
Abstract
The antiepileptic drug valproate (VPA) may be neuroprotective. We treated rats with VPA for 14 days (300 mg/kg twice daily) before intrastriatal injection of 1.5 μmol (1 M) of the succinate dehydrogenase inhibitor malonate. VPA-treated animals developed smaller lesions than control animals: 10 ± 2 mm3 versus 26 ± 8 mm3 (means ± SD; P = 10−4). Injection of NaCl that was equiosmolar with 1 M malonate caused lesions of only 1.2 ± 0.4 mm3 in control animals, whereas physiologic saline produced no lesion. VPA pretreatment reduced the malonate-induced extracellular accumulation of glutamate. This effect paralleled an increase in the striatal level of the glutamate transporter GLT, which augmented high-affinity glutamate uptake by 25%, as determined from the uptake of [3H] glutamate into striatal proteoliposomes. Malonate caused a 76% reduction in striatal adenosine triphosphate (ATP) content, but the glial, ATP-dependent formation of glutamine from radiolabeled glucose or glutamate was intact, indicating that glial ATP production supported uptake of glutamate. Striatal levels of HSP-70 and fos were reduced, and the levels of bcl-2 and phosphorylated extracellular signal-regulated kinase remained unaffected, but histone acetylation was increased by VPA treatment. The results suggest that augmentation of glutamate uptake may contribute importantly to VPA-mediated neuroprotection in striatum.
Inhibition of cerebral energy metabolism may cause accumulation of glutamate extracellularly (Benveniste et al., 1984;Hamann et al., 2002) and excessive stimulation of glutamate receptors, leading to neuronal degeneration. Valproate (VPA), a commonly used antiepileptic and mood-stabilizing drug, has neuroprotective properties (Hashimoto et al., 2002;Mora et al., 1999), and several mechanisms have been proposed to explain this effect, including increases in bcl-2 levels and extracellular signal-regulated kinase (ERK) phosphorylation (for review, see Manji and Chen, 2002). Chronic VPA treatment has been shown to cause an increase in the level of the glutamate transporter GLT in rat brain (Hassel et al., 2001). This transporter, which is the quantitatively most important glutamate transporter in the forebrain, is predominantly expressed in astrocytes (Chaudhry et al., 1995). In the present study we wished to see whether a neuro-protective effect of VPA in vivo would be associated with an increased capacity for removal of extracellular glutamate.
Inhibitors of succinate dehydrogenase, an enzyme of the tricarboxylic acid cycle and the electron transport chain, have been used to cause cerebral energy deficiency. Intrastriatal injection of one such inhibitor, malonate, leads to local energy deficiency (Beal et al., 1993), extracellular accumulation of glutamate (Messam et al., 1995), and an excitotoxic lesion that can be reduced by glutamate receptor antagonists (Beal et al., 1993;Greene and Greenamyre, 1995;Ikonomidou et al., 2000). Inhibition of succinate dehydrogenase in striatum is often used as a model for Huntington's disease; in this disorder, striatal succinate dehydrogenase activity is reduced (Browne et al., 1997;Butterworth et al., 1985;Gu et al., 1996).
We investigated the effect of VPA treatment, 300 mg/kg, twice per day for 14 days, upon malonate-induced striatal degeneration and extracellular glutamate accumulation. We assessed changes in the tissue levels of astrocytic glutamate transporters and other proteins that could be involved in a neuroprotective effect of VPA, and we measured the effect of VPA treatment upon striatal high-affinity glutamate uptake.
MATERIALS AND METHODS
Animal treatment
Male Wistar rats, approximately 350 g bodyweight, were from M & B (Ry, Denmark). The animals were handled in strict accordance with institutional and national ethical guidelines. Twice per day (at approximately 8:00 a.m. and 4:00 p.m.) for 14 days the animals received a subcutaneous injection of VPA (Sigma, St. Louis, MO, U.S.A.), 300 mg/kg, or saline.
Malonate toxicity in striatum: Volumetric measurements
Rats were used for toxicity studies 12 to 18 hours after the last dose of VPA, so that VPA itself would not interfere with the effects of malonate; the half life of VPA in rats is approximately 3 hours (Löscher, 1999). The rats (n =8 in each group) were anesthetized with (per kg bodyweight) fentanyl citrate 0.2 mg, fluanisone 10 mg, and midazolam 5 mg. A solution (1.5 μL) containing sodium malonate, 1 mol/L, pH 7.4, was injected stereotactically into striatum (Hassel et al., 2002) over 4 minutes. Such a high concentration of malonate is needed to produce neurotoxicity (Greene and Greenamyre, 1995) because most of the malonate rapidly diffuses out of the brain, as reported by Koeppen et al. (1978). Body temperature was maintained at 37 to 38°C. Brain pathology was evaluated at 3 days after injection of malonate, as is customary in this model (e.g., Greene and Greenamyre, 1995). The animals were anesthetized with CO2 and decapitated. The brains were frozen with CO2 and cryosectioned coronally in 60 μm sections at −3°C. Every fourth section was mounted on glass slides, and succinate dehydrogenase activity was visualized by incubating the sections in a buffer containing sodium succinate 100 mmol/L, Nitro Blue tetrazolium 5 mg/mL, menadione 3 μmol/L, ethylenedi-aminetetraacetate (EDTA) 1 mmol/L, and sodium azide, pH 8, for 5 minutes at 25°C. The sections were photographed, and the lesion areas were quantified blindly. The volume of the lesion was obtained by multiplying the summed lesion areas (in mm2) by the distance between each measured section surface. Some neighboring sections were stained with the May-Grünewald-Giemsa method, which visualizes structural elements rather than functional mitochondria, to see whether the two staining methods identified the same lesion area.
To evaluate the possible neurotoxic effect of injecting hyperosmolar solutions into the brain, drug-naive control rats (n =5) received 1.5 μL malonate, 1 mol/L, in one striatum and an equiosmolar solution of sodium chloride (2.01 Osm/L) in the other. Osmolarity was measured on an Advanced 3D3 osmometer (Norwood, MA, U.S.A.). Three days after the injection, the rats were killed and the brains processed as described previously in this section.
Microdialysis
Microdialysis was performed in animals (n = 7 for VPA-treated animals and control animals) that were anesthetized and subjected to stereotactic surgery as described previously 12 to 18 hours after the last dose of VPA. A microdialysis probe was inserted into the right striatum. The membrane of the dialysis probe (CMA Microdialysis, Solna, Sweden) was 1 mm long, with an outer diameter of 0.24 mm and a cut-off of 6,000 Da. Three microdialysis buffers were used. A low potassium buffer was (in mmol/L) KCl 5, NaCl 120, NaHCO3 25, NaH2PO4 1.4, MgCl2 2, and CaCl2 2. In the high potassium buffer KCl was 55 mmol/L and NaCl was 70 mmol/L; the other salts were not changed. The malonate-containing buffer was sodium malonate, 1 mol/L. All buffers were pH 7.4 after equilibration with O2:CO2 (95:5) or (in the case of the malonate buffer) 100% O2. The dialysate flow rate was 3 μL/min. After 1 hour of perfusion with the low-potassium buffer, dialysates were collected in 10-minute fractions, and amino acids were analyzed by HPLC and fluorescence detection as described previously (Hassel et al., 1997).
To evaluate the effect of microdialysis with a hyperosmolar solution (the malonate solution was 1 mol/L) drug-naive rats were subjected to striatal microdialysis with a low-potassium buffer of physiologic osmolarity as described previously in this report for 1 hour before dialysis with NaCl that was equiosmolar (2.01 Osm/L) with the 1 mol/L malonate. To evaluate the long-term effect of injection of a hyperosmolar solution upon extracellular levels of glutamate, three rats received 1.5 μL of malonate (1 mol/L) in the right striatum and 1.5 μL equiosmolar NaCl in the left. At 30 minutes, microdialysis probes were inserted into each striatum, and at 60 minutes, sampling of (low potassium) dialysis buffer was begun. The delay from injection of malonate until dialysis was introduced to avoid wash-out of the injected substances.
ATP measurements
Anesthetized, drug-naive rats received 1.5 μL malonate (1 mol/L) or physiologic saline intrastriatally over 4 minutes. One or five minutes after completion of the injection, the brains were frozen in situ by pouring liquid through a plastic N2 funnel onto the exposed skull. The animals were decapitated and the heads kept in liquid N2 for 5 minutes. The tissue was prepared, and ATP was measured fluorimetrically as described by Lowry and Passoneau (1972).
Radiolabeling of brain amino acids from [2-14C]malonate, [U-14C]glucose, or [U-14C]glutamate
To see whether the injected malonate was primarily taken up by neurons or astrocytes, 1.5 μL malonate (1 mol/L) together with 1.5 μCi [2-14C]malonate (54 μCi/μmol; American Radio-labeled Chemicals, St. Louis, MO, U.S.A.; final specific activity 1 μCi/μmol) were injected stereotactically over 4 minutes into the right striatum of anesthetized, drug-naive rats. One minute after completion of the injection the rats were decapitated, the heads were immediately cooled in liquid nitrogen, and the striatal area around the injection site was dissected out on ice and homogenized in 1 mL perchloric acid (3.5% [vol/vol]), with α-aminoadipate (50 μmol/L) as an internal amino acid standard.
To see whether malonate inhibited primarily neuronal or glial metabolism 1.5 μL malonate (1 mol/L) or physiologic saline was injected into one striatum over 4 minutes, and at 1 minute after completion of the injection, 1 μL (corresponding to 0.4 μCi) of [U-14C]glucose (3mCi/mmol; Amersham Pharmacia Biotech) was injected into the striatum over 3 minutes. One minute after completion of this injection, the brain was funnel frozen in situ and processed as described previously in this report.
To see if glial glutamate uptake occurred in malonate-treated striatum, 1.5 μL malonate (1 mol/L) or physiologic saline was injected into one striatum over 4 minutes, and at 1 minute after completion of the injection, 1 μL (corresponding to 1 μCi) of [U-14C]glutamate (200 mCi/mmol; American Radiolabeled Chemicals), was injected. Five minutes after completion of this injection the brain was funnel frozen in situ and processed as described previously in this report. Tissue levels and radiola-beling of amino acids were determined by HPLC, fluorescence detection, and scintillation counting after precolumn derivatization with ophthaldialdehyde as described previously (Hassel et al., 1992).
Immunoblotting
Animals that had received VPA treatment or saline were killed at 4 hours after the last dose of VPA unless otherwise stated, and striatum was dissected out on ice and frozen. The tissue was weighed in the frozen state, homogenized to a 5% (weight/volume) homogenate in sucrose (0.32 mol/L), and prepared for immunoblotting as described previously (Hassel et al., 2001). Sodium dodecylsulphate polyacrylamide gel electrophoresis was performed according to Laemmli (1970). Briefly, 0.6 μg total protein was applied to each lane for the detection of the glutamate transporter GLT, β-actin, and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor subunits GluR 1, 2, and 3. Total protein (2 μg) was applied for the detection of the astrocytic glutamate transporter GLAST, glial fibrillary acidic protein (GFAP), and synaptophysin. For detection of HSP-70 and ERK (phosphorylated and total amount), 7.5 μg protein were applied, and for detection of bcl-2 and fos, 60 μg protein were applied per lane. For detection of acetylated histone H4, the tissue was homogenized to a 10% homogenate (weight/volume) in 0.32 mol/L sucrose containing VPA (30 mmol/L) to inhibit postmortem histone deacetylase activity. Homogenates were acidified with H2SO4 (2 mol/L) to a final concentration of 0.2 mol/L and were centrifuged at 5,000 g. Supernatants, 150 μL, with acid-soluble proteins, were mixed 1:1 with loading buffer (Tris-HCl, 0.2 mol/L, glycerol, 50% [vol/vol], mercaptoethanol, 10 mmol/L, and bromothymol blue), pH was brought to 7 with 10 μL NaOH, and 30 μL of this mixture (corresponding to 1.5 mg of striatal wet tissue) were subjected to gel electrophoresis. ERK phosphorylation and histone H4 acetylation were evaluated at 2, 4, and 12 hours after the last dose of VPA.
Samples were run on 10% polyacrylamide gels (90 V, 2 hours) and were blotted (50 mA, 19 hours) onto 10 × 10 cm2 nitrocellulose paper with 0.2-μm pore size. The antibodies against GLT (anti-B493), which have been extensively characterized (e.g., Trotti et al., 1995), were a gift from Dr. N. C. Danbolt, University of Oslo, Norway. Antibodies against GLAST and synaptophysin were from Chemicon (Temecula, CA, U.S.A.); antibodies against GFAP and GluR 1, 2, and 3 were from Zymed (San Francisco, CA, U.S.A.); antibodies against HSP-70, fos, and acetylated histone H4 were from Upstate (Lake Placid, NY, U.S.A.); antibodies against ERK (phosphorylation-specific and -unspecific) were from Cell Signaling Technology (Beverly, MA, U.S.A.); and antibodies against β-actin and bcl-2, the same as those used to demonstrate an increase in bcl-2 in frontal cortex after VPA treatment (N-19) (Chen et al., 1999), were from Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.). Both primary antibodies and secondary antibodies carrying horseradish peroxidase (Dako, Glostrup, Denmark) were used at a concentration of 0.3 μg/mL buffer. Protein labeling was visualized with the enhanced chemiluminescence method (ECL, Amersham Pharmacia Biotech) and exposure of X-Omat Blue film (Kodak) to the immunoblots. Semiquantitative determination of protein levels was performed by densitometric analysis of the x-ray films (Phoretix 1D Lite, Hewlett-Packard) as described previously (Hassel et al., 2001). β-actin levels were determined to ensure that equal amounts of protein were applied to each lane of the gels. The levels of other proteins were normalized to the level of β-actin, but the results were the same as for nonnormalized data, which are therefore given. The staining of the x-ray film correlated well with the amount of total protein applied to the gel: r = 0.99 (P = 0.008; Pearson Product Moment Correlation), as determined for GLT.
High-affinity uptake of glutamate and dopamine into proteoliposomes
[3,4-3H]Glutamate (60 Ci/mmol) was from American Radio-labeled Chemicals (St. Louis, MO, U.S.A.), and [2,3,6-3H]dopamine (9.1 Ci/mmol) was from Amersham Pharmacia Biotech.
Reconstitution of plasma membrane transporters from striatum into proteoliposomes, as well as high-affinity uptake of glutamate or dopamine into the proteoliposomes, was performed according to Danbolt et al. (1990) with the modifications of Trotti et al. (1995). Briefly, uptake of [3H]glutamate or [3H]dopamine into the proteoliposomes took place at 25°C in a solution containing NaCl (150 mmol/L) and glycerol, 1%, and it was determined in triplicates for samples and in duplicates for blanks. Blank values (approximately 5% of full activity values) were obtained in the presence of nigericin (6 μmol/L), which disrupted the sodium gradient across the proteoliposome membrane. Proteoliposomes were incubated with [3H]gluta-mate for 3 minutes or with [3H]dopamine for 9 minutes. Uptake of dopamine took place in the presence of ascorbate (170 μmol/L) and pargyline (8 mmol/L) to inhibit dopamine oxidation. Proteoliposomes were trapped on filter paper, and radioactivity was measured by scintillation counting in a Packard Tri-Carb 300 scintillation counter after addition of 10 mL Filtercount scintillation fluid (Packard, Meriden, CT, U.S.A.).
Accumulation of [3H]2-deoxyglucose: Measurement of serum VPA
To study the influence of VPA upon uptake of glucose into striatum, we dosed rats with VPA (300 mg/kg twice daily) for 14 days (n = 8 for both VPA-treated animals and controls). Forty-five minutes before the rats were killed, which was 75 minutes after the last dose of VPA, the animals received an intravenous injection of 100 μCi [1-3H]2-deoxyglucose (13.0 Ci/mmol; Amersham Pharmacia Biotech, Little Chalfont, Buckinghamshire, UK) in physiologic saline, so that cerebral accumulation of [3H]2-deoxyglucose could take place for 45 minutes, as recommended by Sokoloff et al. (1977). Because we were interested in differences between treatment groups, calculation of the lumped constant (Sokoloff et al., 1977) was not considered necessary. At 45 minutes the animals were decapitated, and brain radioactivity was measured by scintillation counting. The blood obtained from these rats (2 hours after the last dose of VPA) was centrifuged, and serum was analyzed for VPA by fluorescence polarizing immunoassay (TDX, Abbott, Irving, TX, U.S.A.).
Data presentation and statistics
Data are presented as means ± SD. Values for protein levels are given as percentage of control. Statistical analysis was performed with one-way ANOVA Dunnett's test for multiple comparison or with the unpaired Student's t-test when appropriate. Analysis of microdialysis data on the difference between striata injected with malonate (1 mol/L) or equiosmolar NaCl was performed with ANOVA for repeated measurements using Huynh-Feldt's method.
RESULTS
VPA treatment reduces malonate-induced striatal lesions
Injection of malonate (1.5 μmol) into the striatum of saline-treated animals caused a lesion of 26 ± 8 mm3, which comprised most of the striatum (Fig. 1). In animals pretreated with VPA for 14 days, the lesion was significantly smaller, 10 ± 2 mm3 (P = 10−4). Injection of 1.5 μL of a solution of sodium chloride with the same osmolarity as the 1 M malonate (2.01 Osm/L) caused a much smaller lesion, 1.2 ± 0.4 mm3, than obtained with malonate (P < 10−4), whereas injection of physiologic saline did not produce any striatal lesion (Fig. 1). Staining the brain sections with the May-Grünewald-Giemsa method identified lesion areas identical to those obtained with tetrazolium staining for succinate dehydrogenase activity (data not shown).
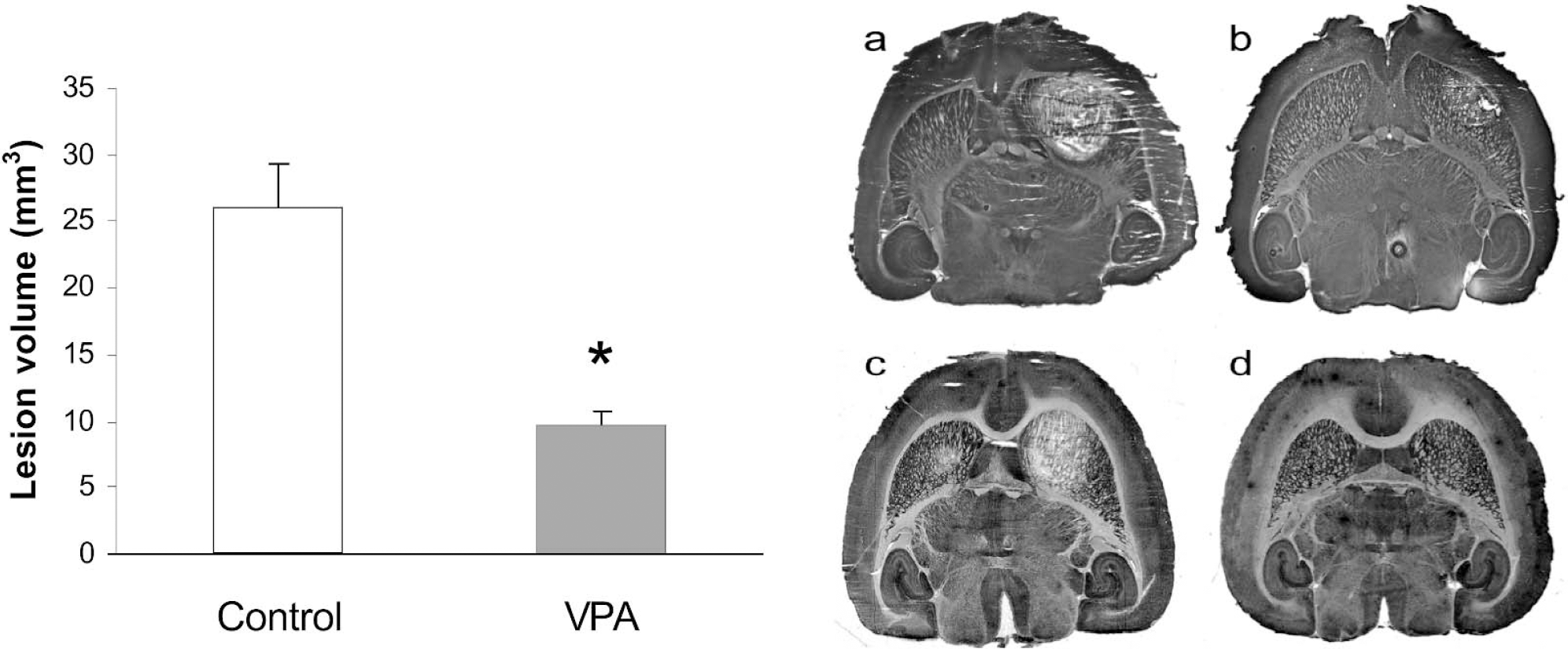
(Left) Effect of VPA treatment upon malonate-induced cell death in striatum. Wistar rats were treated with VPA (300 mg/kg) or physiologic saline twice per day for 14 days. On day 15, 12 to 18 hours after the last dose of VPA, malonate (1.5 μmol) was injected into the right striatum. Three days later, the animals were killed, and the brains were cryosectioned at 60 μm. To visualize the lesion area, brain sections were incubated in sodium succinate 100 mmol/L, Nitro Blue tetrazolium 5 mg/mL, menadione 3 μmol/L, sodium azide 10 mmol/L, pH 8, and lesions were quantified from the lesion area of every fourth section. Data are mm3, means ± SD, n = 7 in each group. *Significantly different from control, P = 10−4. (Right) Sections through the brains of rats that received an intrastriatal injection of 1.5 μL of sodium malonate (1 mol/L) after 14 days of physiologic saline (A) or VPA (300 mg/kg) (B), twice daily. (C) A drug-naive rat that received 1.5 μL of sodium malonate (1 mol/L) in the right striatum and an equiosmolar solution of NaCl (2.01 Osm/L) in the left striatum. (D) A drug-naive rat that received 1.5 μL of physiologic saline in the right striatum.
VPA treatment reduces extracellular accumulation of glutamate
Microdialysis of control striata with a high-potassium buffer (KCl, 55 mmol/L) led to a 4.6-fold increase in glutamate in the dialysate (Fig. 2A) from 0.18 ± 0.08 μmol/L to 0.83 ± 0.26 μmol/L (P = 4 ×10−4). In VPA-treated animals, the high-potassium buffer caused a significantly lower increase of glutamate in the dialysate, to 0.40 ± 0.29 μmol/L (difference from controls:P = 0.012) (Fig. 2A). The basal level of glutamine in control dialysates was 4.9 ± 0.3 μmol/L, a level which was similar in the two groups of animals and which did not change significantly throughout the experiment. These basal levels of glutamate and glutamine are similar to those found in previous studies of rat striatum (e.g., Orwar et al., 1994).
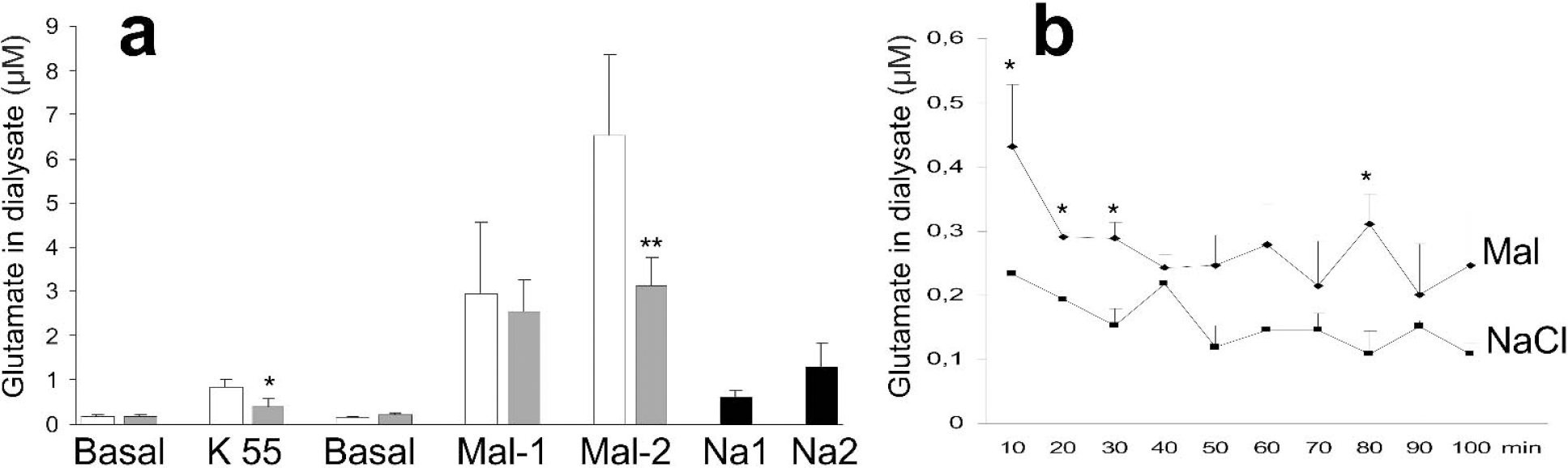
Malonate-induced extracellular accumulation of glutamate. (A) Wistar rats were treated with sodium valproate, 300 mg/kg (white bars), or saline (gray bars) twice per day for 14 days. On day 15, 12 to 18 hours after the last dose of VPA, microdialysis of striatum was performed in anesthetized animals. After 60 minutes of dialysis (3 μL/min) with artificial CSF collection of the dialysate begun. The dialysis buffers were artificial CSF (basal), a high-potassium (55 mmol/L) buffer to induce mild depolarization, or malonate, 1 mol/L. Drug-naive rats were dialyzed with NaCl that was equiosmolar with the 1 mol/L malonate (2.01 Osm/L). Dialysates were collected in 10-minute fractions; malonate and hyperosmolar NaCl were used in two consecutive 10-minute periods (Mal-1 and Mal-2, Na-1 and Na-2). Data are μmol/L glutamate in the dialysate, means ± SD, n = 7 in each group. Significantly different from control: *P = 0.02, **P = 0.003. (B) Drug-naive rats (n = 3) received 1.5 μL of a 1 mol/L sodium malonate solution in the right striatum and an equiosmolar NaCl solution (2.01 Osm/L) in the left striatum. Microdialysis was begun at 60 minutes with collection of the dialysate in 10-minute fractions. There was an overall difference between the malonate- and NaCl-treated striata (P = 0.04). *Difference between malonate- and saline-injected striatum, P < 0.05.
Microdialysis with malonate led to an approximately 15-fold increase in extracellular glutamate in both VPA-treated rats and control animals (Fig. 2A). In control rats, the glutamate level increased further from the first to the second 10 minutes of dialysis with malonate, whereas in VPA-treated rats, no further increase in glutamate was seen.
To see whether hyperosmolarity itself would cause extracellular accumulation of glutamate, drug-naive rats were subjected to striatal microdialysis with 2.01 Osm/L saline, which was equiosmolar to the 1 mol/L malonate. Hyperosmolar saline caused the level of glutamate to increase from a basal level of 0.15 ± 0.13 μmol/L to 0.60 ± 0.16 μmol/L (Fig. 2A) (P < 0.05) during the first 10 minutes of dialysis. During the next 10 minutes, the dialysate level of glutamate increased further to 1.3 ± 0.5 μmol/L (P < 0.05). These values were only approximately 20% of those achieved during dialysis with malonate.
Aspartate was not detected during pre-malonate dialysis. Malonate exposure caused accumulation of aspartate extracellularly; the dialysate levels in VPA-treated rats and controls during the first 10 minutes were 0.21 ± 0.05 μmol/L and 0.27 ± 0.13 μmol/L, respectively (not significantly different). During the second 10 minutes of malonate exposure, the dialysate concentration of aspartate was significantly lower in the VPA-treated animals: 0.14 ± 0.05 μmol/L versus 0.27 ± 0.13 μmol/L in control animals (P = 0.02).
To see if the VPA injections would affect the extra-cellular accumulation of glutamate, we dosed five drug-naive rats with VPA, 300 mg/kg subcutaneously, during dialysis, but, in agreement with a previous study (Biggs et al., 1992), the dialysate level of glutamate was not different from that seen in five control animals that received saline, with either low- or high-potassium buffers (data not shown).
To study the temporal aspect of extracellular accumulation of glutamate after injection of malonate or hyperosmolar saline, we injected sodium malonate (1 mol/L) into one striatum of drug-naive rats and equiosmolar NaCl into the other, both volumes 1.5 μL. Microdialysis 1 hour after injection showed that the extracellular concentration of glutamate was significantly higher in malonate-treated striata (Fig. 2B). The extracellular concentration of glutamate 1 hour after treatment with hyperosmolar saline was not different from that obtained during dialysis with low-potassium buffer of physiologic osmolarity (compared with Fig. 2A).
Malonate inhibits cerebral ATP formation
Injection of malonate, 1.5 μmol, caused the striatal level of ATP to drop by 76%, from approximately 17 nmol/mg protein to approximately 4 nmol/mg protein within 1 minute after completion of the injection (Table 1). At 5 minutes after injection of malonate, the level of ATP was 3.3 ± 0.4 nmol/mg protein.
Levels of ATP and the radiolabeling of glutamine and glutamate in control and malonate-treated striatum

Rats received malonate, 1.5 μmol, or physiologic saline intrastriatally over 4 minutes. At 1 minute, the tissue was funnel frozen in situ to measure ATP (n = 8); alternatively, [U-14C]glucose, 0.4 μCi, was injected over 3 minutes (n = 5). One minute after completion of the [U-14C]glucose injection, the tissue was funnel frozen, and radiolabeling of glutamine and glutamate was measured. Values are mean ± SD.
P = 0.02
P = 10−4
Malonate is primarily taken up by neurons: Radiolabeling from [14C]malonate
To determine whether malonate is taken up predominantly by neurons or glial cells, we injected 14C-labeled malonate together with the 1M solution of malonate and evaluated the labeling of glutamate and glutamine. If a radiolabeled substrate is metabolized predominantly by glial cells, then the specific activity of glutamine exceeds that of glutamate. If the substrate is primarily metabolized by neurons, then the specific activity of glutamate is higher (Hassel et al., 1992, 2002;Van den Berg et al., 1969). 14C-Labeled malonate gave a specific activity of glutamate that was higher than that of glutamine: 4,850 ± 2,730 dpm/μmol versus 2,360 ± 1,100 dpm/μmol. The glutamine/glutamate relative specific activity ratio, as calculated for each animal, was 0.62 ± 0.40, that is, lower than 1, which supports the impression that the injected malonate predominantly entered neurons.
Glial formation of glutamine and uptake of glutamate proceeds in malonate-treated striatum
Because glutamate uptake is a secondarily active process that depends upon ATP, and because we suspected that glial glutamate uptake might account for the lower extracellular accumulation of glutamate in VPA-treated animals, we wanted to know whether glial cells had ATP after malonate treatment. We therefore measured the ability of malonate-treated striatum to form glutamine from radiolabeled glucose. Glutamine synthetase is an ATP-dependent enzyme that is expressed in glia but not in neurons (Martinez-Hernandez et al., 1977). 14C-Labeling of glutamine was unaffected by malonate treatment, whereas 14C-labeling of glutamate was reduced by 60% (Table 1), suggesting inhibition of the tricarboxylic acid cycle in neurons, in which glutamate is concentrated (Storm-Mathisen et al., 1983).
To see if glial cells could take up glutamate in malonate-treated striatum, we injected radiolabeled glutamate and measured the formation of radiolabeled glutamine. [14C]Glutamine was clearly formed in malonate-treated striatum. The specific activity of glutamine was 150 ± 60 dpm/nmol; in control animals, it was 260 ± 200 dpm/nmol. The nonsignificant tendency (P = 0.2) toward a lower specific activity of glutamine in malonate-treated animals was to be expected from the higher extracellular level of glutamate in malonate-treated animals (Fig. 2); this unlabeled glutamate would dilute the injected radiolabeled glutamate. The specific activity of glutamate was the same in malonate-treated animals and controls: 1,330 ± 490 dpm/nmol and 1,580 ± 280 dpm/nmol, respectively. The total level of glutamine and glutamate was the same in the two groups (data not shown).
VPA increases the level of GLT and the high-affinity uptake of glutamate in striatum
The tissue level of the glutamate transporter GLT was approximately 50% higher in VPA-treated striatum than in control striatum, as determined by immunoblotting (Fig. 3). The levels of the astrocytic glutamate transporter GLAST, glial fibrillary acidic protein (GFAP), synaptophysin, and AMPA receptors were the same in VPA-treated and control striatum, indicating that gliosis or alteration in synaptic density had not taken place during VPA treatment.
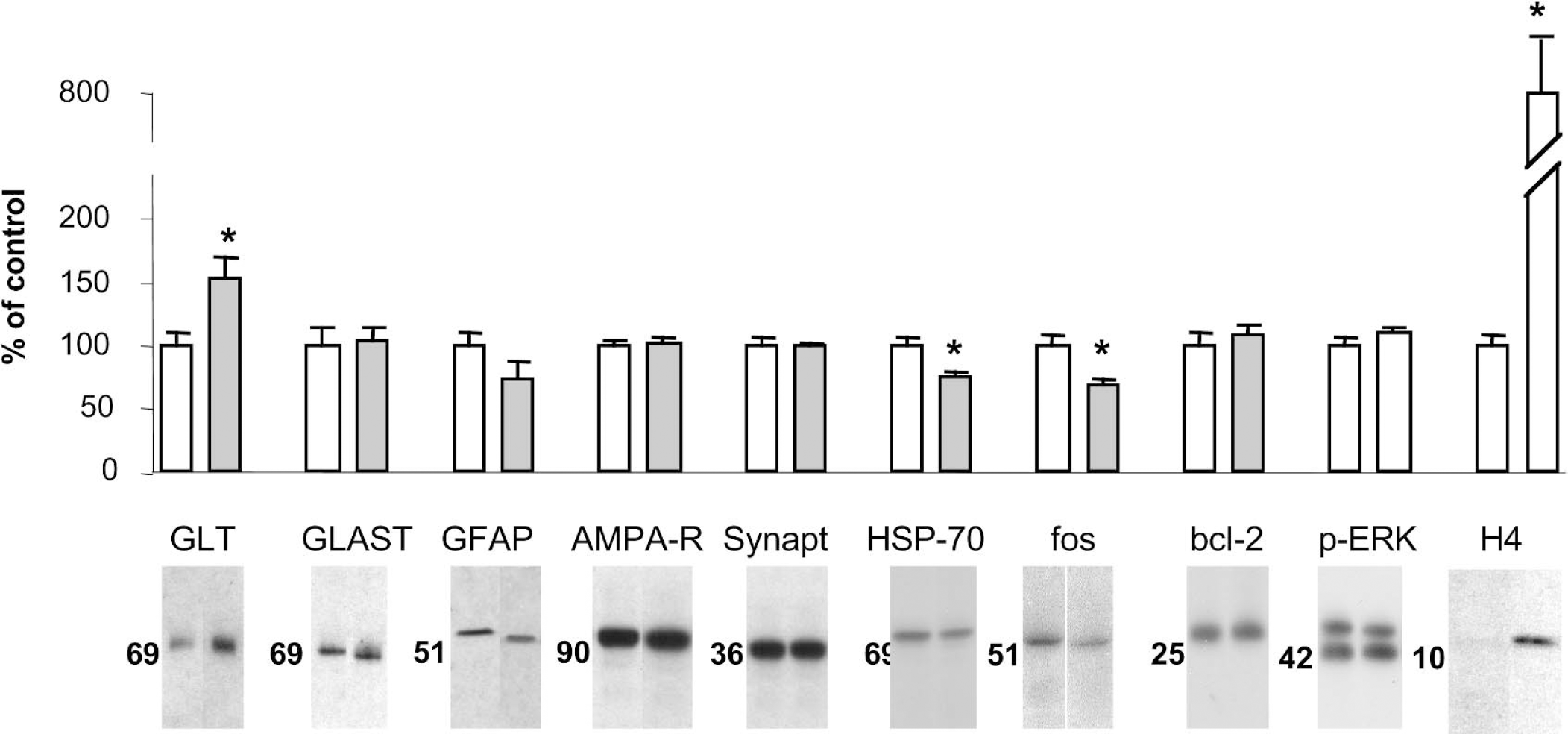
Levels of glutamate transporters GLT and GLAST, GFAP, AMPA receptor, synaptophysin, HSP-70, bcl-2, phospho-ERK, and acetylated histone H4 in striatum after VPA treatment. Wistar rats received VPA, 300 mg/kg, twice per day for 14 days, and were killed 4 hours after the last dose. Brain proteins were separated by gel electrophoresis, blotted onto nitrocellulose paper, visualized by chemiluminescence and exposure to x-ray film, and semiquantified by densitometry. Data are percent of control value, means ± SD, n = 7 in each group. *Significantly different from control, P = 0.01.
Uptake of [3H]glutamate into proteoliposomes made from striatum of VPA-treated animals was 25% higher than in proteoliposomes made from control striatum (Fig. 4), 0.84 ± 0.08 pmol/mg tissue × min−1 tissue vs. 0.68 ± 0.05 pmol/mg tissue × min−1 (P = 0.0003). This finding indicates that VPA treatment led to an increase in functional GLT protein. Treatment with VPA did not affect the uptake of [3H]dopamine into proteoliposomes (Fig. 4).
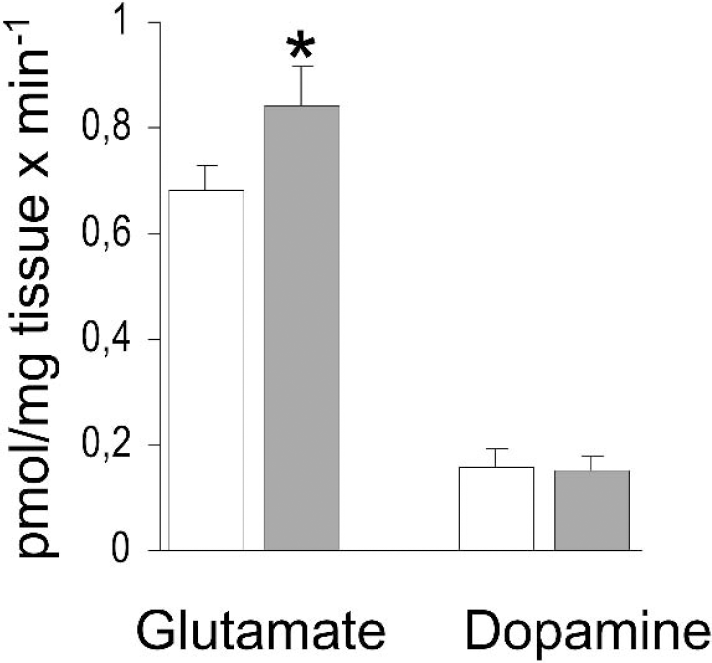
Uptake of glutamate and dopamine into proteoliposomes. Rats received saline (white bars) or VPA, 300 mg/kg (gray bars), twice per day for 14 days. Artificial proteoliposomes with reconstituted membrane-bound proteins were made from striatum and exposed to [3,4−3H]glutamate or [2,3,6−3H]dopa-mine for 3 or 9 minutes, respectively. Data are pmol/mg tissue × min−1, means ± SD, n = 7 in each group. *Significantly different from control, P = 3 × 10−4.
The tissue level of glutamate was unaffected by VPA treatment; it was 11 ± 0.8 nmol/mg tissue in both VPA-treated animals and controls.
Effects of VPA upon HSP-70, fos, bcl-2, ERK phosphorylation, and histone H4 acetylation in striatum
We wanted to see whether VPA treatment could lead to tissue protection through mechanisms other than increased glutamate uptake. We measured tissue levels of HSP-70, because overexpression of HSP-70 was recently shown to confer resistance against malonate toxicity (Dedeoglu et al., 2002). Further, we measured the levels of bcl-2 and the degree of ERK phosphorylation. A neuroprotective effect has previously been attributed to increases in bcl-2 and ERK phosphorylation (for review, see Manji and Chen, 2002). We determined the tissue level of fos as a measure of cellular stress. Surprisingly, VPA treatment reduced the striatal level of HSP-70 by 24 ± 10% and the level of fos by 32 ± 13% (Fig. 3), whereas the levels of bcl-2 or phosphorylated ERK were not different in the two groups at 2, 4, or 12 hours after the last dose of VPA (see Fig. 3 for levels at 4 hours). VPA treatment led to a marked increase in the level of acetylated histone H4 at 2 and 4 hours but not at 12 hours after the last dose of VPA (see Fig. 3 for levels at 4 hours).
Regional cerebral accumulation of 2-deoxyglucose, serum VPA, and weight gain
We measured the accumulation of [3H]2-deoxyglucose in striatum because a previous study had shown that acute VPA treatment caused a reduction in cerebral glucose metabolism (Johannessen et al., 2001), an effect that could influence both malonate toxicity and glutamate uptake. VPA treatment did not cause any difference in striatal accumulation of [3H]2-deoxyglucose, which was 1,194 ± 439 dpm/mg tissue in VPA-treated animals and 1,295 ± 328 dpm/mg tissue in controls.
Two hours after the last dose of VPA, the serum concentration of VPA was 473 ± 267 μM. The VPA-treated animals seemed slightly somnolent approximately 1 hour after dosage but recovered within an hour and did not display ataxia or any abnormal behavior.
VPA treatment inhibited weight gain; control animals had a weight gain of 6.8 ± 2.4% during the treatment period, whereas the VPA-treated animals showed no change in body weight (100 ± 3% of initial weight; difference between groups:P = 2 × 10−4).
DISCUSSION
Long-term VPA treatment may be neuroprotective in striatum
We show here that long-term treatment with VPA before inhibition of neuronal energy metabolism in the striatum may be neuroprotective. The lesion that is caused by malonate poisoning has been shown to be excitotoxic in nature as it can be prevented by glutamate receptor antagonists (Beal et al., 1993;Greene and Greenamyre, 1995;Ikonomidou et al., 2000). Therefore, the extracellular accumulation of glutamate that we and others (Messam et al., 1995) find after malonate treatment probably plays an important role in the neurotoxicity of malonate.
The extracellular accumulation of glutamate in malonate-treated striatum was related to the loss of ATP that follows from inhibition of succinate dehydrogenase (Erecinska and Nelson, 1994). We believe that the accumulated extracellular glutamate was neuronal in origin for two reasons. First, malonate was shown primarily to enter neurons, as could be seen from the higher specific activity of glutamate than of glutamine after injection of radiolabeled malonate (Hassel et al., 2002;Van den Berg et al., 1969). Second, formation of glutamate from radiolabeled glucose, a predominantly neuronal process (Hassel et al., 1997;Storm-Mathisen et al., 1983), was severely reduced in malonate-treated striatum, whereas the formation of glutamine, a strictly glial reaction in the brain (Martinez-Hernandez et al., 1977), was unaffected. A loss of ATP leads to a loss of ion gradients and hence to a reversal of glutamate transporters in ATP-depleted neurons (Erecinska and Nelson, 1994). In contrast, the energy metabolism of astrocytes probably remained unaffected by malonate. This conclusion is based upon two findings. First, glutamine formation from radiolabeled glucose, which is an ATP-dependent reaction (e.g., Powers and Riordan, 1975) in glia, was unaffected after malonate exposure. Second, injection of radiolabeled glutamate into malonate-treated striatum resulted in formation of radiolabeled glutamine, a process that requires glial uptake of glutamate from the extracellular fluid as well as glutamine synthetase activity. These findings indicate that glial cells were capable of ATP formation and glutamate uptake during malonate exposure.
In a previous study, Beal et al. (1993) found an 80% reduction in ATP levels 30 minutes after intrastriatal injection of malonate, similar to our finding at 5 minutes after injection of malonate. The present study thus shows that the effect of malonate on ATP formation occurs within a few minutes of administration.
The ability of VPA to reduce malonate-induced neurodegeneration was associated with a reduction in the accumulation of extracellular glutamate after malonate injection. The reduced glutamate accumulation could be explained by the increase in the striatal level of the glutamate transporter GLT, which augmented high-affinity glutamate uptake, as could be seen in the proteoliposome assay. The importance of the increase in GLT lies in the observation that this glutamate transporter is mainly expressed in astrocytes (Chaudhry et al., 1995). As discussed previously in this report, astrocytes were able to take up extracellular glutamate because of their maintained ability to form ATP during malonate exposure. Also of importance in this context is that astrocytic glutamate uptake appears to be fuelled mainly through glycolysis (Pellerin and Magistretti, 1994;Voutsinos-Porche et al., 2003), which is not inhibited by malonate.
Malonate toxicity in striatum has been shown to involve dopamine (Maragos et al., 1998;Moy et al., 2000). However, in the present study the tissue-protective effect of VPA could not be attributed to increased uptake of dopamine because uptake of dopamine into proteoliposomes was unaffected by VPA treatment.
We also show in this study that the hyperosmolarity of the injected malonate solution is a minor cause of neurotoxicity, in agreement with the lesser and shorter-lasting extracellular accumulation of glutamate after injection of hyperosmolar saline compared with that seen after injection of malonate.
Assessment of various possible neuroprotective effects of VPA
Mice that overexpress HSP-70 have increased striatal resistance to malonate toxicity (Dedeoglu et al., 2002). We therefore wanted to see whether an increased tissue level of HSP-70 could explain the neuroprotective effect of VPA. However, VPA treatment led to a reduction in the tissue level of HSP-70. This finding was unexpected because we would have assumed that a reduction in HSP-70 levels would have rendered the tissue more susceptible to malonate toxicity. An explanation for the lower levels of HSP-70 may be that VPA treatment reduced cellular stress before malonate exposure. It is possible, for instance, that VPA treatment led to a reduction in glutamate receptor activation, as was suggested by the reduction in extracellular glutamate accumulation during potassium-induced depolarization in VPA-treated animals. We investigated the effect of VPA upon striatal levels of fos as another measure of cellular stress. The reduction in the striatal level of fos after VPA treatment, which has been reported previously (Cutrer et al., 1995;Sonnenberg et al., 1989;Tolle et al., 1995), also suggests that VPA treatment entails a reduction in cellular stress.
VPA treatment has been shown to cause an increase in ERK phosphorylation and in cortical levels of bcl-2 (for review, see Manji and Chen, 2002), to which a neuroprotective effect of VPA has been attributed. In the present study, we found no effects upon striatal levels of bcl-2 or ERK phosphorylation. This discrepancy could reflect different responses to VPA in different brain regions. Previously we have seen an increase in GLT in hippocampus, but not in frontal cortex or cerebellum, which illustrates that different brain regions may indeed respond differently to VPA.
The level of histone acetylation after VPA treatment was investigated, because a recent study (Ferrante et al., 2003) found that inhibition of histone deacetylase with butyric acid ameliorated striatal degeneration in a model of Huntington's disease. VPA is a well-known inhibitor of histone deacetylase (Göttlicher et al., 2001;Phiel et al., 2001;Tremolizzo et al., 2002). The increase in his-tone acetylation may have been important for the observed increase in GLT levels, which presumably depended upon increased gene expression; however, increased histone acetylation may have initiated a broader cell-protective response. Therefore, although probably important, augmentation of high affinity glutamate uptake may be one of several neuroprotective effects of VPA treatment.
Footnotes
Acknowledgment
The authors thank Professor N.C. Dan-bolt, University of Oslo, Norway, for the antibodies against GLT.