subject to the epigenetic machinery. Resveratrol, a modulator of sirtuin activity, leads to NRF2 protein acetylation and thereby provides cell protection. 94 Together, these findings indicate a partial contribution of NRF2 in HDACi-mediated protection.
P21 has recently been suggested to be a stabilizer of NRF2 (ref. 95) and is furthermore known to inhibit cell cycle progression and apoptotic pathways. Pretreatment with diverse HDACi enhances p21 promoter acetylation and p21 expression. 51 In neurons, TSA treatment increases the interaction of p21 with ASK1, a regulator of stress activated SAPK/JNK pathways. However, in an in vitro model of oxidative stress, p21-deficient neurons show a comparable protection to wild-type cells upon HDACi administration, which suggests that p21 is not an essential component of oxidative stress-induced cell death and hints at possible compensatory mechanisms. 51
Inflammation
Post stroke inflammation is one of the more delayed mechanisms and yet a major cause of damage (for reviews, see Dirnagl et al., 4 Priller and Dirnagl, 96 and Price et al 97 ). Many different cell types with their characteristic gene expression patterns are involved in the inflammatory response. Accordingly, HDACi have a cell type specific impact on gene expression. Effects can differ extensively, or even be opposed e.g., in microglial cells versus macrophages.98,99 In general, the administration of either HDACi, VPA, or SB, leads to a significant suppression of microglia activation47,53,100 and additionally to the inhibition of further inflammatory markers like monocytes and macrophages in stroke. 47 On the gene level, HDAC inhibition impacts the expression of heat shock protein (HSP)70, a heat shock/stress protein that assists proper protein folding, and is involved in inflammatory processes. Interestingly, HSP70 can show both anti- and pro inflammatory effects depending on cell type, context, and location. 101 In ischemic models, its HDACi-mediated upregulation is observed together with the downregulation of pro-inflammatory molecules such as tumor necrosis factor-α (TNF-α) and interleukin-β (IL-β), both inflammatory cytokines.53,61,102 The elevation of two further molecules is attenuated in this setting: cyclooxygenase 2 (COX2), 47 which accelerates the production of prostanoids and free radicals, as well as iNOS.47,61 iNOS catalyzes the production of nitric oxide and constitutes an essential mediator of the immune response. Its knockdown was shown to ameliorate the overall damage caused by ischemia. 103 All in all, these findings suggest a downregulation of the most important inflammatory players amidst a general rise in gene transcription upon HDACi treatment. On the contrary, a study exists that could not confirm these observations. In this paradigm, the HDACi SAHA was employed and no significant decrease of IL-β, COX2, and iNOS levels could be monitored after 24 hours, in spite of an increase in HSP70 protein levels. 46 Possibly, the application of different HDACi modulates response pathways in various ways.
Apoptosis/Cell Death
Regarding cell death and ischemia, HDAC inhibition seems to shift the ratio of apoptotic versus anti-apoptotic agents to the side of the protective pathways. Upon HDACi, neuroprotective BCL(B cell lymphoma)-2 family members, such as BCL-2 (ref. 46,47) and BCL-XL 48 are upregulated. A coordinated response is induced, which not only includes events of gene induction but also repression. As a consequence of HDACi administration, the downregulation of pro-apoptotic players such p53 (ref. 47) with its downstream actors BCL-2-associated X protein (BAX) 104 and caspase 3 (ref. 49) can be detected. Further, agents that are a typical for endoplasmatic reticulum stress induced cell death, like C/EBP homology protein (CHOP) and caspase 12 (ref. 61), are suppressed. Another example is the protein complex NF-кB, which can consist of various subunits. Different subunit acetylation patterns can either promote binding to pro-apoptotic proteins, such as BCL-2-like protein 11 (BIM), and trigger a neurotoxic cascade, or to the protective BCL-XL promoter. Histone deacetylase inhibitorstreatment prohibits deranged subunit acetylation and promotes survival signaling. 48 The induction of pro-apoptotic BCL-2 family members and diverse caspases is also inhibited by HSP70 upregulation upon HDAC inhibition. HSP70, regarded as one of the key mediators of cell survival pathways, is known to interact with apoptotic protease-activating factor 1 (APAF) and apoptosis-inducing factor (AIF). 5 Under ischemic conditions, HSP70 is expressed,105,106 however, its transcription is highly increased after treatment with diverse HDACi.46,47,53,76,77,107 This increase seems to depend on the AKT pathway and the transcription factor SP1, of which HSP70 constitutes a target. 76 Histone deacetylase inhibition probably augments SP1 protein acetylation, which facilitates SP1 binding to the DNA and promotes SP1-dependent gene expression. 108 Valproate treatment supports the association of SP1 with HAT p300 and the recruitment to the HSP70 promoter, inducing its transcription. The induction is further reflected on the level of histone methylation with increased H3K4 methylation at the HSP70 promoter of neurons as well as astrocytes. 77 The crucial role of HSP70 is confirmed by studies that show that HSP70 overexpression alone attenuates ischemic damage and seizures.109–111
Interestingly, not only the histones at their expected location in the nucleus of the cell are involved in poststroke apoptotic processes. Dead cells release chromatin and as a result, the overall levels of circulating nucleosomes and DNA fragments are increased after an ischemic injury. These floating chromatin pieces turn out to be cytotoxic while the cleavage of extracellular DNA as well as histone neutralisation with antibodies leads to protection in an ischemic mouse model. 112 Whether different histone modifications influence cytotoxicity remains to be investigated.
Poststroke Recovery: Blood Flow and Blood–Brain Barrier
Poststroke angiogenesis correlates with survival. 113 Cellular angiogenic activity is marked by a balance between angiostatic and angiogenic drives. One of the earliest incidences after stroke is the induction of the pro-angiogenic factor, vascular endothelial growth factor (VEGF) mediated by the transcription factor HIF-1. However, postischemic angiogenesis is only a short-lived event. It was suggested in a study with murine cerebral endothelial cells that DNA methylation contributes to the modulation of angiogenesis via epigenetic regulation of thrombospondin-1 (TSP1), an endogenous angiostatic factor, which is expressed in inverse patterns to VEGF. 114 In contrast to VEGF, the TSP1 promoter is methylated and TSP1 expression is downregulated for a brief postischemic period. 114 After 8 hours of re-oxygenation, the expression is already restoring. A prolonged maintenance of repressive epigenetic marks over the TSP1 promoter might lead to an extension of the angiogenic period and hence reduce damage. The human TSP1 promoter region is equally rich in CpG islands and its hypermethylation goes along with reduced expression of TSP1 in several human malignant tumor cell lines.115,116 However, VEGF and TSP1 are not the only factors that concern blood flow. Members of the matrix metalloprotease (MMP) family, especially MMP 2 and 9, display pro-angiogenic properties as well. By degrading extracellular matrix proteins, they set the stage for endothelial cell migration and capillary sprouting. Upon poststroke HDACi treatment, enhanced microvessel density, facilitated endothelial cell proliferation, and increased relative cerebral blood flow were assessed. 52 The protective effect is attributed to HIF-1 activation and subsequent upregulation of its downstream pro-angiogenic factor VEGF, but additionally to highly increased MMP 2 and 9 protein levels and activity upon HDAC inhibition. 52 Hence, in this setting, HDACi are observed to mediate an MMP2 and 9 induction, which is regarded as a protective event.
The role of MMP 2 and 9 however, seems to be more complex. Both are involved in the postischemic disruption of the blood–brain barrier (BBB), which is formed by a neurovascular unit consisting of endothelial cells, pericytes, astrocytes, neurons, and the extracellular matrix forming the basement membrane around the vessels. Loss of the BBB integrity, which is protecting the neuronal microenvironment, allows the extravasation of molecules and fluids, thereby incurring functional disturbance as well as further brain damage. As members of the MMP family, more precisely MMP 2 and 9, have tight junction proteins as substrates, 117 they not only allow for the protective event of capillary sprouting, but constitute key proteases that mediate BBB disruption. Matrix metalloprotease 9 is quickly upregulated 24 hours postinsult. Thus, MMP 9 knockout animals are resistant to BBB opening postischemia.50,118,119 In contrast to the findings mentioned above, 52 where an enhanced upregulation of MMP 2 and 9 was observed upon HDAC inhibition, one study observed a suppression of MMP 9 induction upon HDACi treatment immediately after stroke onset and argues that MMP 9 suppression renders tight junction molecules like claudin 5 and ZO-1 invulnerable to degradation 50 and thereby promotes BBB integrity and protects the tissue.
The paradoxical observations concerning MMP 9 regulation cannot be explained by the inhibition of diverse HDAC subclasses, as in both contradictory studies, valproate was applied directly poststroke. A clue to this question of MMP 9 regulation might be the time point. The angiogenic effects were noticed lately—on day 7 and 14 poststroke—other than the attenuation of blood–brain barrier disruption, which was assessed directly after disease onset.
Thus, angiogenesis as well as the inhibition of BBB disruption constitute key events of poststroke neurovascular remodelling and protection that are influenced by HDAC inhibition.
Neurogenesis and Plasticity
In the healthy brain, neurogenesis mainly takes place in the subventricular zone (SVZ) and the hippocampal dentate gyrus. Cerebral ischemia stimulates cell proliferation in these regions. The newly born cells are witnessed to migrate into damaged regions. 120 A vital factor for neurogenesis, survival signalling, and synaptic plasticity in the ischemic hemisphere is brain-derived neurotrophic factor (BDNF).121–123 Histone deacetylase inhibition induces BDNF-receptor tyrosine kinase (TrkB)-dependent cell proliferation, migration, and differentiation in experimental stroke. 91 Moreover, postischemic application of HDACi triggers cell division outside the subventricular zone and dentate gyrus, for example in the striatum and frontal cortex. However, simultaneous inhibition of the BDNF TrkB abolishes the effects of HDACi sodium butyrate. These findings suggest that HDAC inhibition fosters BDNF-TrkB signalling thereby enhancing postischemic regeneration.
Sex differences
As personalized medicine gains more and more acclaim, epigenetics emerge into the limelight. A look at sex-related differences offers itself. Usually, male gender is considered a risk factor in ischemic injury. Female animals with high circulating estrogen levels, for example, show a less pronounced damage compared with their male counterparts as well as ovariectomized female rats. Matching this result, estradiol treatment protects the cortex from cell death in an estrogen receptor alpha(ERa)– dependent manner. In female rats ERa upregulation is necessary for the protective effect of estradiol. In the ischemic area of female, but not male rodents, estrogen receptora is upregulated. This induction requires the disassociation of the transcriptional repressor MeCP2 and coincides with the corresponding demethylated status of the DNA in the promoter region.124,125
However, not only female hormones influence stroke. Sub-normal testosterone levels in blood correlate with higher stroke incidences, larger infarcts, and poorer recovery. Treatment of castrated mice with testosterone, and especially dihydrotestosterone (DHT), at physiologic levels reduces infarct size in an androgen receptor-dependent manner, which is modulated by epigenetic mechanisms. DHT application induces the androgen-responsive gene salt-induced kinase (SIK1) in neurons. 126 SIK functions as a class IIa HDAC kinase that inhibits HDAC class II activity. Hence, SIK is an endogenous HDACi and an endogenous neuroprotective agent. 127 Knocking down the SIK HDAC kinase leads to a decrease in H3 acetylation levels. Thus, DHT prevents ischemia-induced histone deacetylation after MCAO in castrated male rodents in a dose-dependent manner. 127 Sex-related therapy based on epigenetic differences might be a promising novel treatment strategy in stroke.
Aspects of Translation
The translation of experimental data from animals to humans is difficult. However, the knowledge on similarities between epigenetic mechanisms of animals and humans is accumulating. DNA methylation patterns are highly conserved in CpG-rich gene promoters of mouse and human brains. 128 This is also the case, for example, for the TSP1 gene promoter involved in angiogenesis after ischemia as described above.114,116 Histone modifications and especially colocalizing different epigenetic marks are largely preserved between species where transcription factor-binding sites and gene expression are maintained as well.129,130 Additionally, a great amount of data on human basic epigenetic mechanisms and genes involved in ischemic pathways have been assembled in cancer research. 131 They provide a good basis for stroke research. However, caution is advised before translating cancer data to the pathways of cerebral ischemia, as epigenetic regulation can be very tissue, cell type, and context specific.
Summarizing findings conducted with epigenetic modifiers in experimental stroke, it appears that knowledge on epigenetic marks such as DNA methylation and histone methylation is still too fragmentary to draw any conclusions toward clinical settings. Concerning histone acetylation, however, multiple studies performed in rodent models of stroke suggest neuroprotective and neuroregenerative effects upon treatment with diverse HDACi. Histone deacetylase inhibitor administration improved outcome up to 7 hours post-stroke, 48 which constitutes a relatively broad time window for pharmacological intervention. Regarding clinical practice, a late treatment option represents a major advantage. Currently, only a minority of patients reach the hospital within the 4.5 hours after stroke onset, which would allow intravenous lysis treatment with tissue plasminogen activator, the only approved drug for stroke treatment.
Interestingly, in spite of promising bench findings on the application of HDACi in stroke, any evidence for beneficial effects in human patients is lacking, as no clinical trials with HDACi have been conducted in stroke as yet (sources: clinicaltrials.gov, http://www.ncbi.nlm.nih.gov/pubmed/). One reason might be that the therapeutic application of HDACi in nervous system disorders with human patients still faces significant challenges. A major limitation is the lack of isotype-specific HDAC inhibitors, that are permeable to the blood–brain barrier and display reduced side effects (reviewed in Kazantsev et al. 132 ). However, knowledge from clinical studies on the application of various HDACi in other malignancies, mainly cancer, is accumulating. Recently, the first tests with HDACi in neurologic disorders, such as Huntington and amyotrophic lateral sclerosis have been performed in clinical settings (see Table 1). Based on the results from these trials, it might be possible to anticipate safety risks in stroke patients, or deduce possible trial and treatment paradigms.
CONCLUSION
The amalgamation of the fields of epigenetic research and ischemia promises novel pleiotropic treatment strategies. The great potential of HDAC inhibition lies in the ability to modulate gene expression in all decisive pathways involved in the course of ischemic injury, resulting in neuroprotection and neuroregeneration, as demonstrated in several preclinical stroke models. As neurotoxic effects of single HDACs are being identified, a need for selective subtype inhibitors becomes apparent. Whether the efficacy of the drugs shown in rodent models can be successfully translated to a clinical setting remains to be demonstrated. To take important parameters, such as sex-related differences, as well as age and comorbidities into consideration might be beneficial for a successful translation.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.