
Abstract
To better understand the role of downstream Toll-like receptor (TLR) signaling during acute cerebral ischemia, we performed cDNA microarrays, on brain RNA, and cytokine arrays, on serum, from wild type (WT), MyD88−/− and TRIF-mutant mice, at baseline and following permanent middle cerebral artery occlusion (pMCAO). The acute stress response pathway was among the top pathways identified by Ingenuity Pathway Analysis of microarray data. We used real-time polymerase chain reaction to confirm the expression of four immediate early genes; EGR1, EGR2, ARC, Nurr77, in this pathway, and insulin degrading enzyme (IDE). Compared to WT, baseline immediate early gene expression was increased up to10-fold in MyD88−/− and TRIF-mutant mice. However, following pMCAO, immediate early gene expression remained unchanged, from this elevated baseline in these mice, but increased up to 12-fold in WT. Furthermore, expression of IDE, which also degrades β-amyloid, decreased significantly only in TRIF-mutant mice. Finally, sE-Selectin, sICAM, sVCAM-1, and MMP-9 levels were significantly decreased only in MyD88−/− compared with WT mice. We thus report a new role for downstream TLR signaling in immediate early gene expression during acute cerebral ischemia. We also show that the TRIF pathway regulates IDE expression; a major enzyme that clears β-amyloid from the brain.
INTRODUCTION
Pathogenesis of acute stroke involves molecular and cellular events common to diseases in which ischemia–reperfusion and inflammation play a central role. Understanding the complex events that underlie the inflammatory damage, resulting from ischemia and reperfusion, should lead to effective development of molecularly targeted drugs. Such therapies are needed to limit the damage from the exaggerated inflammatory reactions that typify acute cerebral ischemia. Furthermore, better insight into the timing of such therapies should help preserve the beneficial effects of inflammatory cues that stimulate cellular and tissue repair.
The Toll-like Receptor (TLR) pathway is one of the inflammatory pathways activated following focal cerebral ischemia, but the downstream signaling mechanisms underlying this innate immune response are not completely understood. This pathway, evolutionarily conserved across diverse species, from lower vertebrates to mammals, plays a key role in the innate immune response to infectious stimuli such as exogenous pathogens. 1 In addition, the TLR pathway plays a pivotal role in the innate immune response to endogenous tissue damage resulting from focal cerebral ischemia,2, 3, 4, 5 other types of ischemia,6, 7 and neurological diseases in which inflammation plays a central role. The role of TLRs in the proximal part of the signaling cascade has been well studied and specific TLRs are known to mediate the pathogenesis of cerebral ischemia.8, 9, 10 In contrast, the role of the distal downstream TLR signaling pathway, in focal cerebral ischemia, remains incompletely defined.
Due to the dual role of the TLR receptors in infection and tissue stress, it is imperative to understand the function of the major distal molecular targets that propagate the signaling cascade downstream from these TLR receptors. Such understanding should lead to the identification of targets that selectively affect the innate immune response to cerebral ischemia, while preserving the necessary response to infection.
Our recent studies in a mouse model of focal cerebral ischemia have shed light on the role of these distal signaling events. We demonstrated that adaptor-specific cytokine and chemokine expression is mediated by the downstream TLR signaling adaptors MyD88 and TRIF. We also showed an unexpected abnormal baseline elevation of inflammatory cytokines with disruption of MyD88. 11 These latter findings were not congruent with the known involvement of the MyD88 pathway in the expression of proinflammatory cytokines, and therefore warrant more in-depth investigation to identify additional roles of these downstream adaptors.
The unexpected baseline cytokine kinetics, and the lack of protection against cerebral ischemia in mice with disruption of MyD88 or TRIF,12, 13, 14, 15 led us to hypothesize that these downstream adaptors play additional roles in the optimal response to focal cerebral ischemia. To identify such additional roles and their molecular basis, we sought to determine the temporal changes in differential gene expression that accompanies distal TLR signaling following focal cerebral ischemia in the brains of mice with disruptions of MyD88 and TRIF, and in wild-type (WT) mice. To accomplish this, we used cDNA microarrays, a sensitive and comprehensive method of profiling differences in gene expression. By examining gene expression in the brains of these mice, we are able to investigate key molecular events in the brain—the tissue involved in stroke pathogenesis, an approach; which is limited in humans.
We now show an unexpected role for MyD88 and TRIF pathways in the optimal expression of immediate early genes following focal cerebral ischemia, and explain the abnormal elevation of baseline cytokines in MyD88−/− mice noted in our previous studies. 11 We also showed that the TRIF pathway is necessary for optimal expression of genes important in processing β-amyloid following focal cerebral ischemia. This finding has important implications for linking activation of the innate immune response in neurological diseases, such as Alzheimer's, in which inflammation plays an important role, to specific downstream TLR adaptors. Our present study is the first to show that downstream TLR signaling pathways play a key role in the optimal acute stress response to cerebral ischemia.
MATERIALS AND METHODS
Study Design
Permanent middle cerebral artery occlusion surgery
Permanent middle cerebral artery occlusion (pMCAO) was performed in three groups of mice: MyD88−/−, TRIF mutant, and WT controls, at three time points: baseline (no surgery), and at 3 hours and 24 hours following induction of focal cerebral ischemia.
There were nine mice in each group; three each for each of the three time points and brain homogenates were prepared from the brains of these 27 mice for analysis of gene expression, using cDNA microarrays.
Real-time polymerase chain reaction
Real-time polymerase chain reaction (PCR) experiments were divided into two groups, since the 13 triplicate reactions (test gene primer, loading control primer and three time points per WT, MyD88, and TRIF) required 107 wells and our machine used a 96-well plate. The two real time-PCR groups were as follows:
(1) WT and MyD88 in the same experiment and (2) WT and TRIF in the same experiment. Wild-type samples were from the same animals for both groups.
Detection of serum protein expression
For analysis of serum protein expression, we compared levels of the five different proteins, related to immune activation and inflammation, represented in the Milliplex MAP Cardiovascular Disease (CVD)-Panel-1 kit (Millipore, Billerica, MA, USA), in serum samples from WT, MyD88−/−, and TRIF-mutant mice at baseline, three, and 24 hours following pMCAO.
Animals
Male mice, 12 to 16 weeks old, were used. We excluded female mice, mice with signs of infection or bite wounds, and mice older than 16 weeks of age. MyD88−/− mice, with disruption of MyD88 signaling, due to deletion of exons IV and V of the Toll/Interleukin-1 receptor (TIR) signaling domain, were obtained from the Institute of Systems Biology, Seattle, WA, USA. TRIF-mutant mice, with disruption of TRIF signaling, were obtained from Jackson Laboratories, Bar Harbor, ME, USA. Since both genetically modified mice were on a C56BL/6 background, we used WT mice, also from Jackson Laboratories, as controls for normal comparison. The average weights for mice were as follows: WT, 28.7±2.5 g; MyD, 29.5±2.4 g; and TRIF, 27.1±1.6 g. After obtaining breeder pairs for all three types of mice, progeny were bred from these breeder pairs in the same National Institutes of Neurological Disorders and Stroke (NINDS) animal holding facility with 12 hours of light and dark cycles. Mice were randomly selected from breeder cages, which contained no more than four other littermates, for inclusion in the study. All mice had equal access to food and water before surgery and all protocols were approved by and carried out according to guidelines of the National Institutes of Health, NINDS, Animal Care and Use Committee and comply with the ARRIVE guidelines.
Permanent middle cerebral artery occlusion
Induction of focal cerebral ischemia by pMCAO, blinding (tail color-coding), and pMCAO was carried out as previously described. 11 Briefly, anesthesia was induced with 5% and maintained with 1% to 1.5% isoflurane via face mask, and the distal middle cerebral artery cauterized with an electrocoagulator (ICC 200; ERBE Elektromedizin, Tübingen, Germany). One MyD88−/− mouse was excluded from cytokine and cDNA microarray analysis due to hemorrhage during MCAO surgery. The surgeon excluded this animal without being aware of the group to which the animal belonged. No mice died during or after pMCAO surgery.
Sample Preparation
Brain homogenates
Mice were killed at three time points: baseline (no surgery), 3 hours, and 24 hours following pMCAO. Brains were removed, ipsilateral ischemic cortices separated, and harvested from non-ischemic and from underlying subcortical structures, which were discarded. Separated ipsilateral ischemic cortices were stored at −80°C until further use. Frozen brain tissue was homogenized in buffer (0.2% Triton X, 1X protease inhibitor in 4 to 5 volumes of 1X PBS) and centrifuged at 15,000 g for 30 minutes at 4°C. Some of this supernatant was stored in Trizol, (Qiagen, Valencia, CA, USA) for subsequent extraction of RNA.
RNA isolation
Total RNA was isolated from homogenized ipsilateral cortices using MiRNeasy RNA extraction kits from Qiagen according to the manufacturer's instructions.
Gene Expression Studies
cDNA arrays
The quality and quantity of isolated total RNA were determined using a microfluidics-based Agilent 2,100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and a Nanodrop (Thermo Scientific, Barrington, IL, USA), respectively. Subsequent cRNA synthesis, cDNA amplification, labeling, and hybridization were performed on individual samples.
Briefly, a total of 300 ng of total RNA was reverse transcribed into cDNA and amplified. A total of 10 μg of amplified cDNA was in vitro transcribed into cRNA, and sense strand DNA cleaned up and cRNA hydrolyzed. The resulting hybridization cocktail, containing fragmented and labeled cDNAs, was hybridized to Mouse Gene 1.0 ST array chips (Affymetrix, Santa Clara, CA, USA), according to the manufacturer's instructions. The chips were washed and stained, using the Affymetrix, fluidics station, also according to the manufacturer's instruction. Probe arrays were stained with streptavidin–phycoerythrin solution (Molecular Probes, Carlsbad, CA, USA), and enhanced using an antibody solution containing 0.5 mg/mol of biotinylated anti-streptavidin (Vector Laboratories, Burlingame, CA, USA). An Affymetrix Gene Chip Scanner 3000 was used to scan the probe arrays. Gene expression intensities were calculated using AGCC software (Affymetrix).
Microarray data analysis
A flow diagram of the microarray data analysis is shown in Figure 1. Raw microarray data were analyzed by probe-level summarization and normalization using the Robust Multi-array Analysis Sketch option of the Affymetrix Expression Console (Affymetrix).
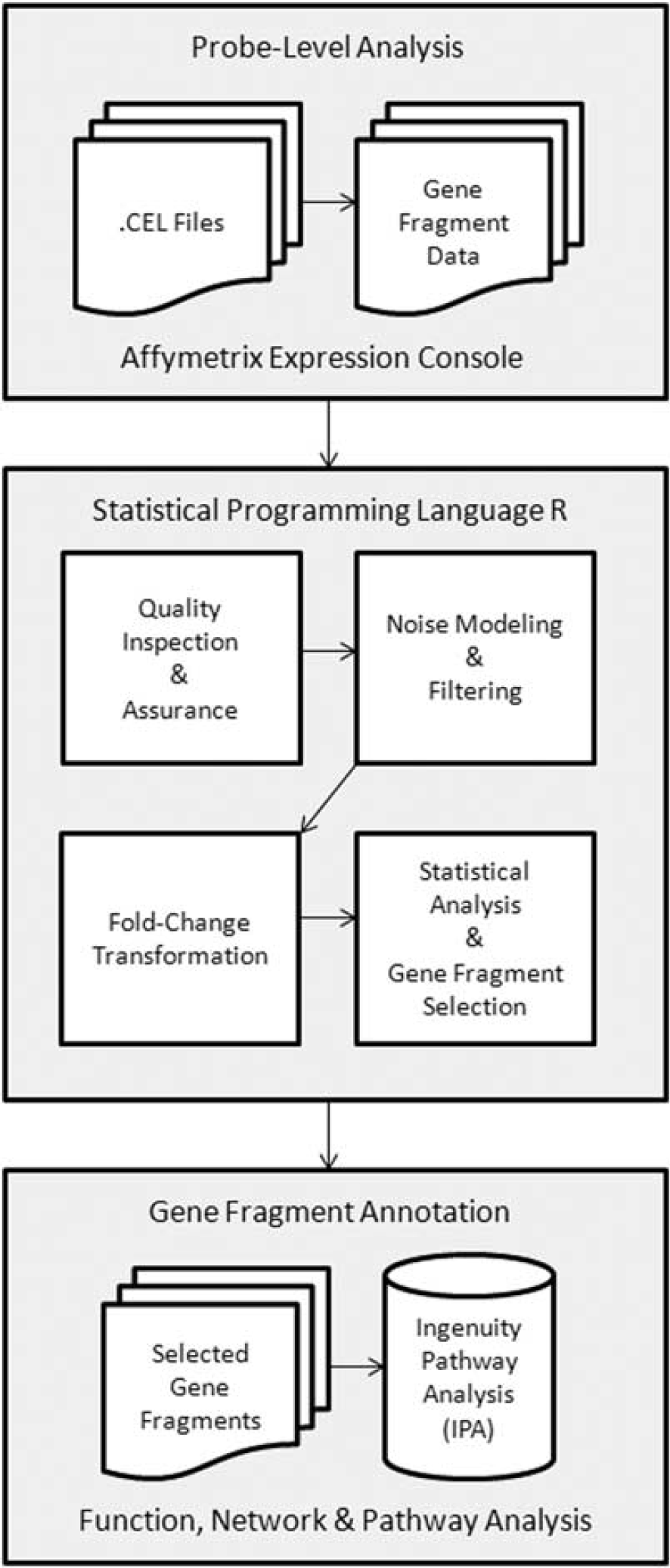
Flow diagram for microarray data analysis.
Subsequent data analysis was performed using the statistical programming language ‘R’ (http://www.cran.r-project.org/) in the following major 7 steps. (1) Data quality was confirmed by three different approaches: (i) Tukey box plot, (ii) covariance-based principal component analysis scatter plot, and (iii) correlation-based heat map. (2) Gene fragments without one or more expression values greater than system noise were discarded. System noise was defined as the expression value, across all samples, at which the locally weighted scatter plot smoothing (LOWESS) fit of CV (coefficient of variation), by mean expression, grossly deviates from linearity. (3) Expression values for the retained gene fragments were floored to system noise if less than system noise. (4) Fold-transformation was applied to each expression value, for each gene fragment, by subtracting, from this value, the corresponding gene fragment mean for the baseline group, on a group by group basis, for each of the three groups of mice. (5) Statistical analysis was performed on the fold-transformed expression values, to identify those gene fragments having differential expression between different time points within any one group or between each of the three groups of mice. (6) Gene fragments identified as having differential expression by statistical analysis were annotated by Ingenuity pathway analysis (IPA) (http://www.ingenuity.com/). (7) Ingenuity pathway analysis was also used to identify enriched biologic functions and pathways for the corresponding genes.
Validation of microarray data with real-time polymerase chain reaction
We confirmed the expression of five genes that were differentially regulated following pMCAO in MyD88−/−, TRIF mutant, and WT mice at the 3-hour time point, compared to the baseline time point (no surgery), for each group, using real-time PCR.
We used four Quantitect primer assays (Qiagen) in real-time PCR analysis: Mm_Egr2_1_SG; Mm_Ide_1_SG; Mm_Arc_1_SG; and Mm_Nr4a1_1_SG. EGR1 primers were synthesized by Sigma-Genosys (Woodlands, TX, USA) and the sequence was as follows: forward, 5′-CTGACCACAGAGTCCTTTTC-3′; and reverse, 5′-CAGGGAGAAGAGGCCAGTAT-3′. Finally, Tubb4_1_SG was used for loading the control.
Real-time PCR reactions were carried out on baseline and 3-hour post pMCAO RNA samples from WT and MyD88 in one experiment using one 96-well plate and samples from WT and TRIF mutant in a second experiment. All real-time PCR reactions were carried out using a total of 50 ng of RNA per triplicate reactions. One-step real-time PCR protocol was carried out using the Quantitect SYBR green RT-PCR kit (Qiagen) on a 7900HT fast real-time PCR machine (Applied Biosystems, Bedford, MA, USA). With the one-step real-time protocol, the PCR reaction was carried out directly on the RNA sample, without the need first to perform a separate cDNA synthesis step.
The PCR conditions were as follows: Stage 1: one step at 50°C, 30 minutes (1 cycle) Stage 2: one step at 95°C, 15 minutes (1 cycle), and Stage 3: three steps at (i) 94°C, 15 seconds, (ii) 55°C, 30 seconds, and (iii) 72°C, 30 seconds (40 cycles).
Results from real-time PCR were analyzed using SDS 2.4 and RQ manager 1.2.1. Data Assist V3.0 (Applied Biosystems) was used to compare WT to MyD88−/− or TRIF-mutant mice within the same experiment using the multigroup comparison feature of this software.
Serum protein analysis
Serum obtained at baseline, 3 hours and 24 hours, following pMCAO, from MyD88−/−, TRIF mutant, and WT mice was used in protein expression studies. The Milliplex Mouse Cardiovascular Disease (CVD) Panel-1 (Millipore) was used to simultaneously determine the levels of five immune activation proteins: (1) soluble E-Selectin (sE-Selectin), (2) matrix metalloproteinase-9 (MMP-9), (3) soluble intercellular adhesion molecule-1 (sICAM-1), (4) soluble vascular cell adhesion molecule-1 (sVCAM-1), and (5) total plasminogen activator inhibitor.
Briefly, the filter plate was prewetted with wash buffer, and 35 μL serum from each of the three groups of mice, at the indicated time points, were diluted 1:50 and 1:100 and added to the wells. Then 25 μL of polystyrene beads conjugated with antibodies to the five proteins listed above, were added to the wells containing the standards and to wells containing serum samples. The mixture was then incubated on a plate shaker for ∼2 hours at room temperature, followed by a wash step (2 × with 200 μL wash buffer). After washing, 25 μL of detection antibodies were added per well, and the plate incubated for an additional 1 hour at room temperature. Incubation with detection antibodies was followed by an addition of 25 μL of streptavidin–phycoerythrin for 30 minutes at room temperature. Finally, the plate was washed with wash buffer twice and 100 μL of sheath fluid added to each well for subsequent reading of the plate. The plate was then stored at 4°C overnight. The next day, the plate was placed on a plate shaker and beads re-suspended for ∼5 minutes prior to reading on a Luminex 100 × MAP technology machine (Luminex Corp., Austin, TX, USA) using Bio-Plex Manager 6.0 software (Bio-RAD, Hercules, CA, USA). Standard curves were generated for each of the five analytes, and median intensity fluorescence values calculated for each analyte using a 4- or 5-point logistic parameter curve.
Statistical analysis
Since we had three different groups of mice and three different time points to analyze, in these studies, we used the nine mice per group; three per time point to obtain statistically analyzable data.
For microarray gene fragment expression analysis, we used the one-factor analysis of variance under Benjamin and Hochberg false discovery rate multiple comparison correction condition, with animal group used as the factor. Gene fragments found to have a corrected P value <0.05 were subjected to the Tukey honestly significant difference (HSD) post hoc test. In addition, the difference of means between all possible pair-wise comparisons of animal groups was calculated. Gene fragments having both a Tukey HSD P value <0.05 and an absolute difference of means 1.5, for a pair-wise group comparison, were deemed to have expression significantly different for that comparison.
For protein expression analysis, the non-parametric Kruskal–Wallis method (Stat View 5.0, SAS Institute, Cary, NC, USA.) was used for comparisons of means among WT, MyD88−/−, and TRIF-mutant mice. For comparison of means between two groups, we used the non-parametric Mann–Whitney test. Differences among groups were expressed as mean±standard deviation and considered significant if P<0.05.
RESULTS
Genomic profiling with cDNA microarrays that represent more than 20,000 genes led to the identification of four immediate early genes representing the acute stress response pathway and insulin degrading enzyme (IDE), representing β-amyloid processing, that were significantly differentially expressed among WT, MyD88−/−, and TRIF-mutant mice, at different time points, following focal cerebral ischemia.
A total of 25,255 genes were represented on the mouse Gene 1.0 ST Array, based on release 32 of the NetAffx annotation file (Affymetrix). Each transcript cluster was detected by 25 bp oligomers, the combination of which depended on the size of the transcript.
Genomic Profiling
A total of 35,557 gene fragment expression values were generated per sample; no outliers were detected. Of these gene fragments, only 3,402 (10%) were noise biased and were discarded, leaving 32,155 gene fragments for significance testing. Following analysis of variance and Benjamin and Hochberg false discovery rate correction, 10,816 gene fragments were available for analysis.
A total of 1,833 gene fragments, representing 754 known genes, were significantly differentially expressed between at least one pair-wise experimental group comparison. These 754 genes included 21 genes that were significantly differentially expressed for the MyD88 versus WT pair-wise comparison and 10 genes that were significantly differentially expressed for the TRIF versus WT pair-wise comparisons. A total of seven groups were used to compare differential gene expression across WT, MyD88−/−, and TRIF-mutant mice for IPA; (Table 1).
Group designations for IPA analysis
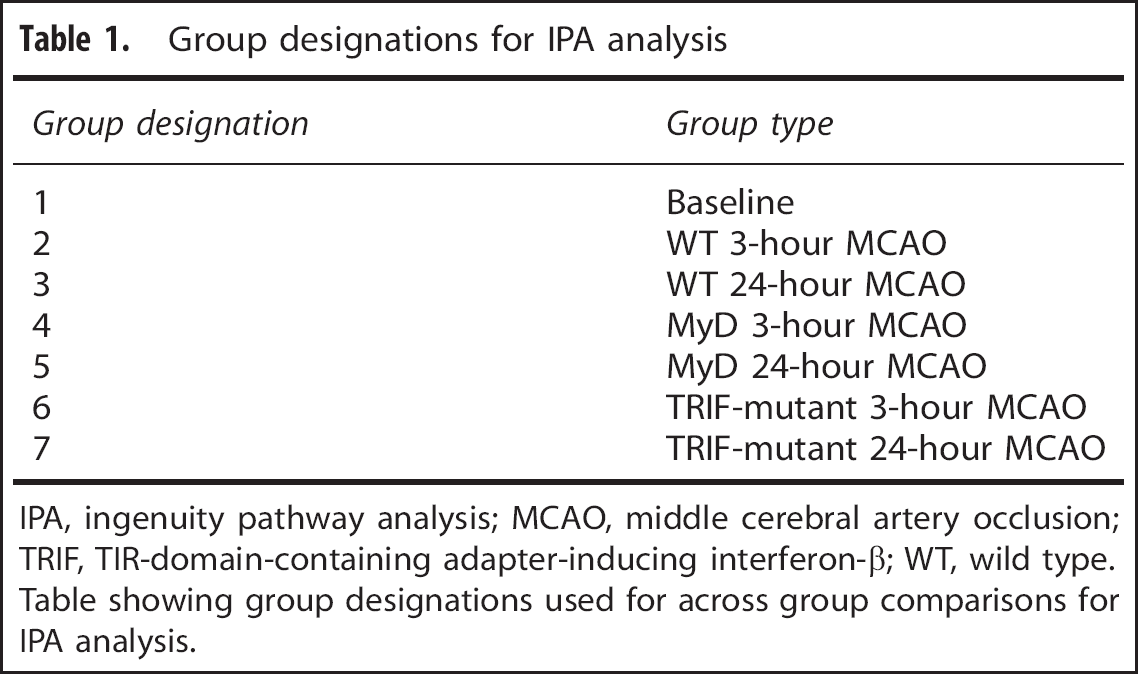
IPA, ingenuity pathway analysis; MCAO, middle cerebral artery occlusion; TRIF, TIR-domain-containing adapter-inducing interferon-β; WT, wild type.
Table showing group designations used for across group comparisons for IPA analysis.
Pathway Analysis
The 754 known genes that were significantly different between at least one pair-wise comparison, and that met criteria for fold change difference of at least 1.5 or greater and P<0.05, were compared to the canonical pathways in the IPA knowledge database. The acute phase response signaling pathway was one of the top five significant canonical pathways identified by IPA analysis using all differentially expressed genes from all possible pair-wise comparisons; Figure 2. Overrepresentation of genes involved in the acute stress response was also shown by network analysis.
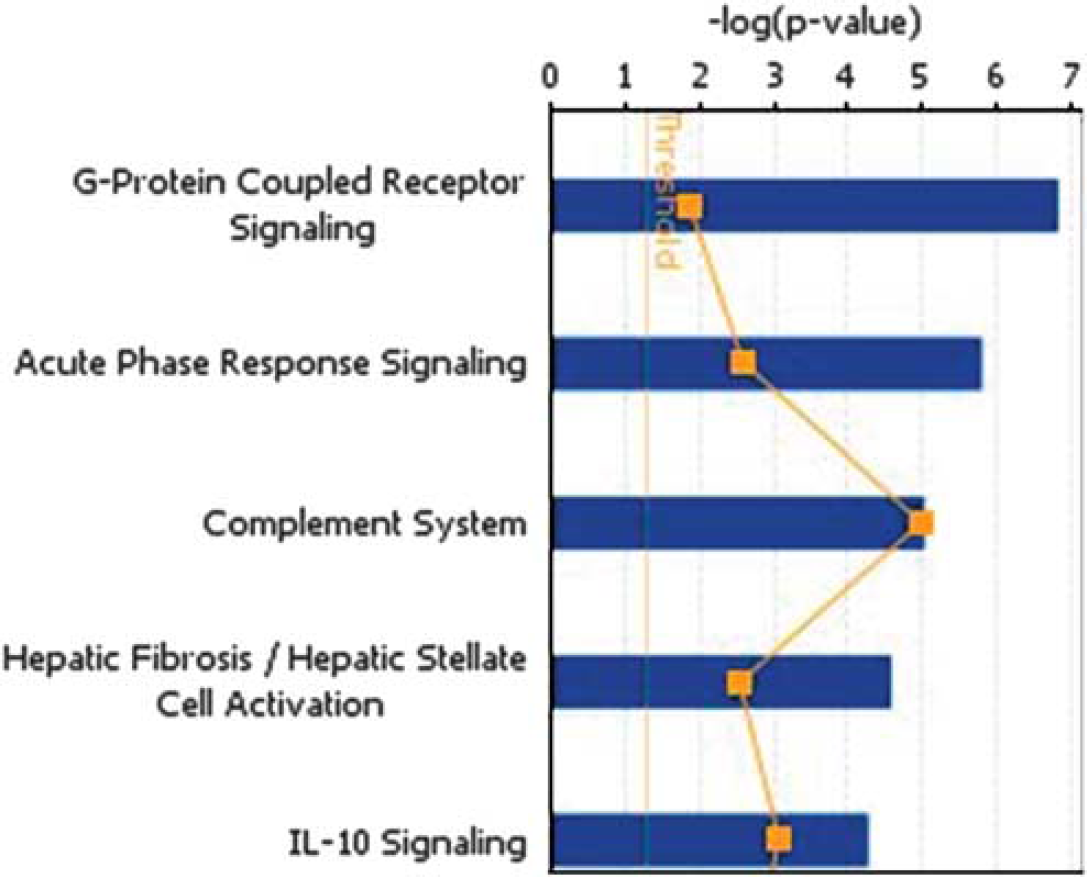
The top five enriched canonical pathways identified by ingenuity pathway analysis using all differentially expressed genes from all possible pair-wise comparisons.
Confirmation of Microarray Data by Real-Time Polymerase Chain Reaction
Using RT-PCR to determine fold gene expression, we confirmed microarray data showing differential expression of five genes, four of which belonged to the acute stress pathway, in mice with deficiency of MyD88 and TRIF compared to WT mice following pMCAO. Of the 21 genes differentially expressed for the specific pair-wise comparison of MyD88−/− versus WT, at 3 hours following pMCAO, we confirmed the expression of two of the three genes with a more than twofold decrease in gene expression. Similarly, of the 10 genes identified by IPA analysis for the specific pair-wise comparison of TRIF versus WT, we confirmed the expression of both of the genes with a more than twofold decrease in gene expression.
We also confirmed the expression of NR4A1/Nurr77, which was an additional gene with a more than twofold expression decrease that was differentially expressed at the later time point in the MyD88−/− versus WT comparison. Finally, we chose to confirm the expression of IDE, which was significantly differentially expressed only with disruption of the TRIF pathway. In total, four of the five genes that we confirmed belonged to the acute stress pathway, which was also one of the top five canonical pathways identified by IPA analysis; Figure 2.
Increased Baseline Expression of Immediate Early Acute Response Genes in Mice with Disruptions of MyD88 and TRIF Compared to Wild-Type Mice
Since our previous studies showed abnormal baseline elevation of some cytokines prior to pMCAO, 11 we evaluated baseline gene expression profiles in all three groups of mice. We made the unexpected observation that expression of immediate early acute response genes were significantly increased, at baseline, in mice with disruptions of MyD88 and TRIF, compared to baseline expression of these same genes in WT mice.
In MyD88−/− mice, baseline expression of immediate early genes were significantly increased, compared to WT baseline, as follows: EGR1 expression was increased 2.3-fold (P<0.05), EGR2, 9.7-fold (P<0.05), ARC, 4.1-fold (P<0.05), Nurr77, 1.3-fold (NS) and IDE 1.4-fold (P<0.05); (Figure 3A). In TRIF-mutant mice, compared to WT mice, baseline EGR1 expression was increased 3.2-fold (P<0.05), EGR2, 8.7-fold (P<0.05), ARC, 2.9-fold (P<0.05), Nurr77, 1.3-fold (NS), and IDE 2.7-fold (P<0.05); (Figure 3B).
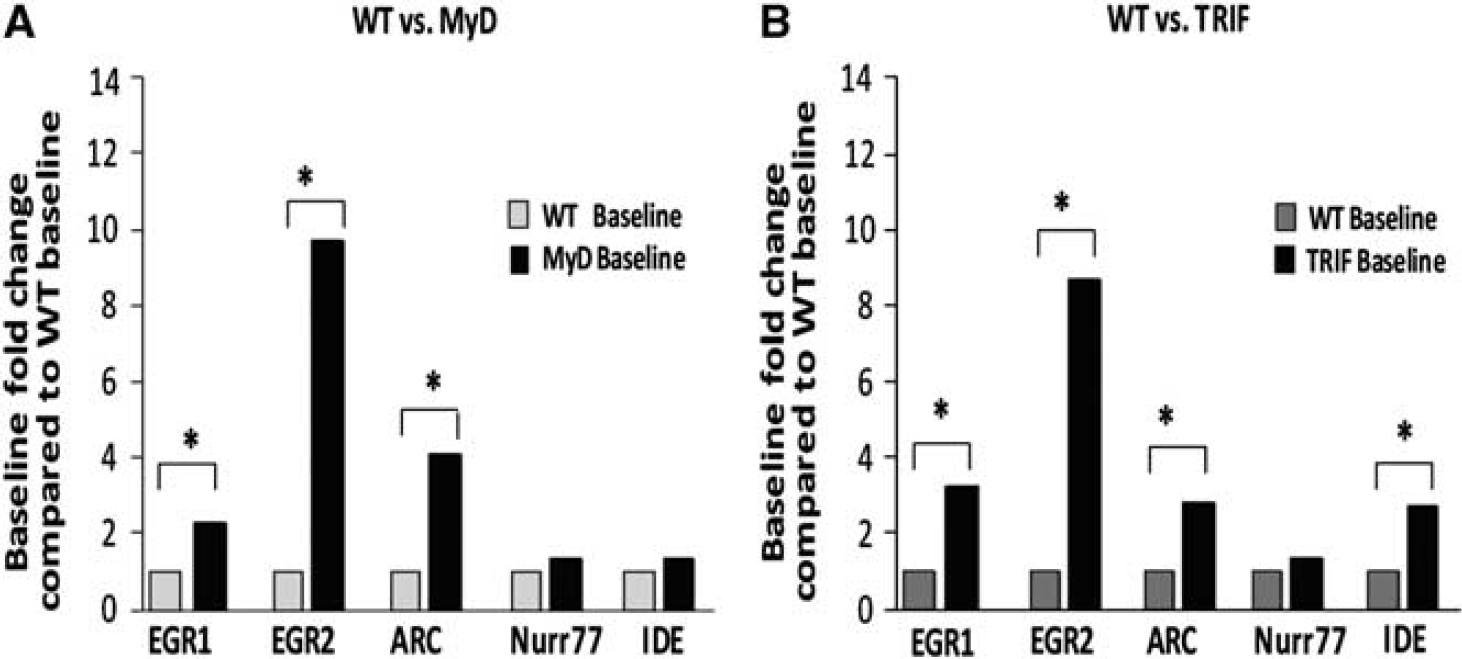
Increased baseline immediate early gene expression in MyD88−/− and TRIF-mutant mice compared to wild-type (WT) baseline. Baseline expression of the above genes was normalized to one in WT mice and compared to baseline expression levels in MyD88−/− mice or TRIF-mutant mice. (
We focused on confirming, by real-time-PCR, differential expression of immediate early genes at baseline and at the 3-hour time point following pMCAO because these two time points showed the greatest difference in gene expression, as indicated by the raw intensity values from microarray data analysis (data not shown).
No Increase in Immediate Early Gene Expression Following Permanent Middle Cerebral Artery Occlusion in MyD88−/− Mice
We observed no significant increase in immediate early gene expression, from baseline, following pMCAO, in mice with deficiency of MyD88, compared to significant changes in expression of these immediate early genes in WT mice under the same conditions.
In WT mice, (n=9 per group; three per time point), we observed significant increases in immediate early gene fold expression from baseline, 3 hours following pMCAO, as follows:
(1) EGR1, 3.6-fold (P<0.01); (2) EGR2, 12.3-fold (P<0.01); (3) ARC, 4.3-fold (P<0.05); (4) Nurr77, 1.8-fold (P<0.05), and (5) IDE, 1.5-fold (NS); (Figure 4A).
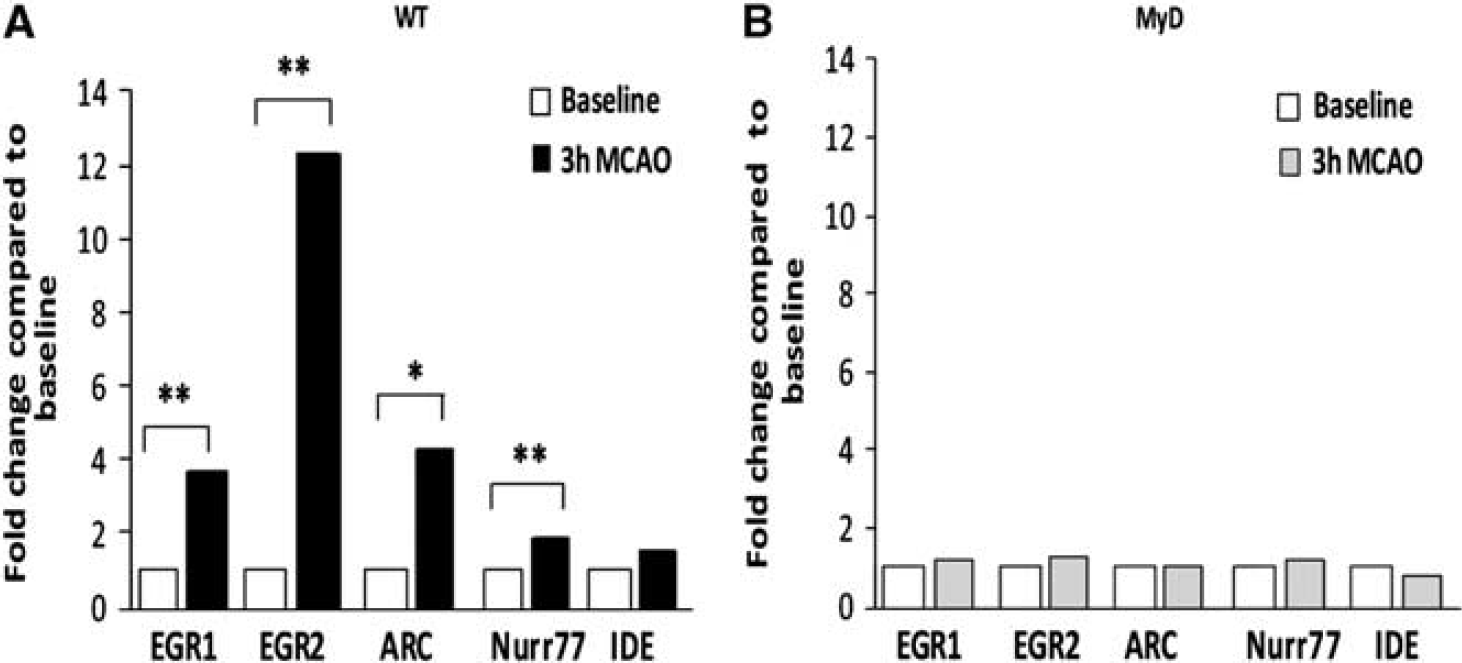
No increase in immediate early gene expression following permanent middle cerebral artery occlusion (pMCAO) in the brains of MyD88−/− mice. Fold change in immediate early gene expression, expressed in arbitrary units, was determined by real-time PCR in wild type (WT) and MyD88−/− in the same experiment. (
In contrast, in MyD88−/− mice, (n=9 per group; three per time point), at 3 hours following pMCAO, we observed no significant differences in fold expression of immediate early genes, compared to baseline, in these group of mice as follows: (1) EGR1, 1.2-fold increase (NS); (2) EGR2, 1.3-fold increase (NS); (3) ARC, 1.02-fold increase (NS); (4) Nurr77, 1.2-fold increase (NS), and (5) IDE, 0.8-fold decrease compared with baseline (NS) (Figure 4B).
These results indicate altered network dynamics regulating immediate early gene expression in mice with disruption of MyD88 following focal cerebral ischemia.
No Increase in Immediate Early Gene Expression in TRIF-mutant Mice following Permanent Middle Cerebral Artery Occlusion
In mice with deficiency of TRIF, similar to the results obtained in mice with disruption of MyD88, we observed no significant increases in immediate early gene expression following pMCAO, compared to significant changes in expression of these immediate early genes in WT mice after pMCAO.
In WT mice, at 3 hours after pMCAO, we observed significant fold increases in brain immediate early gene expression, compared to baseline, using real-time-PCR as follows: EGR2 expression increased 10.9-fold (P<0.01). ARC expression increased 3.9-fold (P<0.05). Insulin degrading enzyme expression increased 1.6-fold (P<0.05); EGR1 and Nurr77 expression increased 4.7- and 1.9-fold respectively; both NS. (Figure 5A).
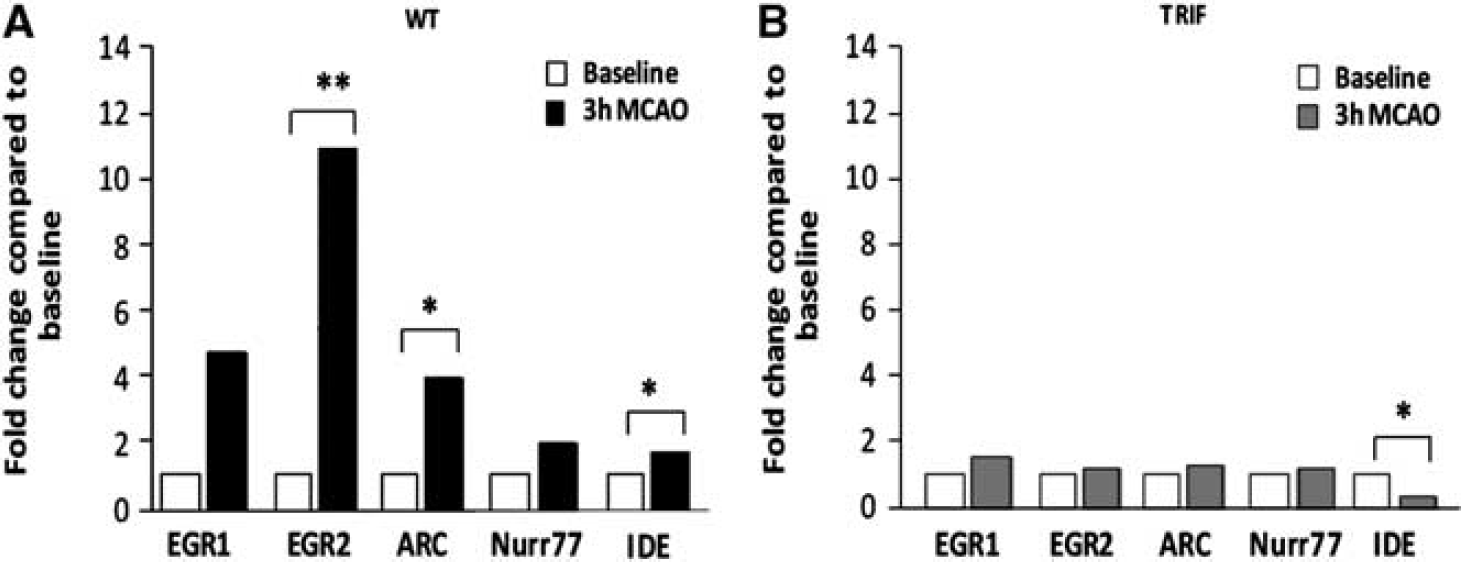
No increase in immediate early gene expression, following permanent middle cerebral artery occlusion (pMCAO) in the brains of TRIF-mutant mice. Fold change in immediate early gene expression in wild-type (WT) and TRIF-mutant mice was again determined by real-time PCR and is expressed in arbitrary units. (
However, in the brains of TRIF-mutant mice, expression of immediate early genes showed no increase and remained unchanged 3 hours following focal cerebral ischemia compared to baseline, except for IDE, which was significantly decreased following pMCAO. EGR1 expression increased 1.5-fold (NS). EGR2 expression increased 1.2-fold (NS.). ARC expression increased 1.2-fold (NS.). Nurr77 expression increased 1.2-fold (NS.). In contrast, IDE expression significantly decreased 0.36-fold (P<0.05) following pMCAO compared with baseline; (Figure 5B).
Decreased Levels of Immune Activation Genes, sICAM, MMP-9, sE-Selectin, and sVCAM-1 noted in the Serum of Mice with Disruption of MyD88 Compared to Wild-Type Mice following Focal Cerebral Ischemia
To determine whether the abnormal immediate early gene expression observed in mice with deficiencies of MyD88 or TRIF had any effect on expression of downstream genes involved in different aspects of immune activation such as ICAM-1,16, 17, 18, 19 E-Selectin, VCAM-1, 20 MMP-9, 21 and tPAI-1,16, 22 we analyzed protein levels of these genes at baseline, and following pMCAO.
Intercellular Adhesion Molecule-1
Serum sICAM-1 levels were significantly decreased in MyD88−/− compared to WT mice, at baseline, and acutely, at 3 hours, following pMCAO.
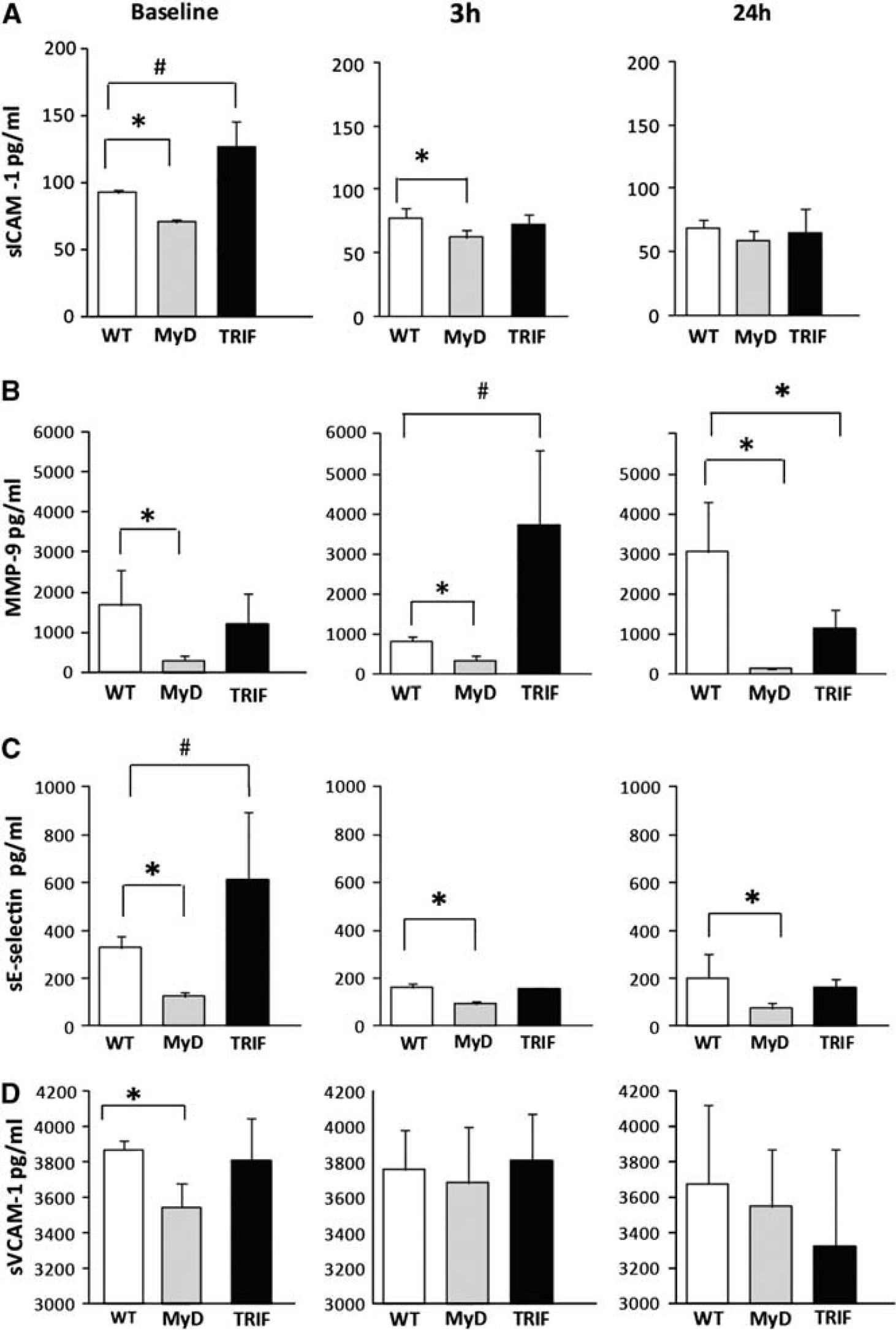
Levels of serum ICAM-1, MMP-9, E-Selectin, and VCAM-1 in wild type (WT), MyD88, and TRIF-mutant mice at baseline and following permanent middle cerebral artery occlusion (pMCAO). (
Matrix Metalloproteinase-9
Serum MMP-9 levels, similar to serum sICAM-1 levels, were significantly decreased in MyD88−/− mice compared to WT mice both at baseline, and following pMCAO. At baseline, serum MMP-9 levels were almost sixfold decreased in MyD88−/− compared to WT mice.
Lastly in TRIF mutant compared to WT, MMP-9 levels were: 1144.78±448 versus 3043. 5±1270 pg/mL; Figure 6B.
E-Selectin
Serum E-Selectin levels followed the same pattern as the other two proteins; sICAM-1 and MMP-9, in being significantly decreased in MyD88−/− compared to WT mice. Serum sE-Selectin levels were significantly decreased in MyD88−/− mice at baseline and at both time points following pMCAO. At baseline, levels of serum sE-Selectin were significantly different among all three groups; P<0.05. At baseline and at 3 hours following pMCAO, serum sE-Selectin levels were more than twofold decreased in MyD88−/− compared to WT as follows:
In comparison, serum sE-Selectin levels were not significantly different in WT compared with TRIF mutant at baseline or at 3 or 24 hours following pMCAO; Figure 6C.
Vascular Cell Adhesion Molecule-1
In addition, serum sVCAM-1 levels, similar to sICAM-1, MMP-9, and sE-Selectin levels, were significantly decreased in MyD88−/− compared to WT mice. However, this decrease was present only at baseline, but not at the 3 and 24 hours time points. At baseline, sVCAM-1 levels in MyD88−/− compared to WT were as follows: 3546.4±134 and 3871.6±52.4 pg/mL, respectively; P<0.05. In contrast, sVCAM-1 levels were not significantly different between WT and TRIF-mutant mice at baseline or at subsequent time points following pMCAO; Figure 6D.
Finally, serum tPA1 levels were not significantly different between the three groups of mice at baseline or following pMCAO in group or pair-wise comparisons at any of the time points.
DISCUSSION
We hypothesized the presence of additional roles for the downstream TLR adaptors, MyD88, and TRIF during cerebral ischemia and have demonstrated previously unidentified roles for these downstream pathways in regulating immediate early gene expression following focal cerebral ischemia. We found that disruption of either downstream pathway leads to an attenuation of the normal increase in expression of these genes following focal cerebral ischemia, possibly due to abnormal baseline kinetics of immediate early gene expression in mice with disruption of these downstream adaptors. We also showed an unexpected role for the TRIF pathway in the expression of IDE, which is known to play a major role in the degradation of β-amyloid. Furthermore, this abnormal immediate early gene expression in mice with disruption of MyD88 and TRIF is associated with abnormal expression of a number of downstream genes such as, MMP-9, 21 E-Selectin, 23 ICAM-1,16, 17, 18, 19 and VCAM-1, 20 important in mediating immune responses to focal cerebral ischemia.
The additional role for the MyD88 adaptor in immediate early gene expression, noted in our current study, is supported by findings in our study of attenuated immediate early gene expression in MyD88−/− compared to WT mice following focal cerebral ischemia. With the results of a baseline group of mice in these studies, we were able to show the significantly increased baseline expression of immediate early genes in these mice. These abnormal baseline kinetics, appear to attenuate the expected increase in the expression of immediate early genes following focal cerebral ischemia and provide a molecular basis for this additional role of the MyD88 pathway in the acute response to focal cerebral ischemia. Other well-known roles of the MyD88 pathway include the fact that all TLRs, except TLR3, signal via MyD88, leading to the expression of proinflammatory cytokines and chemokines.24, 25 In addition, MyD88 is a common TLR/IL-1 downstream adapter for IL-1 signaling and is also required for the expression of IL-1β. 26 However, disruption of MyD88 signaling does not protect against focal cerebral ischemia, in in vitro and in vivo models. 12 Surprisingly, this lack of protection occurs in spite of decreased levels of proinflammatory cytokines in the serum and brains of MyD88−/− mice following focal cerebral ischemia. 11
To date, a molecular basis for these confounding results has not been known. We propose that our current findings of impaired immediate early gene expression and abnormal baseline kinetics of immediate early gene expression, in mice with disruption of MyD88, provides some of this missing molecular evidence. This new molecular evidence and the additional role for the MyD88 adaptor in immediate early gene expression, is supported by published studies showing impaired immediate early gene expression and impaired tissue regeneration, in MyD88−/− mice, following partial hepatectomy. 27 The combination of these published reports and our current results implicate the MyD88 pathway in immediate early gene expression. Therefore, efforts to develop specific MyD88-based pathway treatments will have to be directed at targets further downstream of this adaptor, and the signals that mediate immediate early gene expression.
In addition, we have shown that the TRIF pathway, similar to our findings with the MyD88 pathway, has an additional role in optimal immediate early gene expression following focal cerebral ischemia. In our current study, the role for the TRIF adaptor in immediate early gene expression is supported by evidence of impairment in immediate early gene expression and abnormal immediate early gene baseline kinetics in mice with disruption of TRIF. The mechanism of impairment in immediate early gene expression in TRIF-mutant mice, as was the case with MyD88−/− mice, seemed to be due to the significant increases in the expression of these immediate early genes at baseline compared to WT mice. However, our previous studies showed similar expression of MyD88-mediated cytokines in TRIF-mutant and WT mice, 11 but mice with disruption of TRIF were not protected against focal cerebral ischemia. 12 We postulate that the baseline cytokine changes, reported in our previous study, 11 were limited to the MyD88 versus WT comparison, in part, because immediate early genes, such as EGR1, which was increased at baseline in MyD88−/− and TRIF-mutant mice, compared to WT mice, in our current study, are known to affect the expression of MyD88-specific cytokines, such as IL-6.28, 29
Taken together, these previous results of a cytokine profile similar to WT mice, and the current additional role for TRIF in ischemia-mediated immediate early gene expression, suggest that both optimal cytokine and immediate early gene expression might be essential for the optimal acute phase response to focal cerebral ischemia.
To the best of our knowledge, this is the first report documenting the role of the TRIF pathway in immediate early gene expression following focal cerebral ischemia. These new findings need to be taken into account during attempts to modulate the TRIF pathway for the treatment of focal cerebral ischemia.
In addition to the role of the TRIF pathway in immediate early gene expression, we also found a role for the TRIF pathway in the expression of IDE, an enzyme that degrades both insulin and β-amyloid. 30 We observed increased levels of IDE in the brains of WT mice following pMCAO consistent with previous reports of increased levels of IDE in the brains of rats subjected to focal cerebral ischemia. 31 In contrast, in our study, we found decreased levels of IDE in the brains of mice with disruption of TRIF, following focal cerebral ischemia, consistent with a role for this pathway in IDE expression. These findings also suggest that the plausible physiological role for increased IDE expression following focal cerebral ischemia is the degradation of β-amyloid known to be deposited in response to different types of tissue injury including focal cerebral ischemia. 32 In further support of our findings of a role for the TRIF pathway in IDE expression, are previous reports of elevated plasma insulin (substrate for IDE) levels, and accompanying glucose intolerance, noted in TRIF-mutant mice 33 and elevated levels of insulin as well as β-amyloid in cultured neurons from IDE knockout mice. 34
Furthermore, these findings provide a probable molecular mechanism for the current experimental use of insulin in the treatment of Alzheimer's 35 and for the previously observed, but not explained, increase in IDE expression following intranasal insulin administration. 36 Finally, the involvement of the TRIF pathway in IDE expression, noted in our study, has important implications for understanding the molecular mechanisms of the physiological response to neurological diseases in which β-amyloid deposition plays a central role in disease pathogenesis.
With regards to the altered immediate early gene expression in mice with disruption of MyD88 and TRIF, compared to WT mice, noted in our study, we found possible physiological relevance for these findings in the results of the accompanying protein expression studies. These included findings of abnormally decreased expression of a number of proteins associated with activation of the innate immune response, such as E-Selectin, MMP-9, ICAM-1, and VCAM-1 in MyD88−/−, compared to WT mice, acutely following pMCAO. The lack of significant decreases in the expression of these adhesion molecules and immune activation proteins acutely following pMCAO in mice with disruption of TRIF compared to WT, suggests that expression of these proteins is more MyD88-dependent. Notably, decreased levels of MMP-9 in MyD88−/− compared with WT mice may explain, in part, previous reports of impaired wound healing noted in these mice following tissue injury 37 and tissue regeneration, 27 since MMP-9 is known to play an important role in tissue regeneration 38 and neuronal plasticity. 39 With regards to focal cerebral ischemia, impaired wound healing, noted to occur in MyD88−/− mice, may be detrimental during the tissue remodeling that occurs following this cellular insult. These results show that with disruption of MyD88 and TRIF, there are physiological consequences for the abnormal expression of a number of key genes required for the response to focal cerebral ischemia. Along these lines, in support of our current findings, published reports evaluating immediate early gene expression in the ischemic core and penumbra show a complex temporal pattern of expression of these genes in both transient and permanent cerebral models of ischemia.40, 41, 42, 43, 44, 45, 46 However, evidence as to whether the expression of immediate early genes play a beneficial or detrimental role following tissue injury is just emerging with recent studies showing an association between deletion of some immediate early genes and impaired neurogenesis. 47
Taken together, these combined findings provide further support for the currently reported effect of disruption of either of these two downstream pathways in models of focal cerebral ischemia.12, 13, 14, 15 These studies are the first to show that MMP-9 production occurs via the MyD88 pathway following focal cerebral ischemia. These results are in agreement with previous studies showing MMP-9 production via the TLR4/MyD88 pathway in cells from other tissues. 48
The immediate early genes examined in our study are also known to be induced in response to several types of cellular stress or cellular growth. 49 Our current findings thereby suggest that the downstream TLR signaling adaptors, MyD88 and TRIF, play an important role in translating extracellular stress signals to downstream immediate early gene expression and hence play an important role in the innate response to focal cerebral ischemia as illustrated in the schematic; Figure 7.
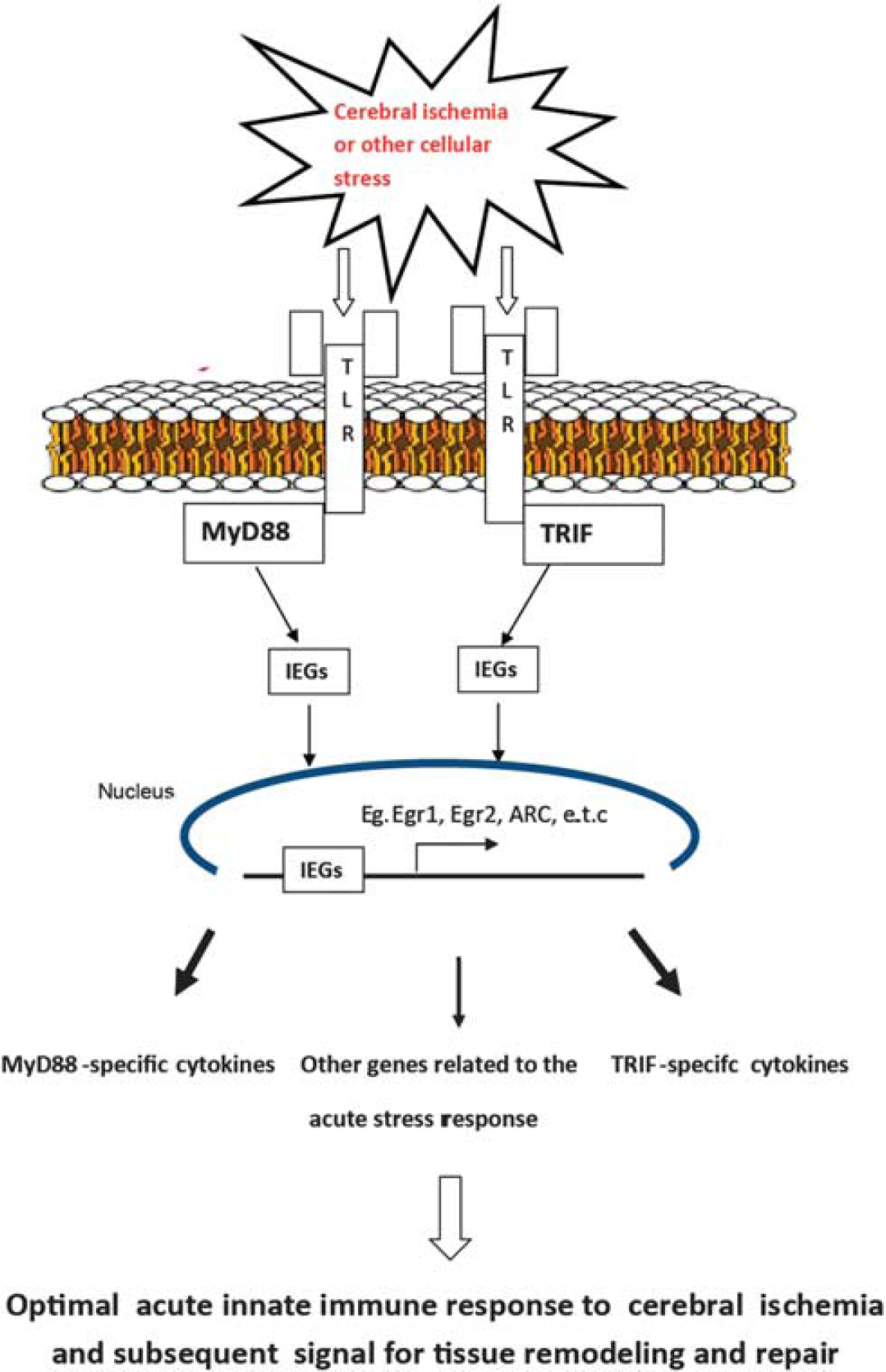
Schematic showing the proposed role of downstream Toll-like receptor (TLR) signaling adaptors, MyD88, and TRIF in the optimal acute stress response to focal cerebral ischemia.
The results of these studies should be taken in context, however, since it is not uncommon for shifts in network dynamics to occur following genetic manipulation of a single gene. However, the strong evolutionary conservation of TLR expression, across several species, makes significant shifts in network dynamics less likely. Along these lines, we found no compensation in cytokine production by the other pathway, when one downstream pathway was disrupted. 11
Further studies are warranted to identify more intricate functions of these downstream pathways to facilitate the development of novel molecularly targeted therapies for focal cerebral ischemia. Such understanding is imperative before therapies can be developed against specific points along the pathway that affect the detrimental inflammatory response to focal cerebral ischemia, but that preserves the beneficial roles of these pathways in the acute response to focal cerebral ischemia.
In conclusion, our findings provide evidence demonstrating additional roles, for the downstream TLR adaptors, MyD88, and TRIF, in modulating the expression of immediate early genes following focal cerebral ischemia. We also provide evidence for the possible role of the TRIF pathway in modulating the expression of IDE required for degradation of β-amyloid in diseases such as Alzheimer's. Overall, these studies provide further compelling and specific molecular evidence supporting additional roles for the downstream adaptors, MyD88 and TRIF, that extends the current state of knowledge regarding the role of these adaptors during focal cerebral ischemia, and supports the emerging paradigm that the innate immune system plays an important role in the acute response to focal cerebral ischemia.
Footnotes
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The authors thank Dr Sungyoung Auh for help with statistical analysis. We also extend our appreciation to Dr Phil McCoy and Leigh Samsel of the National Heart Lung and Blood Institute Flow Cytometry Core Facility, National Institutes of Health, Bethesda, MD, USA, for help with MILLIPLEX assays (Millipore, Billerica, MA, USA). Finally, we thank Dr Abdel Elkahloun of the NINDS–NIMH microarray core facility for help with cDNA microarrays.