
Abstract
Dehydroepiandrosterone (DHEA) has been implicated not only to prevent N-methyl-
Introduction
Dehydroepiandrosterone (DHEA) represents one of the most abundant circulating androgen precursor preferentially produced in the adrenal gland. DHEA is also known to be synthesized de novo in the central nervous system (Baulieu and Robel, 1998). Significant beneficial effects of the DHEA replacement have been reported in patients representing androgen deficiency because of adrenal insufficiency (Cameron and Braunstein, 2005). The age-dependent withdrawal of DHEA levels is likely to be involved in the onset of age-related neuronal diseases including stroke and Alzheimer's disease (Schneider et al, 1992). Both in vivo and in vitro studies have shown that DHEA functions as a neurotrophic or neuroprotective factor to prevent N-methyl-
In contrast, DHEA has been regarded as an excitatory neurosteroid to potentiate selectively the neuronal responses to NMDA (Bergeron et al, 1996), leading to an enhanced Ca2+ influx through the NMDA receptor (NMDAr; Compagnone and Mellon, 1998). Increases in Ca2+ influx through NMDAr by the massive release of glutamate after transient cerebral ischemia has been identified to be one of the major factors to induce delayed neuronal death (Berliocchi et al, 2005). Hypothalamic neurons reportedly undergo apoptosis after exposure to DHEA (Lin et al, 2004). However, to date, little has been studied concerning the DHEA-exerting inconsistent effects on ischemia-induced cerebral damages, notwithstanding outcomes from those studies would be extremely important in clinical practice.
In the present study, we examined the effects of DHEA on the hippocampal neuronal death and deficits in spatial memory induced by transient global cerebral ischemia. Considering possible DHEA benefits in treating ischemic brain damages including those by stroke, we were particularly concerned with how the administration timing of DHEA affects the outcome. To this end, rats subjected to ischemic insult were administered a single injection of DHEA (20 mg/kg) at 1 h before or 1, 3, 6, 12, 24, 36, 48, 72, or 96 h after cerebral ischemia, and neuronal death and learning behaviors were examined after ischemia. In addition, using pharmacologic tools, mechanisms underlying the DHEA neurotoxicity and neuroprotection were investigated.
Materials and methods
Male Sprague—Dawley rats (Oriental Bio Service Inc., Nanjing, China), weighing 200 to 250g before experiments, were used throughout the study. The animals were housed in a light controlled room under a 12 h light—dark cycle starting at 0700 and kept at a temperature of 25°C. Animals were given unrestricted access to food and water. All procedures were in accordance with the guidelines of Institute for Laboratory Animal Research of the Nanjing Medical University.
Preparation of Ischemia Model
We used a 4-vessel occlusion (4VO) method for transient global ischemia as described elsewhere (Kiprianova et al, 1999). Briefly, using sodium pentobarbital anesthesia (60 mg/kg, intraperitoneally, i.p.), both common carotid arteries of the rat were dissected free and then the incision was closed. Immediately after this procedure, both vertebral arteries between the first and second cervical vertebra were exposed and electrocauterized completely using a bipolar cauterizer under an operating microscope (SZHILLB, Olympus, Japan). After 24 h, the common arteries were occluded with aneurysm clips for 10 mins. The clips were then removed, and the blood flow though the arteries was confirmed before the wound was sutured. This was referred to a single ischemia (10 mins). In the case of repeated ischemia, occlusion of the common carotid arteries was repeated once (2× 4VO) with 1 h interval between the two successive occlusions as described earlier (Pu et al, 2004). Rectal temperature was maintained at approximately 37°C, using a heating pad until the animal had fully recovered from anesthesia. Sham operation (control) groups had a permanent bilateral occlusion of vertebral arteries (2VO rat) in the same way as done in 4VO rats except that the arteries were not occluded. All rats were allowed to survive 8 days after the onset of cerebral ischemia. Any rats, which showed either a motor deficit or abnormal spontaneous movements because of ischemia, were discarded (5.72%).
Dehydroepiandrosterone was dissolved in dimethylsulfoxide (DMSO, Sigma, St Louis, MO, USA) and then in saline solution, final concentration being 20 mg/kg. DMSO was used at a final concentration of 2% as vehicle control. DHEA was administered i.p. with a single injection at 1 h before, or 1, 3, 6, 12, 24, 36, 48, 72, or 96 h after ischemia at a dose of 20 mg/kg. We used this concentration based on the report that DHEA at this dosage significantly reduced learning impairments in rats exposed to cocaine (Meunier and Maurice, 2004). In addition, exogenously administered DHEA (20 mg/kg) or DHEA sulfate (DHEAS; 20 mg/kg) reportedly binds to sigma-1 (σ1) receptor in eight brain regions of healthy awake male rats (Waterhouse et al, 2007). To test whether the DHEA effect is produced by its metabolite testosterone (TE), the same dose (20 mg/kg) of TE (i.p.) was used. The NMDAr-channel blocker MK801 was administrated at 2 mg/kg (i.p.), because the dose chosen appears to have therapeutic efficacy on brain injury induced by transient focal ischemia in cats (Miyabe et al, 1997; Moyanova et al, 2007). The administration of the σ1 receptor agonist PRE-084 at 2 mg/kg (i.p.) was selected on the basis of its anti-amnesic effect on Aβ25 to 35 models of amnesia (Meunier et al, 2006). The σ1 receptor antagonist NE100 was injected i.p. at a dose of 4 mg/kg, as our previous study revealed that this dose of NE100 (i.p.) completely prevents the protective effect of DHEAS on ischemia-induced brain damage (Li et al, 2006). Testosterone, DHEAS and PRE-084 were initially dissolved in DMSO and then in saline solution. MK801 and NE100 were dissolved in saline solution. The drugs were prepared freshly on the day of experiment. Control rats were given an equal volume of DMSO or saline.
Histologic Examination
Histologic examinations were made on eighth day after ischemia; the animals were received sodium pentobarbital (60 mg/kg i.p.) and transcardially perfused with 200mL of 0.1M phosphate buffer saline at 4°C (pH 7.4, 100cm H2O), followed by 200mL of 4% buffered paraformaldehyde phosphate. The brains were removed, postfixed in the same fixative for 24 h, and then processed for paraffin embedding. Coronal sections (4 µm in thickness) including the dorsal hippocampus were cut and stained with toluidine blue. The sections were examined under a light microscope (Olympus) at ×100 magnifications, and the numbers of surviving CA1 pyramidal cells per 1 mm length along the extent of CA1 pyramidal layer were counted as neuronal density (cells per mm) as described elsewhere (Guan et al, 2006). We also made supplemental examinations to observe dead cells in slices stained with trypan blue, and obtained essentially the same results as those with toluidine blue-stained slices. To avoid any bias, analyzers were masked to the examination of specimen. Each group data were obtained from five rats throughout experiments. Three slices were prepared from right and left dorsal hippocampus, respectively, and the number of surviving pyramidal neurons was counted (n = 30 slices per 5 rats).
Behavioral Analysis
For the Morris water maze task, a pool (180 cm in diameter) made of black-colored plastic was prepared, and the water temperature was maintained at 20°C. Swimming paths were analyzed by a computer system with a video camera (AXIS-90 Target/2; Neuroscience). In the hidden-platform test, the platform (7cm in diameter) was submerged 1cm below the water surface. Rats did not swim in the pool before training. Three starting positions were used pseudorandomly, and from third day after ischemia each rat was trained with three trials per day for 6 days. After reaching the platform, the rat was allowed to remain on it for 30 secs. If the rat did not find the platform within 90 secs, the trial was terminated and the animal was put on the platform for 30 secs.
Chemicals
Dehydroepiandrosterone, DHEAS, (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK801), and TE were purchased from Sigma Chemical Co. (St Louis, MO, USA). 2-(4-Morpholinethyl) 1-phenylcyclohexanecarboxylate hydrochloride (PRE-084) was from Tocris (Avonmouth, UK). N,N-dipropyl-2-(4-methoxy-3-(2-phenylethox-y)phenyl) ethylamine hydrochloride (NE100) was kindly supplied by Taisho Pharmaceutical Co. Ltd. (Tokyo, Japan).
Data Analysis/Statistics
Data were retrieved and processed with the software Microcal Origin 6.1. The group data were expressed as means±s.e. Each experimental group contained five rats. Number of surviving pyramidal neurons and performance of Morris water maze test in each group were compared by one-way analysis of variance with repeated measures, followed by the Bonferroni post hoc test. Statistical analysis was performed using the software Stata 7 (Stata Corporation, USA). A probability of 0.05 was considered significant.
Results
Effects of DHEA on Ischemia-Induced Neuronal Death
The rats subjected to 10 mins global cerebral ischemia exhibited an approximately 20% loss of pyramidal neurons in the hippocampal CA1 region at day eighth after 4VO (surviving neurons: 78.6%±9.78%; Figure 1A (ii) and 1B) compared with control 2VO rats (100%±8.69%, P < 0.01; Figure 1A (i) and 1B). Surprisingly, a single injection of DHEA at 1 h before or after 4VO significantly increased the neuron death (surviving neurons, 1 h before 4VO: 19.63%±12.64%, 1 h after 4VO: 21.59%±11.81% versus rats treated with DMSO at 1 h after 4VO 80.87%±9.24%, P < 0.01). By contrast, when DHEA was administered between 6 and 36 h after 4VO, the ischemia-induced neuronal loss was significantly reduced (6 h: 102.69%±8.51%, 12 h: 99.08%±4.98%, 24 h: 103.47%±10.47%, 36 h: 99.63%±10.75% versus 4VO rats, P < 0.01), whereas the administration of DHEA at 3 or 48 h after 4VO only partially attenuated the neuron death (3 h: 89.46%±6.48%, 48 h: 90.35%±9.59% versus 4VO rats, P < 0.05). The administration of DHEA after 72 h after 4VO had no effect (72 h: 76.38%±12.84%, 96 h: 75.59%±8.74% versus 4VO rats, P > 0.05). DHEA administration at 1 h after 2VO did not induce neuronal death (102.62%±10.33% versus 2VO rats, P > 0.05). In the dentate gyrus, however, neither 4VO nor the administration of DHEA at 1 or 6 h after 4VO induced granule cells loss (2VO: 100.43%±10.62%, 4VO: 95.94%±9.43%, 4VO/DHEA (1 h): 96.24%±9.67%, 4VO/DHEA (6 h): 101.55%±11.45%, P > 0.05; Figure 1A (ii—iv)). These findings indicate that in the hippocampal CA1 subjected to transient ischemia (10 mins), DHEA exerts not only neuroprotection with a wide efficacious time-window after ischemia (6 to 36 h) but also neurotoxicity when administered during ischemia and early reperfusion.
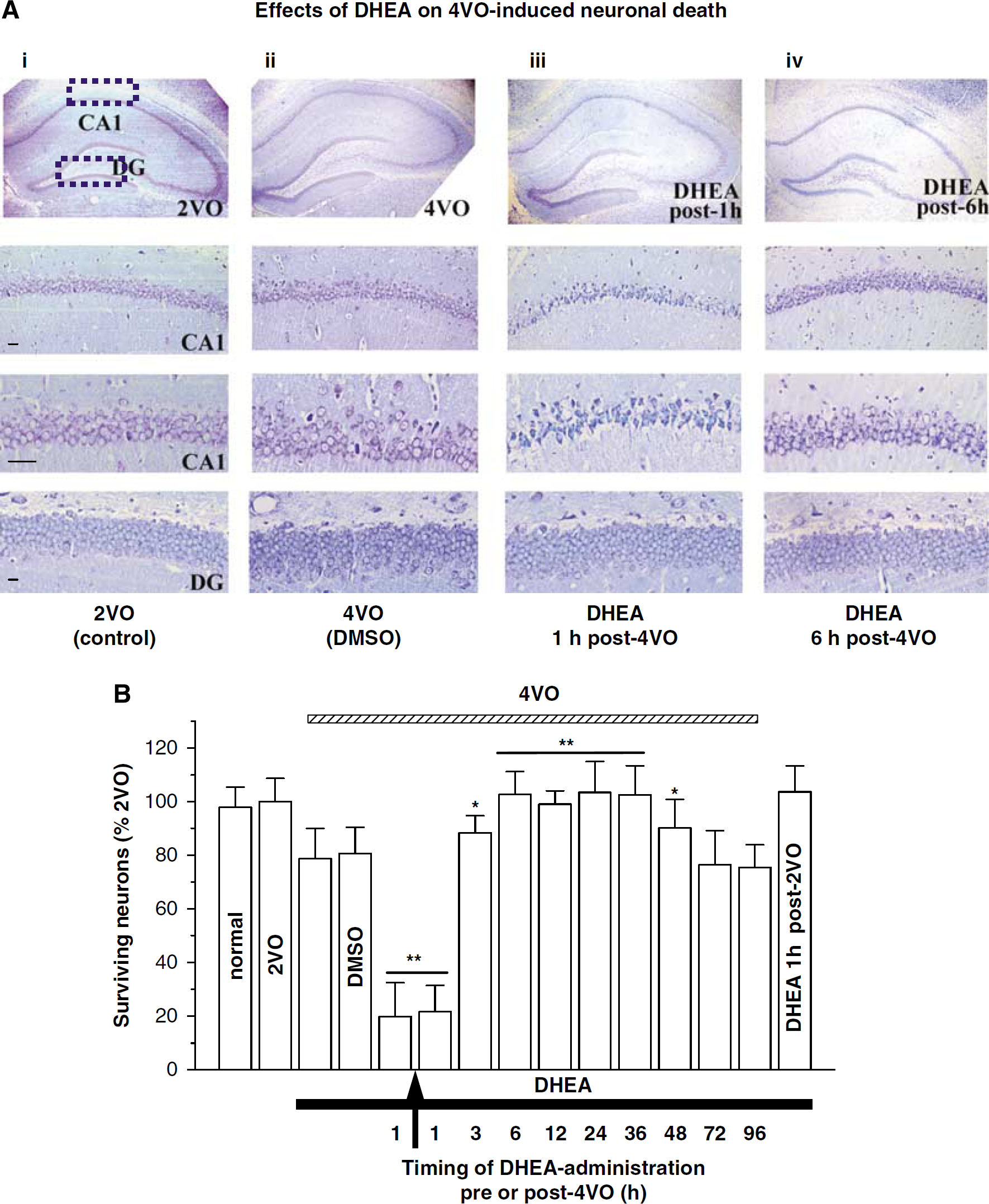
DHEA-neuroprotection and -neurotoxicity in ischemia-induced neuronal damage. (
Effects of DHEA on Ischemia-Induced Deficits in Spatial Memory
On the performance of Morris water maze test, the rats subjected to ischemic insult showed a prolongation of escape latency to the hidden platform (41.57 ± 9.91 secs on sixth day after training) as compared with 2VO rats (7.25 ± 11.35 secs, P < 0.01; Figure 2A), whereas there was no detectable difference in the swimming speed between the two groups (data not shown). The 4VO rats received a single administration of DHEA at 1 h after 4VO required longer time to reach the platform than those treated with vehicle (P < 0.01; Figure 2B). By contrast, when administered at 6 h after 4VO, DHEA could markedly attenuate the ischemia-induced prolongation of the escape latency to the platform (P < 0.01; Figure 2C). However, at 72 h after 4VO, DHEA had no effect on the prolongation of escape latency induced by ischemia (72 h: 39.83 ± 7.61 secs, 96 h: 42.39 ± 16.15 secs versus 4VO rats, P > 0.05; Figure 2D). Figure 2E summarizes the performance of Morris water maze test at sixth day after training. These results provide behavioral evidence that is in harmony with the administration timing-dependent DHEA neuroprotection with a wide efficacious time-window (3 h: 32.69 ± 10.76 secs versus 4VO rats, P < 0.05; 6 h: 15.23 ± 6.58 secs, 12 h: 11.28 ± 9.37 secs, 24 h: 8.44 ± 9.03 secs, 36 h: 12.38± 4.39 secs, 48 h: 10.49 ± 11.23 secs versus 4VO rats, P < 0.01) and DHEA neurotoxicity (1 h before 4VO: 78.21 ± 9.57 secs, 1 h after 4VO: 71.69 ± 12.07 secs versus 4VO rats, P < 0.01) observed in the ischemic brain.
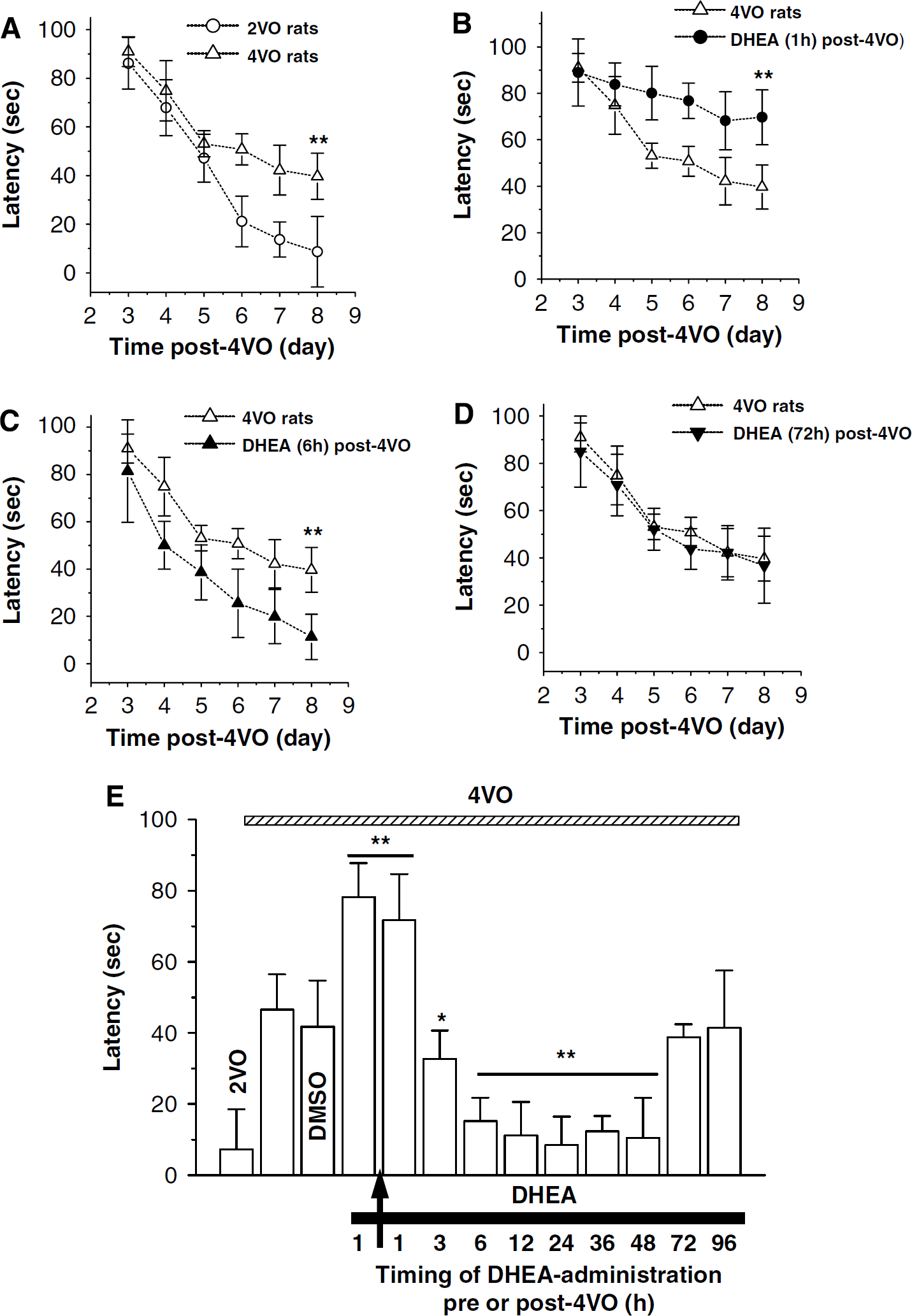
DHEA-neuroprotection and -neurotoxicity in ischemia-impaired spatial memory. (
Characteristics of DHEA Neurotoxicity
Dehydroepiandrosterone and its sulfated derivative, DHEAS, are the most abundant products in the human adrenal cortex and regarded as relatively inert precursors of the sex steroid TE (Labrie et al, 2003). Previous study reported that TE increases the lesion size of brain induced by middle cerebral artery occlusion (Hawk et al, 1998). To determine whether the DHEA neurotoxicity is caused specifically by DHEA or its metabolite TE, the 4VO rats were given a single injection of TE or DHEAS at 1 h after 4VO. As shown in Figure 3A, TE at 20 mg/kg did not affect the ischemia-induced neuronal death (4VO/TE, 80.32%±10.24% versus 4VO rats, P > 0.05), whereas DHEAS (20 mg/kg) appeared to have a DHEA-like neurotoxic effect (39.84%±8.25% versus 4VO rats, P < 0.01).
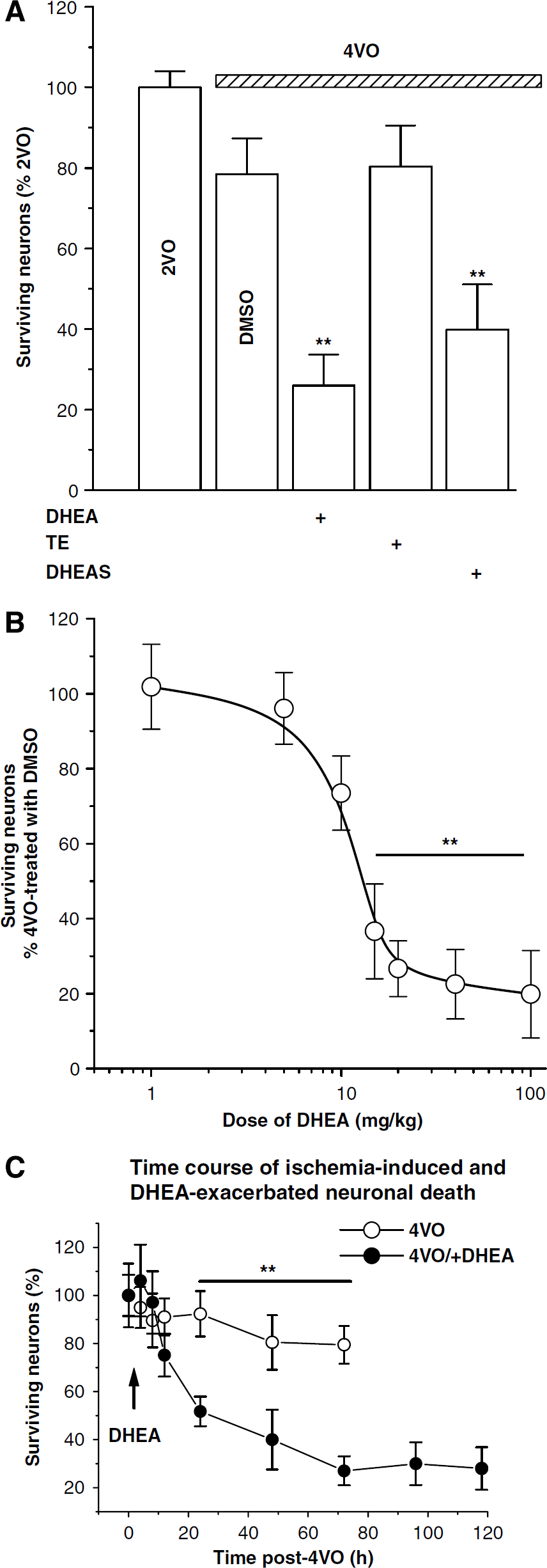
Dose dependency of DHEA-neurotoxicity. (
To further confirm the DHEA neurotoxicity on the ischemic brain damages, the effects of DHEA at various concentrations (1, 5, 10, 20, 40, or 100 mg/kg) when administrated at 1 h after 4VO were examined. The results in Figure 3B show that DHEA exacerbated the ischemia-induced neuronal death in a dose-dependent manner with an ED50 value of 10.58 mg/kg (at each dose, n = 5 rats). Next, we examined the time course of ischemia-induced and DHEA-aggravated neuronal death in the hippocampal CA1 region subjected to ischemia. The 4VO ischemia (10 mins) caused 20% CA1 neuronal death within 48 h after 4VO, whereas the administration of DHEA at 1 h after 4VO (Figure 3C) remarkably exacerbated neuronal death during 12 to 48 h after 4VO, reaching to maximal (80%) neuronal death at 72 h after 4VO.
Involvement of σ1 Receptors and NMDAr in DHEA Neurotoxicity
Dehydroepiandrosterone has been identified as one of the most potent activators of σ1 receptor (Phan et al, 2003). We have recently reported that the σ1 receptor antagonist NE100 prevents neuronal cell death induced by middle cerebral artery occlusion (Cai et al, 2008). It is likely that the DHEA effects are mediated by an activation of σ1 receptor. Thus, we conducted an experiment to examine the effects of blocking σ1 receptor on the ischemia-induced and DHEA-aggravated neuronal death. As shown in Figure 4A, pretreatment with NE100 (4 mg/kg) at 30 mins before the administration of DHEA not only prevented the DHEA-aggravated neuronal death but also the 4VO-induced cell death (lane 5: 96.70%±10.74% versus lane 4: 4VO/DHEA rats, P < 0.01). Consistent with this observation, NE100 alone could attenuate the 4VO-induced cell death (lane 3: 86.60%±8.23% versus lane 2: 4VO rats, P < 0.05; Figure 4A), suggesting that σ1 receptor might be involved in the 4VO-induced cell death. Finally, a single administration of the σ1 receptor agonist PRE-084 (2 mg/kg) at 1 h after 4VO appeared to have a DHEA-like neurotoxic effect (lane 6: 43.61%±8.37% versus lane 2: 4VO rats, P < 0.01).
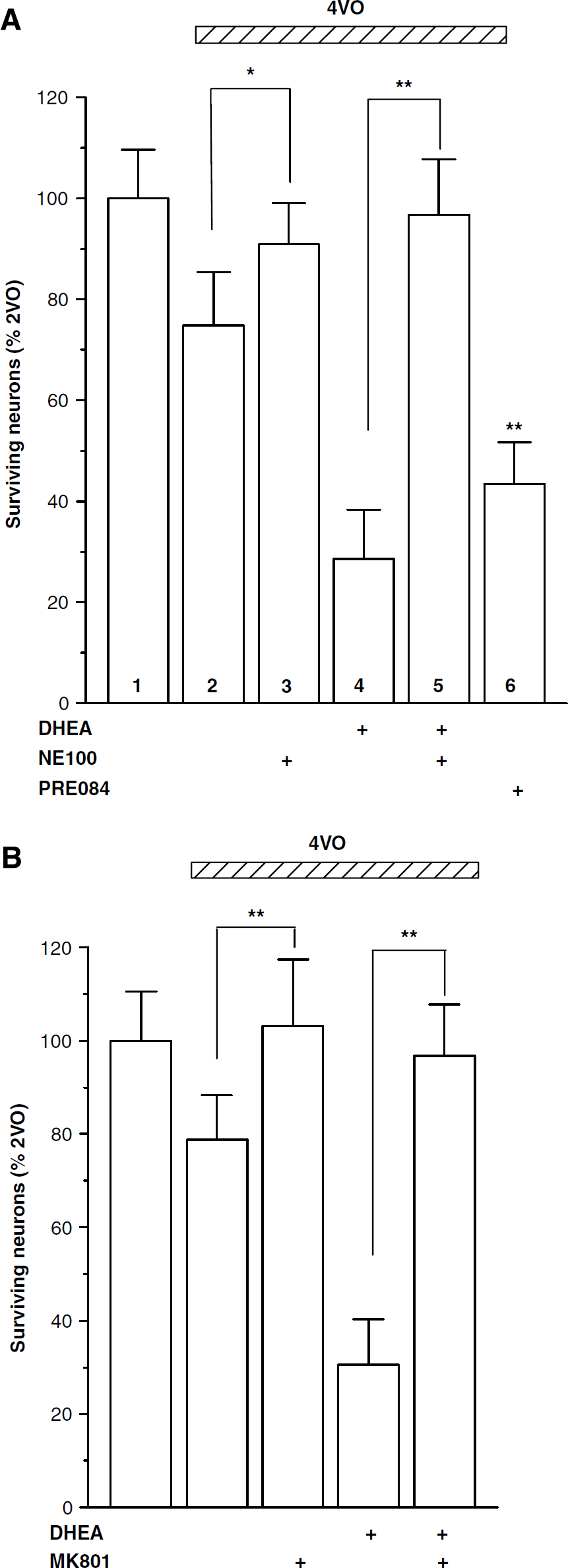
DHEA-neurotoxicity is σ1 receptor- and NMDAr-dependent. (
We observed that the administration of NMDAr-channel blocker MK801 (2 mg/kg) alone at 1 h after 4VO significantly reduced the ischemia-induced neuronal death (103.17%±11.14% versus 4VO rats, P < 0.01, lane 3 in Figure 4B). Similarly, treatment with MK801 (2 mg/kg) at 30 mins before the DHEA administration could prevent the DHEA-aggravated neuronal death in the ischemic brain (97.26%±10.48% versus 4VO/DHEA rats, P < 0.01, lane 4 in Figure 4B). These results suggest that the DHEA neurotoxicity is caused by the activation of NMDAr and σ1 receptor.
Effects of DHEA on Severe Cerebral Damages by Repeated Ischemia
The neuroprotection of DHEA was further evaluated in a rat model with repeated cerebral ischemia, which shows severe impairment of spatial memory and hippocampal CA1 cell death (Pu et al, 2004). The twice cerebral ischemia (2×10 mins 4VO) with 60 mins interval resulted in approximately 50% loss of CA1 pyramidal neurons (surviving neurons: 46.11%±8.85% versus 2VO rats, P < 0.01; Figure 5A) on eighth day after 2× 4VO. Similar to the case of one-time ischemia, a single administration of DHEA (20 mg/kg) at 6 h after 2× 4VO significantly reduced the pyramidal neurons death (89.73%±8.64% versus 2×4VO rats, P < 0.01), whereas DHEA treatment after 72 h after 2× 4VO had no protective effect on ischemia-induced neuronal death (50.69%±9.56% versus 2×4VO rats, P > 0.05). Furthermore, the rats subjected to the repeated cerebral ischemia showed a significant disruption of spatial cognition (51.77 ± 9.78 secs on eighth day after 2× 4VO) compared with 2VO rats (8.17 ± 10.28 secs, P < 0.01; Figure 5B). Importantly, the administration of DHEA at 6 h after 2× 4VO significantly improved the ischemia-induced prolongation of escape latency to the hidden platform (17.74 ± 12.24 secs, P < 0.01), but showed no effect at 72 h after 2× 4VO (45.48 ± 7.48 secs, P > 0.05), further supporting the neuroprotective effect of DHEA against ischemia-induced cerebral damages.
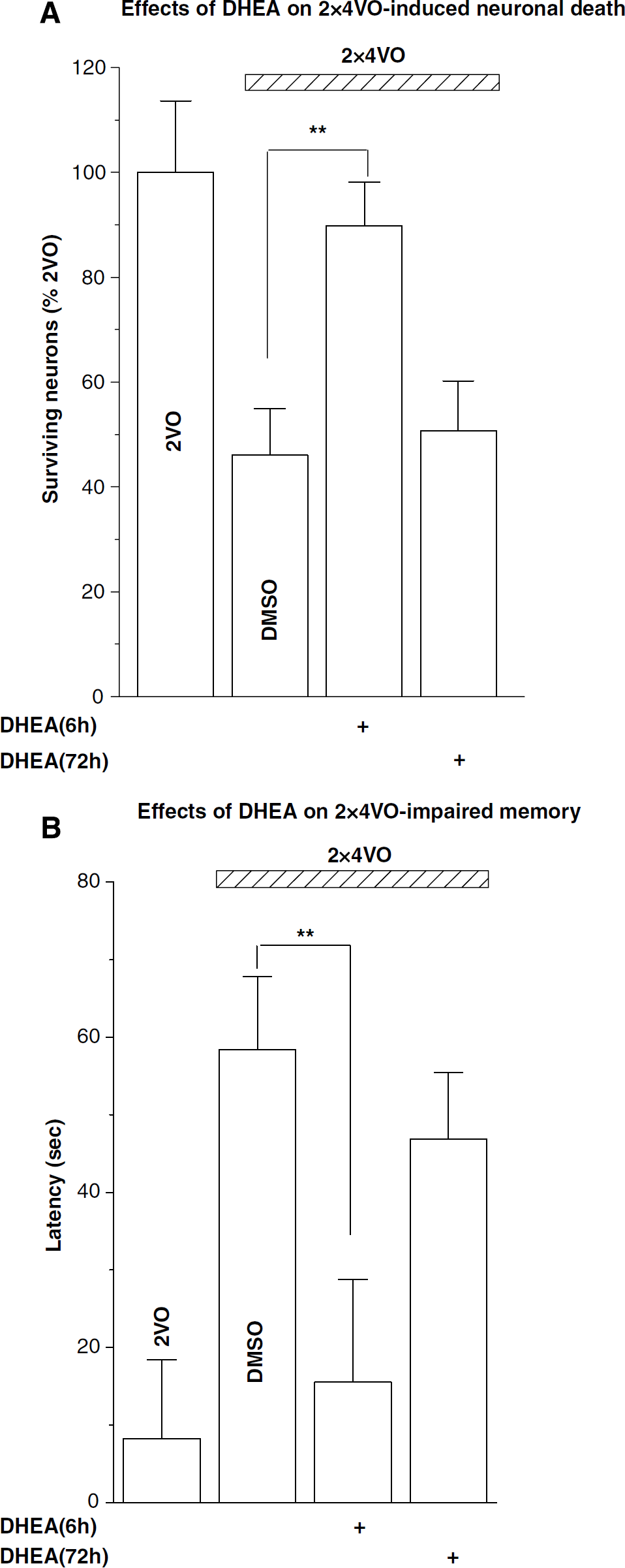
DHEA protects neuronal death induced by repeated ischemia. Repeated ischemia (2×4VO) induced severe CA1 neuronal death (
Involvement of σ1 Receptor in DHEA Neuroprotection
Given the result that σ1 receptor was involved in the DHEA neurotoxicity, an experiment was designed to determine whether the DHEA neuroprotection is also related to σ1 receptor. The pretreatment with NE100 (4 mg/kg) suppressed the neuroprotective effect of DHEA administered at 6 h after 4VO on the ischemia-induced cell death (lane 4: 85.68%±9.17% versus lane 3: DHEA/4VO rats, P < 0.05; Figure 6). By contrast, the pretreatment with NMDAr-channel blocker MK801 (2 mg/kg) did not affect the DHEA neuroprotection (lane 5: 103.05%±9.45% versus lane 3: 4VO/DHEA rats, P > 0.05), and the administration of MK801 (2 mg/kg) alone at 6 h after 4VO failed to reduce the ischemia-induced neuronal death (lane 6: 82.44%±8.15% versus lane 2: 4VO rats, P > 0.05), suggesting that the DHEA-exerting neuroprotection involves σ1 receptor but not NMDAr.
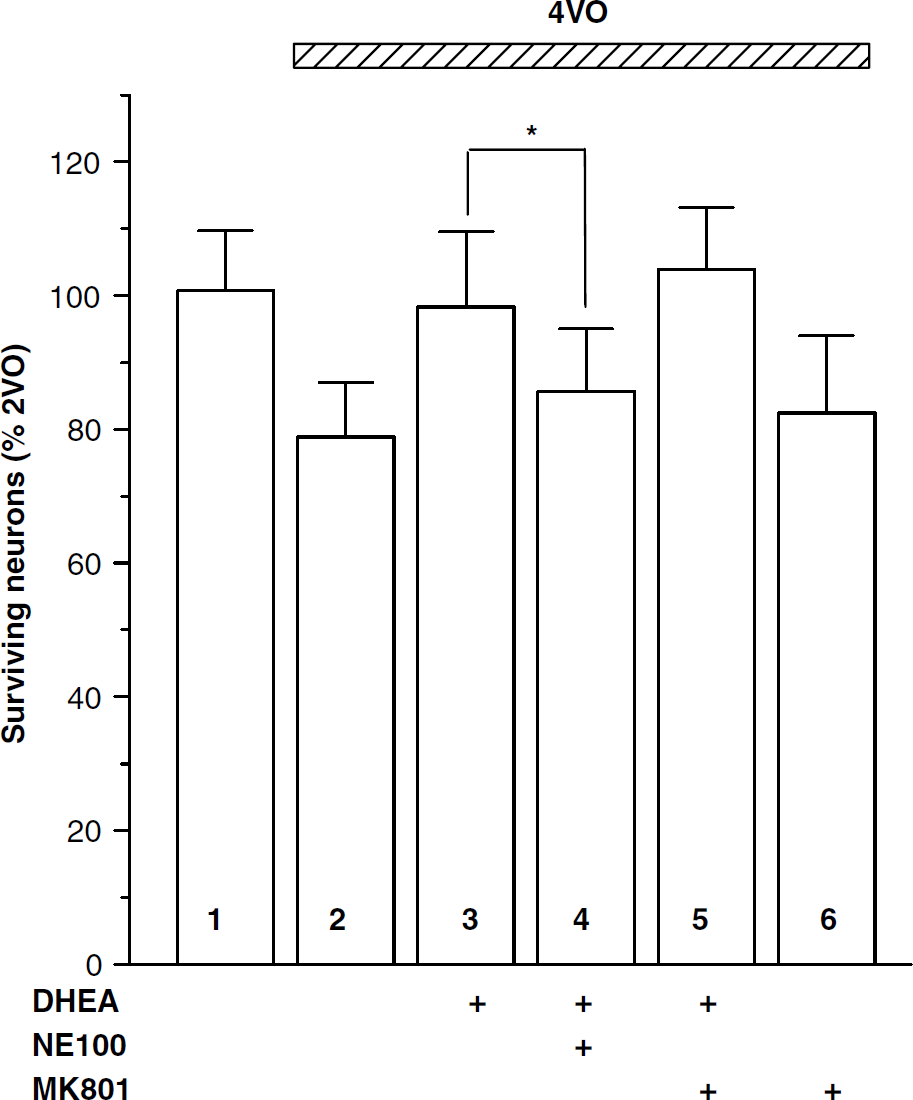
DHEA neuroprotection is σ1 receptor-dependent. Effects of NE100 and MK801 on the ischemia-induced neuronal death and DHEA neuroprotection. NE100 or MK801 was administrated before 30 mins of DHEA or at 6 h after 4VO. Note that NE100 blocks the DHEA neuroprotection, although MK801 does not affect the ischemia-induced neuronal death. *P<0.05.
Discussion
Using a rat model of transient global cerebral ischemia, the present study shows for the first time that the contradictory effects of DHEA on ischemia-induced cerebral damages arise from the difference in the administration timing of DHEA: when administered during 3 to 48h, time-window after ischemia DHEA exerts neuroprotection, although it shows neurotoxicity when administered during ischemia or early reperfusion, which implicates the importance of the administration timing of DHEA in treating ischemic brain damages including those by stroke.
Hippocampal CA1 pyramidal neurons are particularly vulnerable to transient ischemia, being used as a model for ischemic neuronal damages. Although the underlying precise mechanisms remain to be clarified, an excessive increase in the intracellular Ca2+ concentration ([Ca2+]i) has been implicated as one of the pivotal events to induce ischemic neuronal death (Choi, 1995; Mitani et al, 1993). The Ca2+ overload may cascade excitotoxic and proapoptotic signaling to produce cysteine proteases including caspase-12 (Berliocchi et al, 2005; Bano and Nicotera, 2007). Increases in the [Ca2+]i during ischemia and early reperfusion are mainly brought about by massive Ca2+ influx through NMDAr (Bano and Nicotera, 2007) or voltage-gated calcium channels (Choi, 1995). We show here that the NMDAr-channel blocker MK801 prevents nearly perfectly the DHEA neurotoxicity, strongly suggesting that the DHEA neurotoxicity here is attributable mainly to an increase in the Ca2+ influx through NMDAr. Then a question arises how DHEA increases the Ca2+ influx through NMDAr. DHEA and DHEAS have been identified to be one the most potent activators of σ1 receptor (Phan et al, 2003). Actually our study here showed that the σ1 receptor agonist PRE-084 could mimic the DHEA neurotoxicity, whereas the σ1 receptor antagonist NE100 prevented the DHEA neurotoxicity, suggesting that σ1 receptor is the major target of DHEA. DHEA interacts with σ1 receptor, leading to enhanced NMDA-evoked responses (Bergeron et al, 1996). We have reported that DHEAS enhances NMDAr-mediated Ca2+ influx via upregulating the tyrosine phosphorylation of NR2B (Chen et al, 2006), which is σ1 receptor-dependent (Li et al, 2006). Furthermore, a large body of evidence has been accumulated that the activation of σ1 receptor facilitates the Ca2+ influx across NMDAr (Monnet et al, 1995, 2003; Compagnone and Mellon, 1998; Cai et al, 2008). Collectively, we conclude that the cascade, DHEA-σ1 receptor-NMDAr-enhanced [Ca2+]i increase, would mainly contribute to the DHEA neurotoxicity. There is, however, an apparently conflicting report describing that the σ1 receptor agonist (+) SKF10047 inhibits the rise in [Ca2+]i evoked by glutamate (Kume et al, 2002). This discrepancy regarding the role of σ1 receptor in the regulation of [Ca2+]i levels may be because of differences in the experimental systems. Further studies are needed to evaluate the effects of DHEA/σ1 receptor on the regulation of [Ca2+]i during ischemia and early reperfusion.
Another principal observation in this study is that the DHEA treatment after 3 h reperfusion produced a potent neuroprotection with a wide efficacious time-window. The DHEA neuroprotection observed here is unlikely to be produced through a blockade of NMDA neurotoxicity as reported previously (Kimonides et al, 1998), because the administration of MK801 alone at 6 h after 4VO failed to reduce the ischemia-induced neuronal cell death as shown in Figure 6 (lane 6). Our results showed that the neuroprotective effect of DHEA was sensitive to NE100, a selective σ1 receptor antagonist. Bucolo and Drago (2004) reported that the σ1 receptor antagonist BD1047 blocked similarly the neuroprotective effect of PRE-084 and DHEAS, suggesting that most of the neuroprotective action of the steroid involved in its interaction with the σ1 receptor. Related to this, neuroprotective effects of the σ1 receptor agonist 4-phenyl-1-(4-phenylbutyl)-piperidine have been reported in a model of focal cerebral ischemia (Takahashi et al, 1995, 1996; Harukuni et al, 2000). DHEA at micromolar concentrations has an ability to scavenge free radicals via the activation of σ1 receptor (DeCoster et al, 1995). DHEA inhibits NMDA-induced nitric oxide synthase activity and reduces nitric oxide production (Kurata et al, 2004). Furthermore, DHEA protects sympathoadrenal medulla cells from apoptosis through a phosphatidylinositol 3-kinase/Akt-cascaded anti-apoptotic Bcl-2 (Charalampopoulos et al, 2004), a potential pathway of neuroprotection against brain ischemic insult (Kawano et al, 2006). DHEA stimulates PI3-kinase activity to protect ischemia-induced neuronal apoptosis (Raval et al, 2003). Another candidate molecule involved in the DHEA neuroprotection may be the glial glutamate transporter, GLT-1. A recent study has provided evidence that the cerebral ischemia/reperfusion results in a delayed downregulation of GLT-1 in the hippocampus even after a recovery from depletion of oxygen glucose (Li et al, 2008). Interestingly, the time-profile of the GLT-1 downregulation by ischemia/reperfusion in vivo (Yeh et al, 2005; Chen et al, 2005) and in vitro (Brongholi et al, 2006) studies looks very similar to the time window (3 to 48 h post-ischemia) of the DHEA neuroprotection observed here. Furthermore, DHEA increases glucose transport rate to enhance 2-[3H]deoxy glucose uptake (Kajita et al, 2000; Perrini et al, 2004) and upregulates the function of GLT-1 via the activation of σ1 receptor (Sokabe et al, 2007). More recently, Shen et al (2008) have provided supporting evidence that the σ1 receptor agonist dimemorfan protects brain damage against ischemic stroke through decreasing glutamate accumulation. Thus, it would be interesting to examine whether DHEA rescues the deficit of GLT-1 functioning observed several hours after reperfusion.
Despite the fact that the underlying mechanisms have not yet been fully elucidated, the present study clearly shows a DHEA-exerting neuroprotection with a long therapeutic opportunity (at least 48 h post-ischemia) against ischemic brain damages. At the same time, our results provide evidence that DHEA negatively impacts the hippocampal neurons during or shortly after ischemic attack, which implicates the importance of administration timing of DHEA in treating brain ischemia including stroke.
Disclosure
We declare that there is no competing financial that could be construed as influencing the results or interpretation of the reported study.