
Abstract
Schönfeld and Reiser recently hypothesized that fatty acid β-oxidation is a source of oxidative stress in the brain. To test this hypothesis, we inhibited brain mitochondrial β-oxidation with methyl palmoxirate (MEP) and measured oxidative polyunsaturated fatty acid (PUFA) metabolites in the rat brain. Upon MEP treatment, levels of several nonenzymatic auto-oxidative PUFA metabolites were reduced with few effects on enzymatically derived metabolites. Our finding confirms the hypothesis that reduced fatty acid β-oxidation decreases oxidative stress in the brain and β-oxidation inhibitors may be a novel therapeutic approach for brain disorders associated with oxidative stress.
INTRODUCTION
While the brain constitutes only 2% of total body mass, it consumes 20% of total body energy. In contrast to the heart and the liver, the brain is largely fueled by the oxidation of glucose rather than fatty acids, leading to a respiratory quotient of 0.97 to 1.1, 2 Even though glucose yields 20% less ATP per carbon than palmitate, glucose oxidation consumes 15% less oxygen per ATP, suggesting that the brain's dependence on glucose for energy over fatty acids reduces the risk of hypoxia in neurons where oxygen is limited. 3 According to Schönfeld and Reiser, the selection of glucose over fatty acids may be evolutionarily beneficial to the brain as mitochondrial fatty acid β-oxidation may generate superoxides via multiple pathways including; (1) increased binding to complexes I and III of electron transport chain and (2) increased flavoprotein-ubiquinone oxidoreductase activity due to the high FADH2/NADH ratio generated by fatty acid β-oxidation.4, 5, 6 Fatty acid β-oxidation might increase the peroxidation of polyunsaturated fatty acids (PUFA). Peroxidized PUFA, which are present in several brain disorders, may be especially problematic in the brain given the high levels of arachidonic acid (ARA; 20:4n−6) and docosahexaenoic acid (DHA; 22:6n−3) in the phospholipid membrane.7, 8 Finally, given the relatively low expression of superoxide dismutase and glutathione peroxidase, the brain may not have the capacity to handle oxidative stress induced by fatty acid β-oxidation. 9
Therefore, to test the hypothesis of Schönfeld and Reiser that fatty acid β-oxidation increases the oxidative stress in the brain, 10 we administered a carnitine palmitoyltransferase I inhibitor, methyl palmoxirate (MEP), to rats and analyzed the brain levels of nonenzymatic auto-oxidative PUFA metabolites and enzymatically derived metabolites. Upon MEP treatment, there was a selective decrease in brain basal levels of nonenzymatic auto-oxidative PUFA metabolites.
MATERIALS AND METHODS
Animals and Surgery
All procedures were performed in accordance with the policies of the Canadian Council on Animal Care and were approved by the Animal Ethics Committee at the University of Toronto. Male Sprague Dawley rats were purchased from Charles Rivers (Saint-Constant, QC, Canada) at 12 weeks of age and kept at the animal facility within an automated 12 hours light–dark cycle and a constant temperature of 22°C. The rats received ad libitum access to standard chow (Teklad 2018; Harlan, Madison, WI, USA) and water. At 15 weeks of age, six rats were subjected to either high-energy, head-focused microwave irradiation or CO2 asphyxiation. A separate group of 11 rats were implanted with a tail vein catheter (intravenous catheter 24 gauge/0.75 inch, Angiocath; Becton Dickinson, Mississauga, ON, Canada) and received either an intravenous injection of vehicle or 10 mg/kg of MEP (donated by S.I. Rapoport). Fifteen minutes after injection, rats were rapidly euthanized by high-energy, head-focused microwave irradiation (13.5 kW for 1.6 seconds; Cober Electronics Inc., Stratford, CT, USA) to avert ischemia for accurate quantification of in vivo basal levels of nonenzymatic auto-oxidative PUFA metabolites and enzymatically derived metabolites. Previously, we reported that this method reduced β-oxidation of fatty acid by 23% to 74%. 11 Methyl palmoxirate readily crosses the blood–brain barrier with a plasma half-life of 0.6 minute in the rat. The brain was excised and stored at −80°C for lipidomics profiling.
Eicosanoid/Docosanoid Preparation and Extraction
Composite standards of lipid metabolites (natural or deuterated; Cayman Chemicals Company, Ann Arbor, MI, USA) were diluted from stock solutions in ethanol for performing an eight-point calibration curve (0.05 to 5 ng). The internal standard mixtures were prepared in ethanol and added to all composite standards and samples before extraction. Siliconized glassware was used for extraction and sample preparations. Auto-oxidation of PUFA was minimized by extracting on ice, in reduced light conditions and with solvents containing 0.1% butylated hydroxytoluene. Frozen brain hemispheres were homogenized in methanol. Aliquots of 250 mg of homogenized brain were combined with 1 ng of internal standard mixture. In addition, external ARA, eicosapentaenoic acid (EPA), and DHA standards were prepared along with homogenized brain samples. Samples were mixed for 1 minute, incubated on ice for 30 minutes, and then centrifuged at 1000 g for 10 minutes. Supernatants were collected; while the pellet was resuspended and mixed in ethanol for 1 minute, then centrifuged again. The ethanolic supernatants were collected and combined with previously extracted methanolic supernatant. The supernatants were evaporated under nitrogen gas, suspended in 10% ethanol, acidified to pH 3 with 1 N HCl and extracted three times with ethyl acetate. The ethyl acetate layer was washed to neutrality with water and dried under nitrogen gas. Residues from brain and external standard samples were reconstituted in acetonitrile to water (1:1 by volume), transferred to inserts in amber vials, caped and immediately analyzed by LC-MS-MS.
LC-MS-MS Based Lipidomics
LC-MS-MS was performed on a 1290 UHPLC System (Agilent Technologies, Santa Clara, CA, USA) and a QTRAP5500 Mass Spectrometer (ABSciex, Framingham, MA, USA). Chromatography ran at a flow rate of 600 μL/min on a Zorbax SB-Phenyl column (Agilent Technologies; 3.0 × 50 mm, 3.5 μm) with a gradient initiating at 80% water and ramping up to 100% acetonitrile over 9 minutes. The mass spectrometer was operated in negative electrospray ionization mode with a source voltage setting of 4,500 V and a source temperature setting of 600°C. Precursor to product ion mass transitions was acquired by scheduled multiple reaction monitoring. Quantitative analysis was performed by the Analyst 1.5.2 Software (ABSciex). Area ratios of integrated peaks (natural to deuterated standard) were plotted against standard curves for quantification. The limit of quantification was 0.025 ng per sample and values below this point down to ∼0.005 ng per sample were semiquantitative. Measured lipid mediators were classified as nonenzymatic or enzymatic metabolites based on the appearance of auto-oxidative metabolites in external PUFA standards (ARA, EPA, and DHA) upon extraction with ethanol. Metabolites exceeding the threshold of 0.005 ng were classified as nonenzymatic auto-oxidative metabolites while those below are considered as enzymatically derived metabolites. Because 8-iso-PGF3α is nonenzymatically produced from free-radical-induced peroxidation, it is categorized as a non-auto-oxidative metabolite.
Statistics
Data are presented as the percent change in concentration relative to vehicle-injected rats and are expressed as mean±s.d. Statistical comparisons of eicosanoids/docosanoids between microwave fixed and CO2 asphyxiated rats as well as vehicle-injected and MEP-treated rats were performed, a priori, using two-tailed Student's t-test. Statistically significant differences are set at ∗P<0.05, ∗∗P<0.01, and ∗∗∗P<0.001.
RESULTS
Brain Lipidomics of Microwave Fixed and CO2 Asphyxiated Rats
When rats were asphyxiated with CO2, there were significant increases in the brain levels of both nonenzymatic auto-oxidative metabolites and enzymatically derived metabolites as compared with the brains of microwave fixed rats (Figure 1). This elevation ranged from a 5-fold increase in 5-hydroxyeicosatetraenoic acid (5-HETE) to a 633-fold increase in prostaglandin D2.
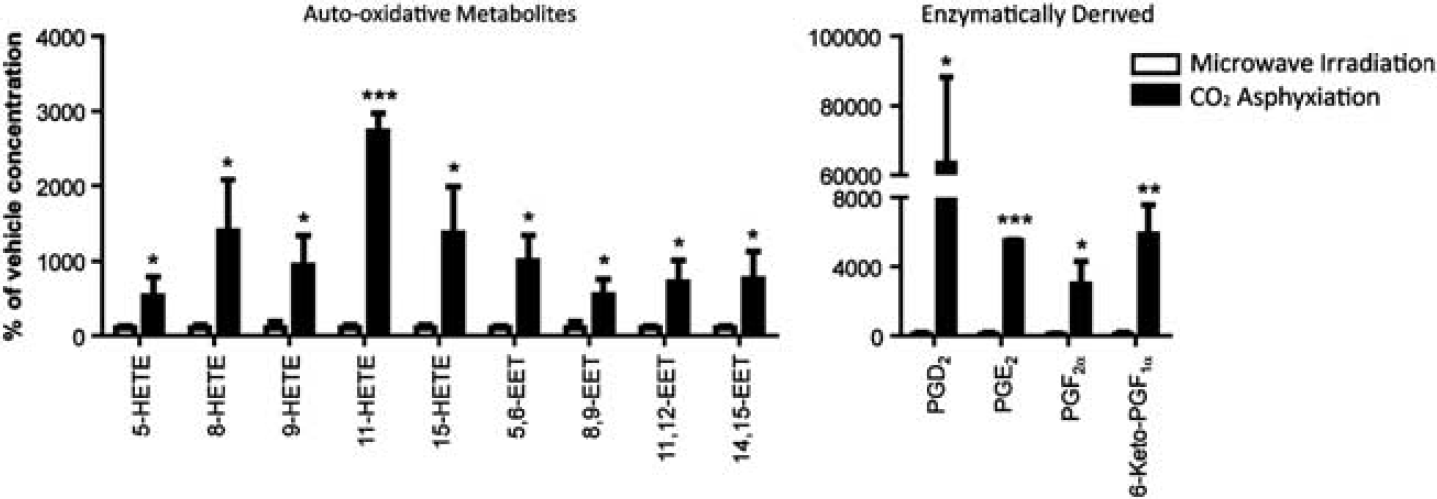
Arachidonic acid (ARA)-derived eicosanoid profiles of rat brains fixed with high-energy, head-focused microwave irradiation (white; n=3) and CO2 asphyxiation (black; n=3). Nonenzymatic auto-oxidative polyunsaturated fatty acid (PUFA) metabolites and enzymatically derived metabolites for ARA were expressed as percent of vehicle concentration. P values denote significant differences compared with vehicle injection at ∗P<0.05, ∗∗P<0.01, and ∗∗∗P<0.001. Refer to methods for classification of nonenzymatic auto-oxidative PUFA metabolites. HETE, hydroxyeicosatetraenoic acids; EET, epoxytrienoic acid; PG, prostaglandin.
Brain Lipidomics of Vehicle-Injected and Methyl Palmoxirate-Treated Rats
Inhibition of brain mitochondrial β-oxidation by MEP significantly reduced the levels of all measured HETE and epoxytrienoic acids (EET), nonenzymatic auto-oxidative metabolites of ARA, by 23% to 44% and 32% to 50% compared with vehicle-injected rats, respectively, except for 15-HETE which was unaffected (Figure 2A). In contrast, there were no significant changes in the enzymatically derived series 2 prostaglandins between the brains of vehicle-injected and MEP-treated rats. However, there was a significant 34% reduction in the level of 6-keto-PGF1α, a byproduct of PGI2 (prostacyclin) in MEP-treated rats. Similarly, the brain level of hydroxyeicosapentaenoic acids, nonenzymatic auto-oxidative metabolites of EPA, was reduced by 35% to 76% upon MEP treatment relative to vehicle. There was also a significant 30% reduction in 17(18)-epoxyeicosatetraenoic acids with MEP treatment. No significant differences were detected in the level of non-auto-oxidative EPA isoprostane, 8-iso-PGF3α, and enzymatically derived D17-6-keto-PGF1α, a byproduct of PGI3 (Figure 2B). Finally, with inhibition of brain fatty acid β-oxidation via MEP, the levels of 7- and 14-hydroxydocosahexaenoic acid were reduced by 49% and 28%, respectively; whereas 4-hydroxydocosahexaenoic acid and 17-hydroxydocosahexaenoic acid levels were not affected upon MEP treatment relative to vehicle injection (Figure 2C).
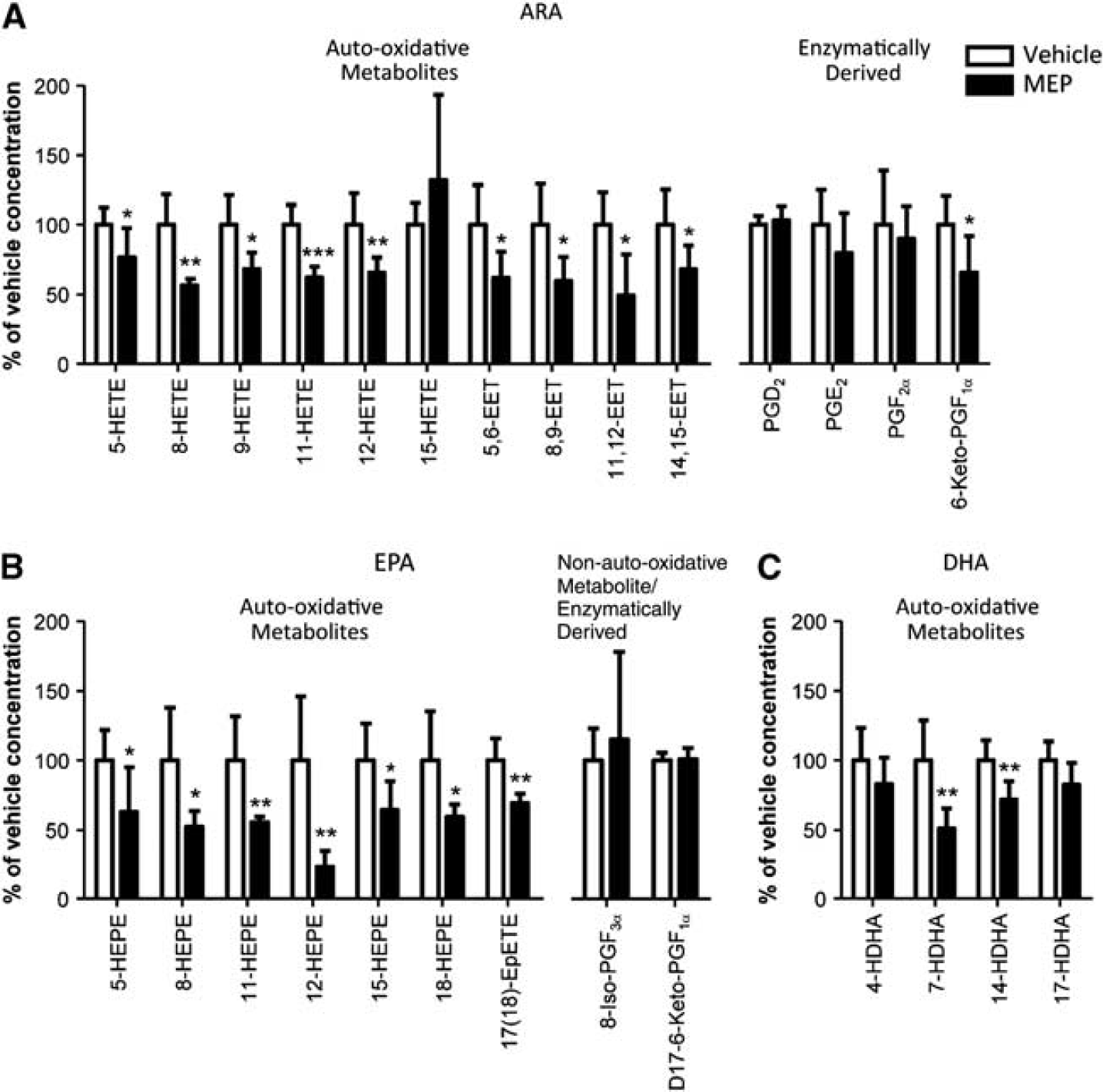
Brain eicosanoid and docosanoid profiles with vehicle injection (white; n=5) or methyl palmoxirate (MEP) treatment (black; n=6). Nonenzymatic auto-oxidative polyunsaturated fatty acid (PUFA) metabolites and enzymatically derived metabolites for (
DISCUSSION
Schönfeld and Reiser 10 proposed that fatty acid β-oxidation leads to increased oxidative stress in the brain. Therefore, we examined the role of mitochondrial β-oxidation in the brain on concentrations of oxidative metabolites of ARA, EPA, and DHA, which are susceptible to formation of lipid radicals due to their multiple double bonds. The presence of auto-oxidative PUFA metabolites from pure PUFA standards were used to classify nonenzymatic auto-oxidative PUFA metabolites and enzymatically derived lipid metabolites extracted from microwaved brains. Consistent with the literature, we found that microwave fixation is important to avert artifacts from ischemia-induced synthesis of nonenzymatic auto-oxidative PUFA metabolites and enzymatically derived metabolites (Figure 1). 12 Thus, using microwave fixation, we were able to measure in vivo basal levels of oxidative lipid metabolites in the brain. We then observed that inhibition of brain mitochondrial β-oxidation with MEP largely reduced the levels of nonenzymatic auto-oxidative PUFA metabolites of ARA, EPA, and DHA (Figure 2), suggesting that reduced brain fatty acid β-oxidation decreased oxidative stress. It is unclear why 15-HETE was unaffected, but the MEP-treated rats exhibited relatively large variation in 15-HETE concentration as compared with other lipidomics measures. In addition, this study is limited to a static picture of HETE metabolism; therefore, it is possible that while no difference was observed with 15-HETE levels, there may be difference in its turnover or longer inhibition of β-oxidation is necessary to significantly reduce 15-HETE levels. We previously reported that MEP inhibition of β-oxidation in the brain does not alter brain concentrations of unesterified ARA and DHA while it increased unesterified EPA. 11 Furthermore, MEP does not affect the uptake of DHA and EPA into the brain. 11 Thus, the reduction in nonenzymatic oxidative PUFA metabolites is unlikely due to decreases in levels of unesterified PUFA.
A limitation of this study was the use of the whole brain for lipidomics profiling. The circumventricular organs including the neurohypophysis and adenohypophysis of pituitary gland and pineal gland β-oxidize fatty acids instead of glucose. 13 Therefore, it would be of interest to see if subregions of the brain that rely on fatty acid β-oxidation have increased oxidative stress or higher antioxidant capacity.
The novel finding that MEP decreased markers of oxidative stress in the brain may have potential therapeutic implications in traumatic brain injury, epilepsy, stroke, Alzheimer's disease, and Parkinson's disease where, following the initial insult, the production of lipid peroxides increases.14, 15 Inhibition of mitochondrial fatty acid β-oxidation in the brain, and decreasing oxidized PUFA may prevent the subsequent generation of lipid peroxides which can inflict secondary damage to the brain. However, the link between mitochondrial fatty acid β-oxidation and oxidative stress requires further detailed investigation. Especially, it would be interesting to examine whether rotenone or paraquate-induced oxidative stress would increase PUFA auto-oxidative metabolites and whether MEP is protective in these models.
In conclusion, consistent with the hypothesis of Schönfeld and Reiser, inhibiting brain fatty acid β-oxidation decreased nonenzymatic auto-oxidative PUFA metabolites in the rat brain. Therefore, mitochondrial fatty acid β-oxidation could be a novel therapeutic target for brain disorders associated with increased oxidative stress.
Footnotes
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The authors wish to thank Michael Leadley and Dr. Denis Reynaud of the Analytical Facility for Bioactive Molecules of The Centre for the Study of Complex Childhood Diseases, The Hospital for Sick Children, Toronto, Canada for assistance with LC-MS-MS based lipidomics.