
Abstract
Acidosis is one of the key components in cerebral ischemic postconditioning that has emerged recently as an endogenous strategy for neuroprotection. We set out to test whether acidosis treatment at reperfusion can protect against cerebral ischemia/reperfusion injury. Adult male C57BL/6 J mice were subjected to 60-minute middle cerebral arterial occlusion followed by 24-hour reperfusion. Acidosis treatment by inhaling 10%, 20%, or 30% CO2 for 5 or 10 minutes at 5, 50, or 100 minutes after reperfusion was applied. Our results showed that inhaling 20% CO2 for 5 minutes at 5 minutes after reperfusion-induced optimal neuroprotection, as revealed by reduced infarct volume. Attenuating brain acidosis with NaHCO3 significantly compromised the acidosis or ischemic postconditioning-induced neuroprotection. Consistently, both acidosis-treated primary cultured cortical neurons and acute corticostriatal slices were more resistant to oxygen–glucose deprivation/reperfusion insult. In addition, acidosis inhibited ischemia/reperfusion-induced apoptosis, caspase-3 expression, cytochrome c release to cytoplasm, and mitochondrial permeability transition pore (mPTP) opening. The neuroprotection of acidosis was inhibited by the mPTP opener atractyloside both in vivo and in vitro. Taken together, these findings indicate that transient mild acidosis treatment at reperfusion protects against cerebral ischemia/reperfusion injury. This neuroprotection is likely achieved, at least partly, by inhibiting mPTP opening and mitochondria-dependent apoptosis.
INTRODUCTION
Ischemic postconditioning, a neuroprotective strategy for ischemic stroke, has attracted much attention over the past 7 years. It is defined as a series of cycles of brief reperfusion and ischemia applied at the onset of reperfusion, which can mobilize the brain's own endogenous adaptive mechanisms and limit the extent of reperfusion injury. In 2006, Zhao et al 1 first reported that ischemic postconditioning reduces infarct volume in experimental cerebral ischemia and reperfusion (I/R) injury. Since then, its cerebral protective effects have been confirmed in several distinct ischemia models.2, 3, 4, 5 However, the clinical application of ischemic postconditioning may be limited because of the difficulties in achieving well-controlled interruptions of reperfusion and the risks associated with these manipulations. 6 Hypoxia, as a key component of ischemia, is a feasible treatment for ischemic stroke in theory. Surprisingly, we have recently found that an early application of hypoxic postconditioning after reperfusion does not induce neuroprotection against cerebral I/R injury. 7 Thus, attempts should be undertaken to identify the other key factors, which are responsible for the protection afforded by ischemic postconditioning, and to develop a new therapeutic strategy, which is relatively safe and practical.
Acidosis is another key component of ischemia besides hypoxia and glucose depletion. Extracellular pH can fall from 7.2 to below 6.5 in the core during focal cerebral ischemia.8, 9 Acidosis treatment at reperfusion induces cardioprotection.10, 11, 12, 13 Cohen et al 11 provided the experimental evidence, in the isolated rabbit heart, that alkalotic buffer blocked ischemic postconditioning-conferred protection, and transient acidic reperfusion immediately applied at the onset of reperfusion protected against cardiac ischemia. The studies of acidosis treatment on hearts have provided much theoretical support for the acidosis therapeutic strategy toward ischemic stroke.
Recently, we showed that mild acidosis pretreatment may produce a preconditioning-like neuroprotective effect in a forebrain ischemia model. 14 Although transient acidosis treatment before cerebral ischemia may be beneficial to the ischemic brain, the unpredictable occurrence of stroke greatly limits the translational relevance of this treatment. Therefore, the transient mild acidosis treatment at reperfusion after ischemia would be more feasible and have wider clinical applications. However, only indirect evidence was presented about severe acidosis treatment during recovery, which was performed in a metabolic inhibition model in cultured neurons. 15
The present study was designed to investigate the neuroprotective effects of transient mild acidosis at reperfusion in cerebral I/R models both in vivo and in vitro. We further tested the hypothesis that this acidosis treatment reduces I/R-induced neuronal death by inhibiting mitochondrial permeability transition pore (mPTP) opening and mitochondria-dependent apoptosis.
MATERIALS AND METHODS
Animals
Male adult C57BL/6 mice (8 to 10 weeks) weighing 22 to 25 g were provided by the Experimental Animal Centre of the Zhejiang University and housed under diurnal lighting conditions (12 hours darkness/light). Our manuscript was written according to ARRIVE guidelines. All experimental protocols and animal handling procedures were in complete compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the experimental protocols were approved by the Zhejiang University Animal Experimentation Committee. Every effort was made to minimize any pain or discomfort, and the minimum number of animals was used.
Transient Focal Cerebral Ischemia
Mice were anesthetized with an intraperitoneal injection of choral hydrate (400 mg/kg). Transient focal cerebral ischemia was induced by middle cerebral artery occlusion (MCAO), as previously described.
16
Briefly, a 6-0 nylon monofilament suture, blunted at the tip and coated with 1% poly-
Infarct volume was determined 24 hours or 72 hours after reperfusion by 2,3,5-triphenyltetrazolium hydrochloride (0.25%) staining, and the extent of the normal and infarcted areas was analyzed with ImageJ software and determined by the indirect method, which corrects for edema (contralateral hemisphere volume minus nonischemic ipsilateral hemisphere volume). The percentage of the corrected infarct volume was calculated by dividing the infarct volume by the total contralateral hemispheric volume, and this ratio was then multiplied by 100.
Neurologic deficit scores were evaluated, as previously described, 17 at 24 hours of reperfusion. 0, no deficit; 1, flexion of contralateral forelimb upon lifting of the whole animal by the tail; 2, circling to the contralateral side; 3, falling to contralateral side; and 4, no spontaneous motor activity.
Protocols of Acidosis Treatment and Ischemic Postconditioning In Vivo
For postconditioning experiments, mice were randomly assigned to each group. For acidosis treatment, the mice were maintained by inhaling CO2 (21% O2 supplemented with N2), and the flow rate was 1 L/minute. For time-window experiments, after 60 minutes of MCAO, reperfusion was established for 5, 50, or 100 minutes and then mice were subjected to 5-minute 20% CO2 inhalation. For time course experiments, inhaling 20% CO2 for 5 or 10 minutes was applied at 5 minutes after 60-minute MCAO. For concentration-dependence experiments, inhaling 10%, 20%, or 30% CO2 for 5 minutes was applied at 5 minutes after 60-minute MCAO. For ischemic postconditioning, the bilateral common carotid arteries were transiently occluded for 5 minutes at 5 or 50 minutes after reperfusion using aneurysm clips. In the NaHCO3 treatment groups, NaHCO3 (2 μL/animal, 78 mg/mL) was intracerebroventricularly injected at 3 minutes after reperfusion. Corresponding control groups were injected with artificial cerebrospinal fluid (ACSF; 2 μL/animal, intracerebroventricularly). The mPTP opener, atractyloside (2 μL per animal, 3 mmol/L, intracerebroventricularly) was injected 5 minutes before reperfusion.
Intracerebral pH Measurement after Ischemia
For continuous measurement of pH in the brain, a fiberoptic pH micro system (pH-Optica, WPI, Sarasota, FL, USA) was used, as previously described. 18 A glass fiber with its pH-sensitive tip (140 μm OD) was stereotaxically implanted into the brain immediately after MCAO at the following coordinates: 0.1 mm caudal to bregma, 2.4 mm lateral to midline, and 2.2 mm deep. The pH value was continuously measured for 30 minutes. The instrument was calibrated with standard solutions (pH 4.0, 5.0, 6.0, 7.0, 8.0, and 9.0) before and after the experiments. To mimic the brain environment, the standard solutions were prewarmed to 37°C and the calibration was performed in the absence of light. Acidosis treatment-induced changes in pH were evaluated.
Preparation of Brain Slices, Oxygen–Glucose Deprivation Procedures, and Slice Viability Determination
Acute brain slices were prepared from adult male C57BL/6 mice and oxygen–glucose deprivation (OGD) procedures were carried out, as we previously described. 6 Briefly, corticostriatal slices (400-μm thick) were cut in ice-cold ACSF bubbled with 5% CO2 and 95% O2 (pH 7.4). The slices were immersed in oxygenated ACSF for 1 hour and then at 37°C for 15 minutes before experiments. For OGD, slices were transferred into glucose-free ACSF bubbled with 5% CO2 and 95% N2 for 15 minutes, then returned to oxygenated ACSF for 1 hour. Control slices were kept in oxygenated ACSF. For acidosis treatment, slices were transferred to acidic buffer equilibrated with 80% O2 and 20% CO2 (pH 6.8) for 1, 3, or 5 minutes at 0, 5, 15, or 30 minutes after simulated reperfusion. Slice viability was determined by 0.25% 2,3,5-triphenyltetrazolium hydrochloride staining. Formazan extracted with ethanol/dimethylsulfoxide (1:1) was measured at 490 nm. Viability was determined by absorbance normalized to the dry weight of the slice and expressed as the percentage of control slices included in each experiment.
Cell Culture, Oxygen–Glucose Deprivation Procedures, and Cell Viability Determination
Primary cortical neuronal cultures were made with embryonic day 17 Sprague–Dawley rat fetuses, as previously described.
14
After trypsin digestion, separated cells were seeded onto poly-
Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling Assays
Apoptosis was determined by terminal deoxynucleotidyl transferase dUTP nick end labeling assays using an in situ cell death detection kit (Roche Diagnostics Corporation, Indianapolis, IN, USA), according to the manufacturer's protocol. Images of five random fields were captured from each sample. The results were expressed as percentage of terminal deoxynucleotidyl transferase dUTP nick end labeling-positive cells to total 4′,6-diamidino-2-phenylindole-stained cells. Each experiment was repeated at least six times.
Assessment of Mitochondrial Membrane Potential (ΔΨ m) and Mitochondrial Permeability Transition Pore Opening
JC-1 staining indicates the changes in relative ΔΨm. 19 Cells were incubated with 2.5 μg/mL JC-1 (Beyotime, Jiangsu, China) dissolved in Dulbecco's Modified Eagle Medium for 15 minutes at 5 minutes after reperfusion and excited at 488 nm under a fluorescence microscope. In the acidosis treatment group, the JC-1-containing Dulbecco's Modified Eagle Medium was equilibrated with normoxic mixed gas containing 20% CO2. Images of five random fields were captured from each sample. Green JC-1 staining indicated impaired and red indicated normalΔΨm. Data were calculated as the average green/red ratio using Image-Pro Plus 5.0 (Media Cybernetics, MD, USA). The results are expressed as the percentage of control. Each experiment was repeated at least six times.
To assay mPTP opening, mice were killed at 3 hours after reperfusion, the brain was quickly removed, and immediately frozen in liquid nitrogen. Brains were cut at a 30-μm thickness on a freezing microtome. Then, the mPTP opening of the slices was measured using an mPTP fluorescent assay kit (GENMED, Shanghai, China) according to the manufacturer's instructions. The fluorescent dye is calcein-AM, which is applied in the presence of cobalt chloride to quench cytoplasmic signal. Opening of mPTP is marked by a loss in green fluorescence from mitochondria. 20 Images of five random fields were captured from each sample. Data were calculated as the integrated intensity of green fluorescence using Image-Pro Plus 5.0 (Media Cybernetics). The results are expressed as the percentage of control.
Western Blot Analysis
For Western blot, protein samples were separated on 12% SDS-PAGE and blotted onto nitrocellulose membranes. The blocked membranes were incubated with anti-cleaved caspase-3 (1:1,000; CST, Danvers, MA, USA), anti-cytochrome C (1:1,000; CST) anti-glyceraldehyde 3-phosphate dehydrogenase (1:3000; Kangcheng Biotech, Shanghai, China), and further incubated with either IRDye 800 anti-rabbit (1:10,000; LI-COR Biosciences, Lincoln, NE, USA), or IRDye 700 anti-mouse (1:5000, LI-COR Biosciences). Band density was analyzed. The results are presented as the anti-caspase-3/glyceraldehyde 3-phosphate dehydrogenase or cytochrome c/glyceraldehyde 3-phosphate dehydrogenase ratio and normalized to that in the control groups.
Statistical Analysis
All data were collected and analyzed in a masked fashion. Data are presented as mean±s.e.m. One-way analysis of variance with least significant difference or Dunnett's T3 post hoc test (where equal variances were not assumed) was applied for multiple comparisons, whereas Student's t-test was used for comparisons between two groups. Neurologic deficit scores were analyzed with the nonparametric Mann–Whitney U test. P<0.05 was considered statistically significant.
RESULTS
Acidosis Treatment at Reperfusion Reduced Infarct Volume
To determine the effect of acidosis treatment, mice were maintained under 20% CO2 with various permutations after transient MCAO. Five minutes of acidosis treatment initiated at 5 minutes after the onset of reperfusion reduced infarct volume by ∼50% (P<0.01, Figure 1A), and the animals exhibited lower neurologic deficit scores at 24 hours (MCAO alone group: 2.33±0.17, n=9; acidosis treatment group: 1.44±0.18, n=9; P<0.01). The neuroprotection was still robust, even when the onset time was delayed to 50 minutes after reperfusion. However, acidosis treatment initiated at 100 minutes did not block the I/R injury. In addition, the neuroprotection disappeared when the duration of acidosis treatment was prolonged to 10 minutes (Figure 1B). Furthermore, 10% or 20% CO2 inhalation for 5 minutes applied at 5 minutes after reperfusion significantly reduced infarct volume (P<0.01) whereas more severe acidosis treatment (30% CO2) failed to induce neuroprotection. We measured the pH in the ischemic cortex of mice and found that breathing 10%, 20%, and 30% CO2 for 5 minutes reduced pH by ∼0.16, ∼0.24, and ∼0.36, respectively, whereas the pH increased by ∼0.19 in the MCAO alone group at the same time point (Figure 1C). Correspondingly, the arterial pCO2 significantly increased after 20% CO2 inhalation (MCAO alone group: 54.58±4.62 mm Hg, n=4; acidosis treatment group: 140.50±11.92 mm Hg, n=4; P<0.01), and the arterial pH value also significantly decreased (MCAO alone group: 7.20±0.02, n=4; acidosis treatment group: 6.81±0.03, n=4; P<0.001). The pH value of ischemic brain tissue at 50 minutes after reperfusion in the MCAO alone group still did not completely recovery to normal pH (6.92±0.07, n=6), whereas an alkalosis was observed at 100 minutes after reperfusion (7.19±0.06, n=6). To further confirm that the protection by CO2 is attributable to the acidosis of brain tissue, an alkaline solution (NaHCO3) was infused intracerebroventricularly before 20% CO2 inhalation. NaHCO3 reversed the neuroprotection induced by 20% CO2 inhalation for 5 minutes at 5 minutes after reperfusion (P<0.01), whereas NaHCO3 alone had no effect on I/R injury (Figure 1D). During the 20% CO2 inhalation period, a significant elevation of CBF in the core and peripheral regions of the MCA territory was observed compared with MCAO alone group, and the CBF quickly recovered at 10 minutes after withdrawing CO2 (Figure 1E). NaHCO3 did not attenuate the elevation of CBF induced by CO2 inhalation. The 20% CO2 inhalation also induced a slight decrease of mean arterial blood pressure and heart rate, whereas NaHCO3 (intracerebroventricularly) did not inhibit this decrease (Supplementary Figure 1). In addition, 5-minute acidosis treatment (20% CO2) initiated at 5 minutes after reperfusion also significantly decreased infarct volume at 72 hours after reperfusion (P<0.01, Figure 1F).
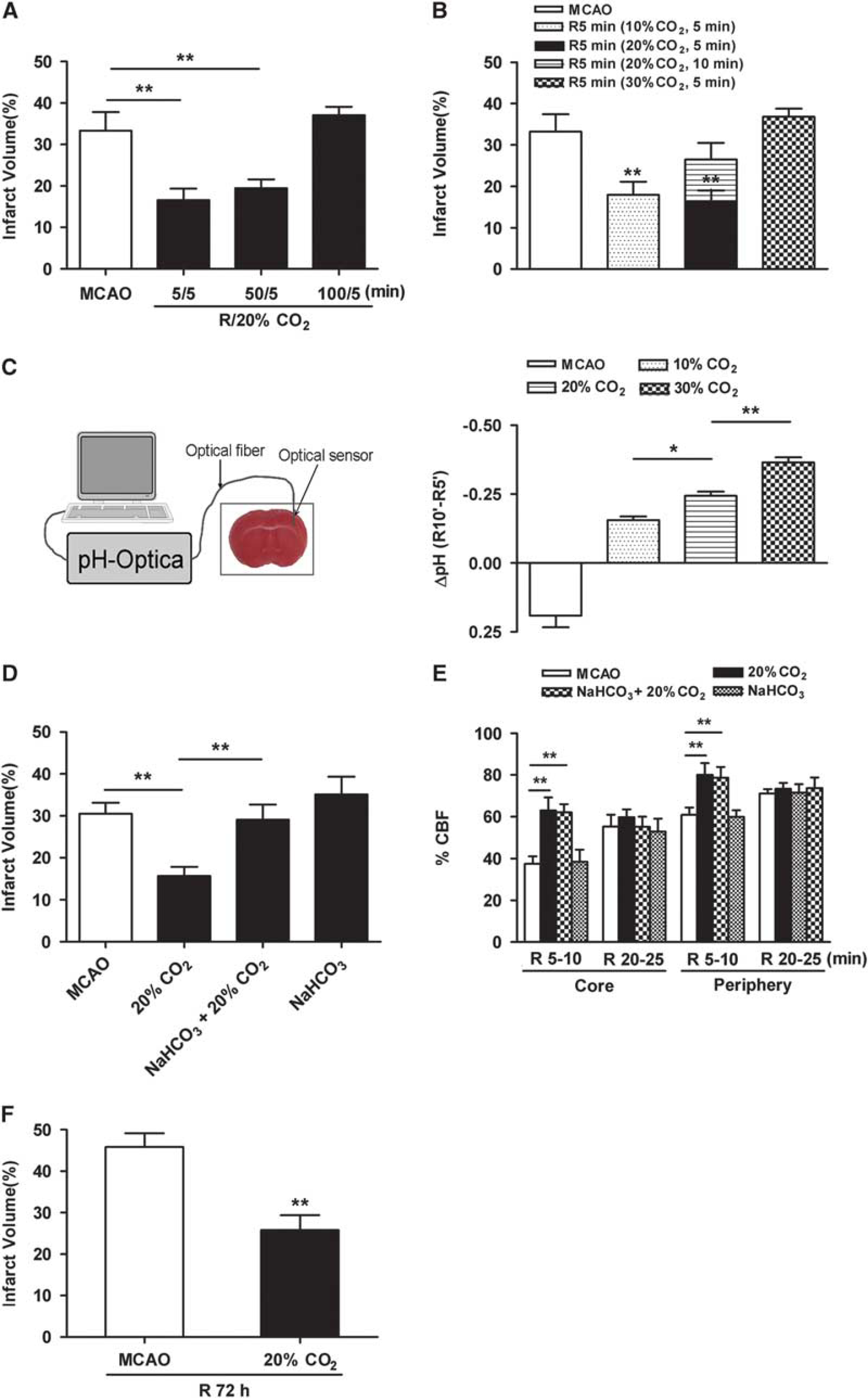
Effects of acidosis treatment on infarct volume after transient middle cerebral artery occlusion (MCAO). Animals were subjected to 60-minute MCAO. Infarct volume was quantified by 2,3,5-triphenyltetrazolium hydrochloride staining at 24 hours after reperfusion. (
NaHCO3 Reversed Ischemic Postconditioning-Induced Neuroprotection
To compare the time-windows of ischemic postconditioning with that of acidosis treatment, animals were subjected to postconditioning with 5 minutes of bilateral common carotid artery occlusions at 5 or 50 minutes after 60-minute MCAO (Figure 2). Ischemic postconditioning initiated at 5 but not 50 minutes after the onset of reperfusion reduced infarct volume (P<0.01). To assess the contribution of acidosis in ischemic postconditioning-induced neuroprotection, alkalization of the ischemic brain with NaHCO3 (intracerebroventricularly) was applied before ischemic postconditioning. Indeed, NaHCO3 reversed the neuroprotection induced by ischemic postconditioning (P<0.01).
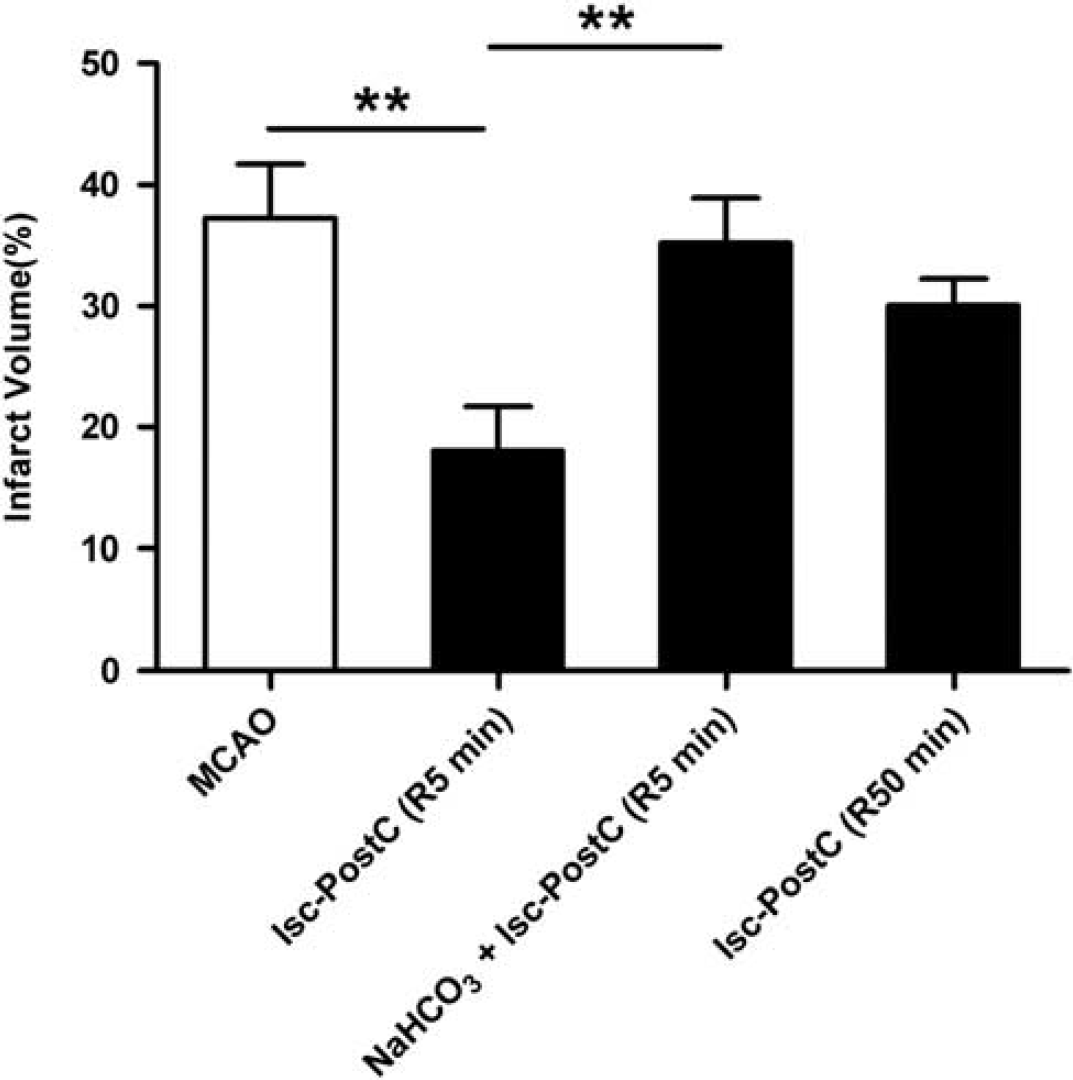
Effects of NaHCO3 on ischemic postconditioning-induced neuroprotection. Animals were subjected to 60-minute middle cerebral artery occlusion (MCAO). In the ischemic postconditioning group, bilateral common carotid arteries were occluded for 5 minutes at 5 or 50 minutes after reperfusion. NaHCO3 (2 μL per animal, 78 mg/mL, intracerebroventricularly) was infused at 3 minutes before ischemic postconditioning. Infarct volume was quantified by 2,3,5-triphenyltetrazolium hydrochloride(TTC) staining at 24 hours after reperfusion. Values show mean±s.e.m. n=8 to 10 for each group; ∗∗P<0.01. Isc-PostC, ischemic postconditioning; R, reperfusion.
Acidosis Treatment at Reperfusion-Induced Neuroprotection in vitro
We also established in vitro models with OGD-treated primary cultured neurons and acute brain slices. In cultured neurons, both 5 and 15 minutes of acidosis treatment significantly improved viability at 24 hours after 120-minute OGD, whereas 30 minutes did not (Figure 3A). The neuroprotection by acidosis treatment remained protective within 15 minutes after reperfusion, whereas 30 minutes did not (Figure 3B). Five minutes of recovery and then 15 minutes of acidosis treatment induced the optimal neuroprotection, which was used in further studies. In brain slices, acidosis treatment also protected against OGD/reperfusion-induced injury in an onset time- and duration-dependent manner (Figures 3C and 3D). In detail, 3 minutes of acidosis treatment initiated at 5 minutes after the onset of reperfusion achieved the maximum neuroprotection.
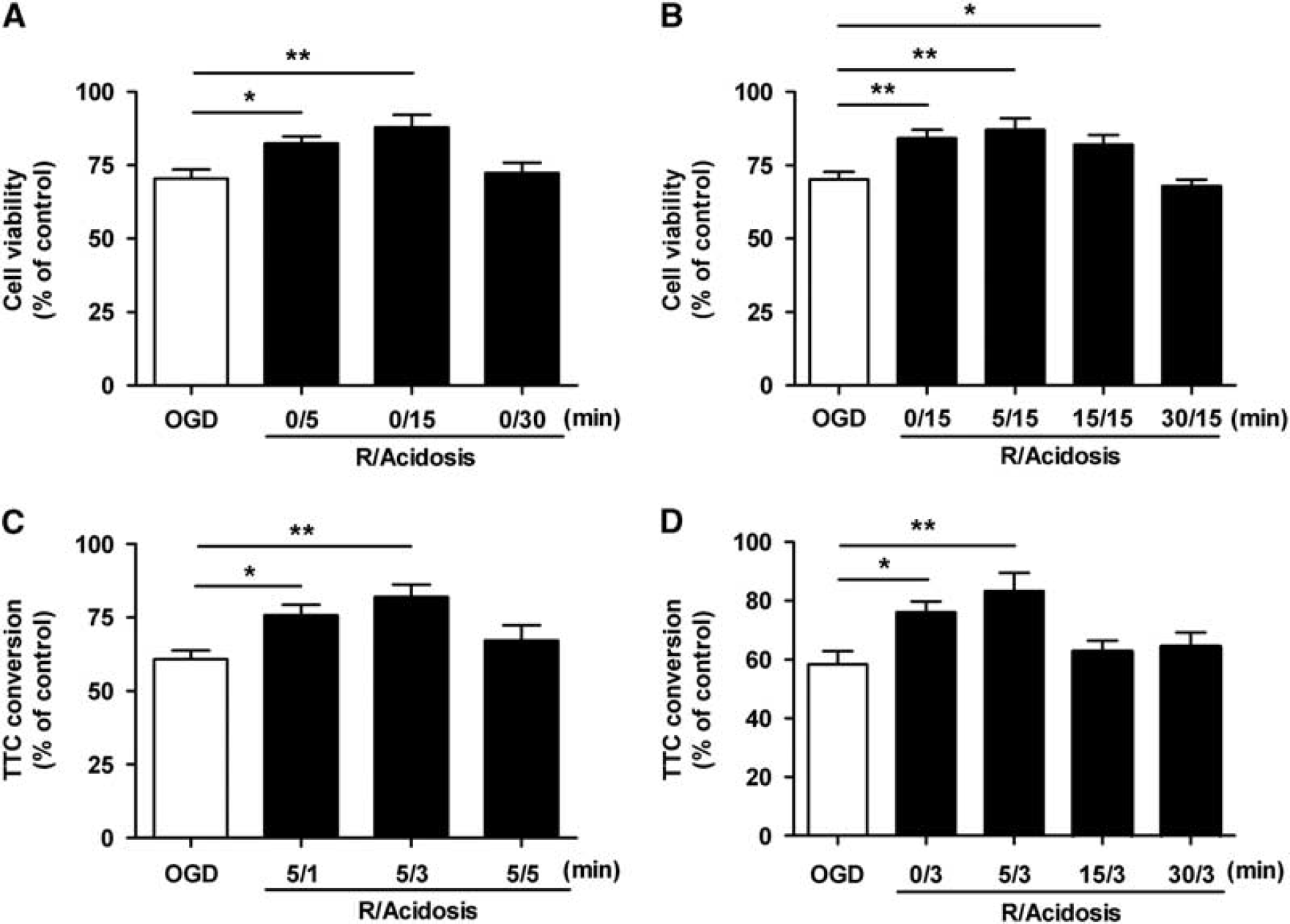
Effects of acidosis treatment on oxygen–glucose deprivation (OGD)/reperfusion-induced injury in cultured neurons and corticostriatal slices. Cultured neurons (
Acidosis Treatment Inhibited Ischemia and Reperfusion-Induced Apoptosis and Mitochondrial Permeability Transition Pore Opening
To test whether acidosis treatment prevents ischemia-induced neuronal apoptosis, the apoptotic ratio was determined by terminal deoxynucleotidyl transferase dUTP nick end labeling staining. Acidosis treatment reduced the OGD/reperfusion-induced apoptotic ratio from 42.4±4.6 to 23.2±3.0% (P<0.01; Figure 4A). The cleaved caspase-3 expression was also inhibited by acidosis treatment in both primary cultured neurons and ischemic brain (P<0.01 versus OGD group and P<0.05 versus MCAO group; Figures 4B and 4C). In addition, cytochrome c release to the cytoplasm was also attenuated by acidosis treatment in cultured neurons (P<0.05 versus OGD group; Figure 4D). These data suggest that acidosis treatment protects against ischemia-induced neuronal injury by, at least partly, reducing mitochondria-dependent apoptosis.
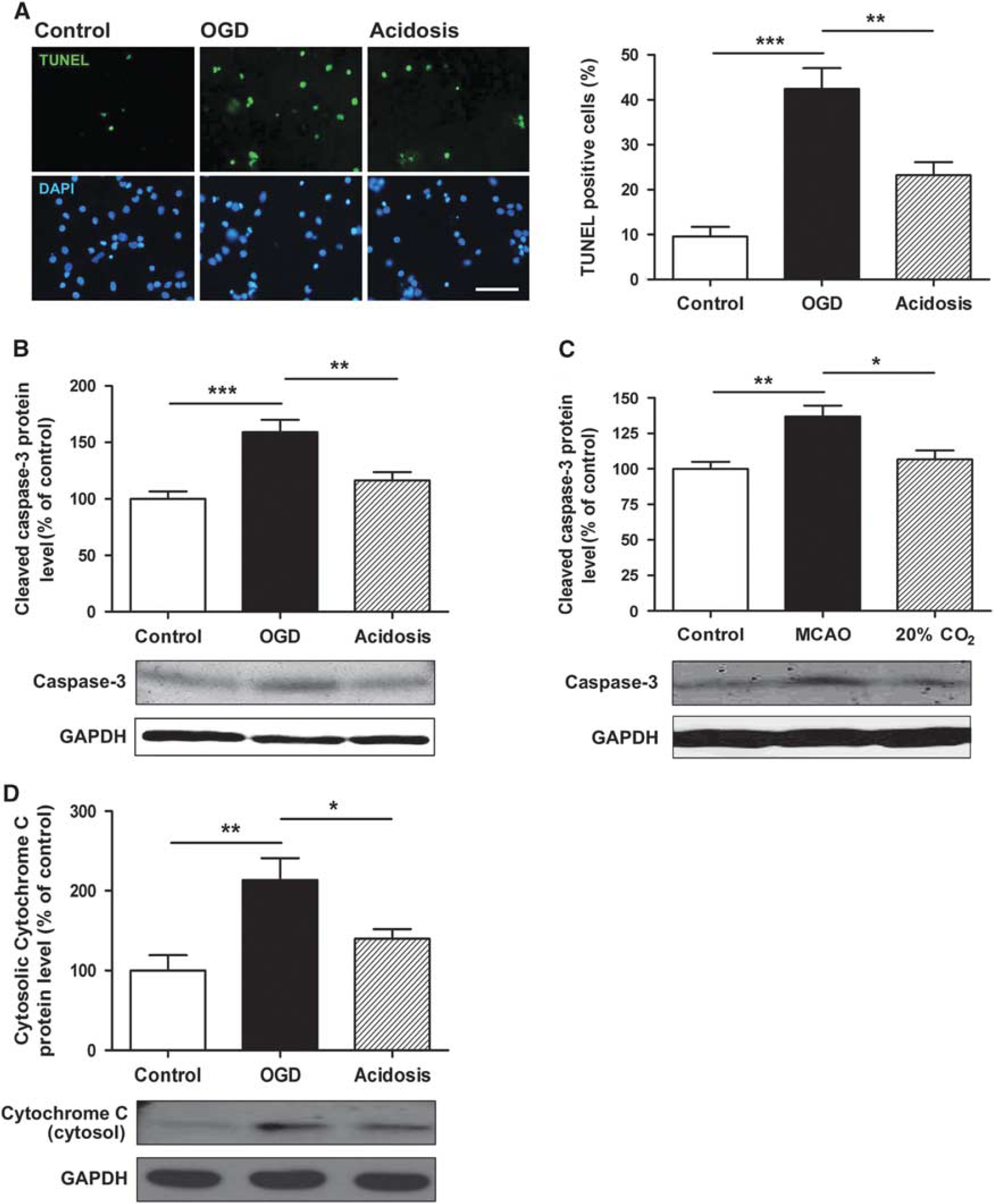
Effects of acidosis treatment on ischemia and reperfusion-induced apoptosis. Cultured neurons were treated with acidosis (pH 6.8) for 15 minutes at 5 minutes after reperfusion, and animals were treated with acidosis by inhaling 20% CO2 for 5 minutes at 5 minutes after reperfusion. Apoptosis was determined at 24 hours after reperfusion using terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) staining. (
Mitochondria-dependent apoptosis is triggered by opening of the mPTP as marked by dissipation of ΔΨm and loss of the mitochondrial calcein fluorescence.20, 21, 22 Dissipation of the ΔΨm was observed at 20 minutes after reperfusion in the cultured neurons (P<0.001 versus control group; Figure 5A), and this was attenuated by acidosis treatment (P<0.05 versus OGD group). In in vivo model, after 1-hour MCAO and 3-hour reperfusion, green fluorescence of calcein in the periphery of the MCA territory strongly attenuated (P<0.001 versus control group), which means the openning of mPTP, whereas acidosis treatment significantly inhibited the I/R-induced mPTP opening (P<0.05) as shown by increased green calcein fluorescence compared with the MCAO group (Figure 5B). Further, the neuroprotection by acidosis treatment was blocked when the mPTP opener atractyloside was applied to cultured neurons and the ischemic brain (Figures 5C and 5D).
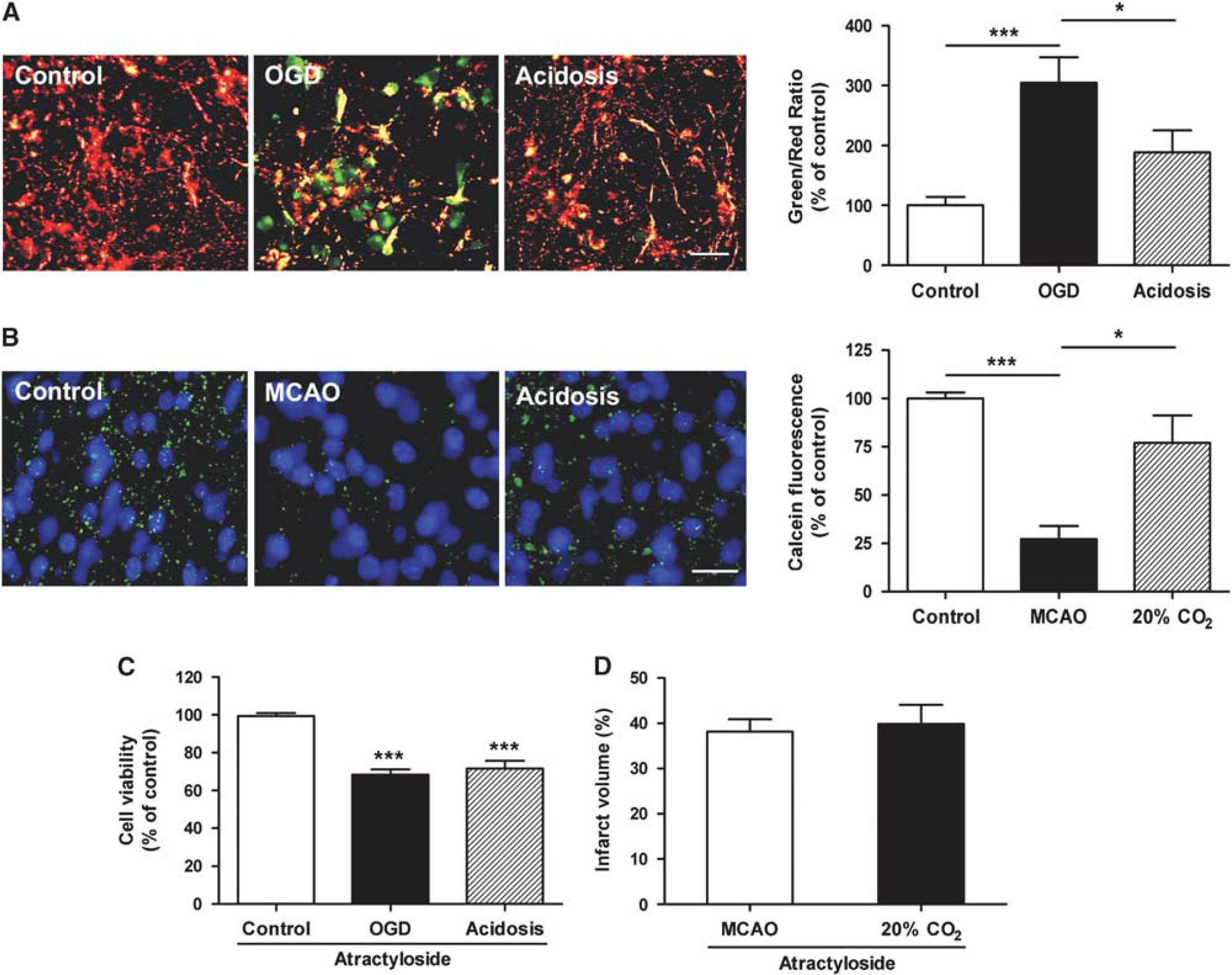
Effects of acidosis treatment on ischemia and reperfusion-induced mitochondrial permeability transition pore (mPTP) opening. Cultured neurons were treated with acidosis (pH 6.8) for 15 minutes at 5 minutes after reperfusion, and animals were treated with acidosis by inhaling 20% CO2 for 5 minutes at 5 minutes after reperfusion. (
DISCUSSION
In the present study, we showed for the first time that transient mild acidosis treatment at reperfusion protected I/R-induced neuronal injury both in vivo and in vitro. The acidosis treatment by briefly inhaling CO2 afforded a better outcome in adult mice, as shown by a remarkable reduction in infarct volume. This protection was comparable with that achieved by ischemic postconditioning, and the time-window was as long as 50 minutes. The protection was robust, as we observe less infarction in the brain 72 hours after reperfusion, which suggests that acidosis treatment results in long-lasting neuroprotection. Interestingly, we found that alkalosis by intracerebroventricularly injection of NaHCO3 completely aborted the protection by ischemic postconditioning or acidosis treatment, which implies that acidosis might be a main contributor to ischemic postconditioning-induced neuroprotection. Furthermore, the effect of acidosis treatment was supported by our in vitro models, in which acidosis treatment with acidic medium equilibrated with normoxic mixed gas containing 20% CO2 protected both acute corticostriatal slices and primary cultured neurons against OGD/R-induced injury. These data strongly suggest that acidosis treatment by briefly inhaling CO2 could be a feasible and promising therapeutic strategy for ischemic stroke.
It has been reported that in the transient focal cerebral ischemic injury model, the initial time-window of ischemic postconditioning is no more than 10 minutes. 3 This narrow time-window may limit the application of ischemic postconditioning. Here, we found that optimal protection was obtained with a single 5-minute period of acidosis treatment administered 5 minutes after the 60-minute MCAO. Interestingly, even when the onset of acidosis treatment was delayed for 50 minutes, the neuroprotection was still robust and reduced the infarct volume by ∼42%, whereas, at the same time point, ischemic postconditioning did not induce neuroprotection. These results indicate that the time-window of acidosis treatment is much wider than that of ischemic postconditioning. The wide time-window implies that patients can easily receive effective therapy without a lot of risk. The discrepancy between acidosis treatment and ischemic postconditioning in time-window implies that the acidosis applied at 50 minutes after reperfusion confers its neuroprotection by mechanisms distinct from that of delayed ischemic postconditioning. In addition, the time-windows are also different between brain and heart. It has been shown that acidosis treatment applied immediately at the onset of reperfusion is optimal in the ischemic heart. 11 Delaying treatment for even 1 minute eliminates protection in the isolated heart, which implies that acidosis at reperfusion for treatment of ischemic stroke might be a more practical therapeutic strategy than for treatment of cardiac ischemia.
Besides the onset time, the change of pH and duration of acidosis are also critical for neuroprotection. Interestingly, we found that acidosis treatment showed pH-dependent neuroprotection against MCAO/reperfusion. Both 10% and 20% CO2 inhalation induced a robust reduction in the infarct volume, and the corresponding pH measured by pH-Optica in the ischemic cortex decreased by ∼0.16 and ∼0.24, respectively (Figure 1C). However, 30% CO2 inhalation, in which the pH decreased by ∼0.36, failed to induce neuroprotection. These results suggest that excessive downregulation of pH may not be necessary or beneficial for effective clinical acidosis treatment. The 10% CO2 level may be a better dose, because this concentration is closer to the 5% CO2 in oxygen supplies for medicine, and would lower the risk of hypercapnic acidosis in the clinic. In addition, excessive duration is also not beneficial for inducing neuroprotection, as a 5-minute acidosis treatment was more effective in preventing MCAO/reperfusion-induced brain injury than a prolonged 10-minute acidosis treatment. Consistently, in our in vitro models, prolonged acidosis in neural cells (30 minutes) and brain slices (5 minutes) did not improve the survival rate. These results are in accord with those in acidosis treatment-protected cardiac cells and acidic pretreatment in brain slices in our previous report.12, 14 Therefore, the duration of acidosis might be a crucial factor contributing to acidosis treatment-induced neuroprotection.
It has been known that CO2 enhances vasodilatation and increases CBF.23, 24 In this study, a slight elevation of CBF was also observed during CO2 inhalation, and the CBF quickly recovered at 10 minutes after withdrawing CO2. The transient elevation of CBF induced by CO2 inhalation may alleviate I/R injury. However, alkalization of the ischemic brain with NaHCO3 attenuated the neuroprotection of CO2 inhalation but had no effect on the CBF. In addition, in vitro, substrate availability is always perfect in contrast with milieu in vivo where it depends on the CBF level, and the neuroprotection of acidosis treatment can still be found in acute corticostriatal slices and primary cultured neurons. These data eliminate the possibility that the neuroprotection induced by acidosis treatment is solely due to a modification of CBF, so it is reasonable to propose that there is additional neuroprotective mechanism besides the elevation of CBF.
In the reperfusion phase, the rapid return of pH values from acidic to normal worsens the injury, which is called ‘pH paradox’. 10 The mPTP regulation is one of the mechanisms involved in the ‘pH paradox’ phenomenon, which has been found in myocardium but still uncertain in neurons.11, 25 Rapid relative alkalosis at the early phase of reperfusion may facilitate mPTP opening, whereas low pH is a potent blocker.11, 26, 27 Here, it was observed that acidosis treatment at reperfusion prevented OGD/reperfusion-induced ΔΨm loss in vitro and inhibited mPTP opening in vivo. Importantly, the mPTP opener atractyloside almost compeletly reversed the neuroprotection of acidosis treatment, suggesting that acidosis treatment ameliorated I/R-induced injury mainly by, if not all, inhibiting mPTP opening. The mPTP opening is a prerequisite for mitochondria-dependent apoptosis.21, 22 As a result, we further found that acidosis treatment decreased apoptotic cell number, caspase-3 expression and cytochrome c release to cytosol, which is similar with the previously reported ischemic postconditioning-induced anti-apoptotic effects.1, 2 These data indicate that acidosis treatment retard reperfusion-induced pH increase, subsequently inhibit mPTP opening and mitochondria-dependent apoptosis.
Overall, the current data indicate that mild acidosis treatment at reperfusion protected against I/R-induced brain injury both in vivo and in vitro, and inhibiting mPTP opening and mitochondria-dependent apoptosis is involved in the acidosis-induced neuroprotection. Taking into account the practical advantages, this research may provide a novel and promising strategy to reduce or even prevent cerebral I/R injury in patients treated with intravenous thrombolytic agents or suffering from surgically induced circulation arrest, as acidosis treatment by inhaling CO2 may be easily translated into clinical practice if the onset time, duration, and pH change can be well controlled.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.