
Abstract
Some 40 years ago it was recognized by Furchgott and colleagues that the endothelium releases a vasodilator, endothelium-derived relaxing factor (EDRF). Later on, several groups identified EDRF to be a gas, nitric oxide (NO). Since then, NO was identified as one of the most versatile and unique molecules in animal and human biology. Nitric oxide mediates a plethora of physiological functions, for example, maintenance of vascular tone and inflammation. Apart from these physiological functions, NO is also involved in the pathophysiology of various disorders, specifically those in which regulation of blood flow and inflammation has a key role. The aim of the current review is to summarize the role of NO in cerebral ischemia, the most common cause of stroke.
Introduction
In the late 1970s, it was recognized that the endothelium releases a factor that relaxes vascular smooth muscle cells thereby causing vasodilatation (Furchgott and Zawadzki, 1980). Since the chemical structure of this factor was unknown at the time, it was named EDRF (endothelium-derived relaxing factor). Later on, EDRF was identified to be a color and odorless gas, nitric oxide (NO) (Furchgott et al, 1987; Ignarro et al, 1987a, 1987b). Since then, NO, the most convincing gaseous signaling molecule in vertebrates, was increasingly recognized as one of the most versatile and unique molecules mediating such diverse physiological functions as the maintenance of vascular tone, thrombotic-thrombolytic homeostasis, cell growth, and inflammation. Apart from its physiological functions, NO has also an important role in the pathophysiology of various disorders, specifically those in which regulation of blood flow and inflammatory reactions are key pathophysiological events.
Nitric Oxide—Physiology
Nitric oxide is an uncharged gas that can easily cross biological membranes. It is, however, quickly deactivated by oxidation with a biological half-life of a few seconds thereby limiting its spatial extent in biological tissues (Gibson and Roughton, 1957; Stamler et al, 1992). Nitric oxide is synthesized by a group of three NO synthases (NOS) from
Nitric oxide toxicity usually occurs through a direct interaction with various protein moieties. This may result in the reversible formation of mixed disulfides between cysteines and glutathione, termed
Apart from these direct actions, NO can also promote intercellular signaling via activation of soluble guanylate cyclase and subsequent formation of cyclic guanosine monophosphate (cGMP). The best documented signaling activity using this mechanism is the migration of NO generated by the endothelium into neighboring smooth muscle cells whereupon NO relaxes vascular smooth cell through Ca2+-mediated formation of cGMP and phosphorylation of downstream kinases that ultimately impact calcium availability (Murad et al, 1987).
Endothelial NOS-derived NO has furthermore been demonstrated to have a key role in vascular remodeling and angiogenesis (Papapetropoulos et al, 1997; Rudic et al, 1998; Murohara et al, 1998).
In the brain, NO is mainly formed by nNOS and eNOS. Accordingly, it serves as a neurotransmitter and neuromodulator (Garthwaite et al, 1988; Dawson and Snyder, 1994; Dawson, 1995) and—among many other functions—is concerned with the maintenance of basal cerebral blood flow (CBF) (Tanaka, 1996). Specifically, NO has been implicated in cerebral autoregulation (Dirnagl et al, 1994; Richards et al, 1997; White et al, 2000), chemoregulation of CBF (Iadecola, 1992; Thompson et al, 1996; Lavi et al, 2003), and neurovascular coupling (Dirnagl et al, 1994).
More recent evidence suggests a role of NO in neuronal proliferation and differentiation. Tanaka et al (1994) showed that NO inhibits neural proliferation and differentiation in the developing brain;
Neurogenesis, which in the adult brain seems restricted to the subventricular zone, the olfactory bulb, and the subgranular zone of the hippocampus (Zhao et al, 2008), persists in the adult brain and can be induced after cerebral insults (Zhao et al, 2008; Imayoshi et al, 2009). Nitric oxide exerts a dual effect on neurogenesis: While nNOS-derived NO decreases neurogenesis (Packer et al, 2003; Moreno-Lopez et al, 2004; Zhu et al, 2006), NO produced by endothelial (Reif et al, 2004) or inducible (Luo et al, 2007; Bechade et al, 2011) NOS seems to stimulate it.
Nitric Oxide—Role in Cerebral Ischemic Preconditioning
It was discovered in the early 1990s (Kitagawa et al, 1990) that exposing the brain to sublethal injury increases its tolerance for subsequent insults (Gidday, 2006). Among the stimuli investigated for induction of tolerance are sublethal ischemia and proinflammatory mediators (Stagliano et al, 1999; Dawson and Dawson, 2000; Dirnagl et al, 2003; Kunz et al, 2007). Nitric oxide seems to have a major role in mediating ischemic tolerance induced by preconditioning. While there is mounting evidence that iNOS-derived NO is a key effector of ischemic preconditioning (Park et al, 2003; Cho et al, 2005), the contribution of constitutive NOS is not fully elucidated yet (Gidday et al, 1999; Gonzalez-Zulueta et al, 2000; Atochin et al, 2003).
Nitric Oxide—Pathophysiology in Stroke
Cerebral ischemia induces multiple and distinct changes in cerebral NO content and signaling. Occlusion of the middle cerebral artery (MCAo) results in an increased production of NO by 20-fold for up to 30 minutes (Malinski et al, 1993; Kader et al, 1993) most likely through increased calcium availability and activation of nNOS (Huang et al, 1994). Thereafter, the brain tissue NO is reduced below detectable levels for up to 7 days (Malinski et al, 1993; Sugimura et al, 1998), indicating a long-lasting NO deficiency in the ischemic brain. If reperfusion occurs, NO concentration may transiently increase by 50% for about 30 minutes (Fassbender et al, 2000; Uetsuka et al, 2002). Concomitant with changes in NO levels, the activities of eNOS and nNOS increases within the first few minutes after MCAo, but decrease significantly thereafter (Kader et al, 1993). In contrast to the constitutive NOS isoforms, iNO becomes upregulated from 12 hours after MCAo for up to 7 days (Niwa et al, 2001).
In the chronic phase after cerebral ischemia, angiogenesis has been shown to occur (Beck and Plate, 2009); the extent of collateralization is a predictor for neurological outcome after stroke (Christoforidis et al, 2005). Nitric oxide has a crucial role in angiogenesis after ischemic stroke: NO donor DETA-NONOate increased angiogenesis after experimental cerebral ischemia (Zhang et al, 2003; Chen et al, 2004). Endothelial NOS-deficient mice revealed significantly impaired neovascularization after stroke, indicating that endothelial-derived NO mediates this effect (Cui et al, 2009).
Based on the time course of ischemia-induced changes in NO levels and NOS regulation in the brain and cerebral blood vessels, several strategies were suggested to manipulate the NO system for the treatment of stroke.
When the cationic amino-acid substrate
Another restorative mechanism after stroke that is NO dependent is neurogenesis (see Zhang et al, 2005, 2008 for reviews). While iNOS-derived NO was shown to be neurotoxic in the acute phase after stroke, several studies suggest an important role of the inducible isoform for cell proliferation after experimental ischemia: iNOS-positive cells increased significantly in the periinfarct zone 1 and 3 days after focal ischemia (Sehara et al, 2006), the number of iNOS-positive cells increased with increasing survival time (Corsani et al, 2008). Treatment with iNOS inhibitor aminoguanidine prevented postischemic neurogenesis (Zhu et al, 2003); the phenomenon could not be detected in iNOS null mice, either (Zhu et al, 2003). Nitric oxide produced by nNOS exerts negative effects on neurogenesis (Packer et al, 2003; Moreno-Lopez et al, 2004; Luo et al, 2007), nNOS inhibition has been shown to increase neurogenesis after experimental ischemia (Sun et al, 2005). Neuronal NOS and iNOS therefore seem to have opposite roles in postischemic neurogenesis. There are hints that there is crosstalk between the neuronal and inducible NOS isoform: genetic deletion of nNOS or nNOS downregulation after cerebral ischemia-induced iNOS upregulation and—thus—neurogenesis while the protective effect of pharmacological nNOS inhibition was abolished in iNOS-deficient mice (Luo et al, 2007).
Given the ample evidence for NO-mediated neuroprotection after cerebral ischemia, several treatment strategies have been examined that increase postischemic NO content specifically generated by eNOS activity to augment CBF to the penumbra. One putative strategy is to replenish the early postischemic NO deficit by application of NO or NO precursors; another is to increase or modulate endogenous NO production/bioavailability by enhancing eNOS activity (Endres et al, 2004); for example, via the phosphatidylinositol 3-kinase (PI3-kinase)/protein B kinase (Akt) pathway (Dimmeler et al, 1999). Another potential mechanism is to directly or indirectly inhibit NOS, preferably nNOS and/or iNOS. Putative treatment strategies are summarized in Figure 1.
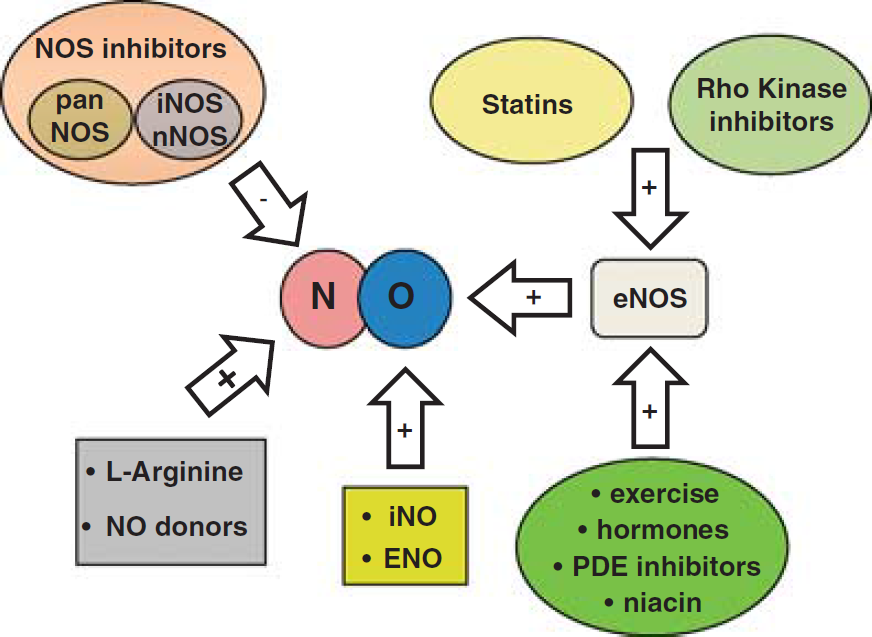
Summary of therapeutical strategies influencing nitric oxide signaling in ischemic stroke. eNOS, endothelial nitric oxide synthases; iNOS, inducible NOS; nNOS, neuron NOS; PDE, phosphodiesterase.
Therapeutic Approaches
L -Arginine and Nitric Oxide Donors
After encouraging preclinical data following application of the NO precursor
Nitric Oxide Synthase Inhibitors
The excessive production of NO by brain parenchyma is a characteristic of ischemic stroke. Hence, inhibition of NOS was viewed as an alternative strategy to reducing NO availability in cells expressing the neuronal isoform. In a first approach, nonselective inhibition of NOS by
More selective approaches using nNOS and iNOS inhibitors seem to be more promising. Pharmacological inhibition of nNOS with 7-nitroindazole given 5 minutes after MCAo reduced infarct volume in rats by up to 27% (Yoshida et al, 1994; Zhang et al, 1996b) and the application of aminoguanidine (Zhang et al, 1996a; Cash et al, 2001),
However, NOS inhibitors currently are not in use clinically and are not evaluated in any ongoing clinical stroke trial because most are nonselective and may thus cause deleterious side effects due to their actions on eNOS. The development of highly specific inhibitors of NOS subtypes might make this therapeutic approach feasible (Salerno et al, 2002).
Statins (HMG-CoA-Reductase Inhibitors)
HMG-CoA-reductase inhibitors, so called statins, completely inhibit the conversion of HMG coenzyme A to mevalonate thereby blocking cholesterol and isoprenoid intermediate synthesis (Liao and Laufs, 2005). Isoprenoid intermediates such as farnesylpyrophosphate and geranylgeranylpyrophosphate are responsible for modulation of activity of several small GTPases such as Ras, Rho, and Rac, which are blocked by statins.
Originally developed to treat and prevent coronary heart disease, statins were found to be effective also in primary and secondary prevention of ischemic stroke in several randomized clinical trials (Amarenco et al, 2004, 2006; Kennedy et al, 2007; Ridker et al, 2008). Initially designed to treat hypercholesterinemia, it soon became evident that the beneficial effect of statins in stroke patients was independent of the serum cholesterol lowering effect of the compounds (Treasure et al, 1995; Laufs et al, 2001). Statins have a number of pleiotropic effects that contribute to vascular integrity and health of the blood vessel wall. Experimental stroke studies revealed that long-term treatment with HMG-CoA-reductase inhibitors induced eNOS upregulation (Endres et al, 1998; Laufs et al, 2000b, 2002; Amin-Hanjani et al, 2001) and blocked hypoxia induced downregulation of eNOS in rodents (Laufs et al, 1997), thus increasing NO bioavailability and CBF (Endres et al, 1998) and saving brain tissue from ischemic injury (Endres et al, 1998; Laufs et al, 2000b, 2002; Amin-Hanjani et al, 2001). The effects were dose dependent and observed with a number of HMG-CoA-reductase inhibitors. That statins mediate their neuroprotective effect through the upregulation of eNOS and consecutive improvement of penumbral blood flow was further validated by experiments showing that treatment was ineffective in mutant mice deficient in eNOS expression (Endres et al, 1998; Laufs et al, 2000b, 2002).
In addition to the eNOS-dependent effect of statins
To evaluate the value of statins for the acute treatment of stroke, several studies applied HMG-CoA-reductase inhibitors after the initiation of cerebral ischemia. Surprisingly, also under these conditions, statins significantly reduced infarct size and brain edema formation (Sironi et al, 2003; Kilic et al, 2005; Nagaraja et al, 2006; Sugiura et al, 2007; Prinz et al, 2008; Mariucci et al, 2011). Interestingly, neuroprotection could be achieved by statin application as late as 2 days after the onset of MCAo (Sugiura et al, 2007). The main neuroprotective mechanisms are thought to be mediated via enhancement of eNOS activity (Mariucci et al, 2011), either by phosphorylation of eNOS at the S1177 site via the PI3 k/Akt pathway (Kureishi et al, 2000; Dimmeler et al, 2001) leading to reduction of inflammation (Sugiura et al, 2007) and induction of angiogenesis and neurogenesis (Chen et al, 2003) or via mRNA stabilization by inhibition of the Rho Kinase (ROCK) pathway (see below). This was proven by measurements of CBF, which showed that immediate postischemic CBF was not affected by acute statin treatment (Kilic et al, 2005; Berger et al, 2008), but that 5 days later—consistent with angiogenesis—CBF was improved (Berger et al, 2008).
Given their widely proven neuroprotective actions in experimental as well as in clinical trials, statins are recommended for primary as well as for secondary prevention of stroke (Heart Protection Study Collaborative Group, 2002; Adams et al, 2007). Despite these clear recommendations, it remains unclear whether HMG-CoA-reductase inhibitors indeed improve cerebral perfusion when given in the acute postischemic phase (Kennedy et al, 2007). Furthermore, most recent data suggest that statins exhibit untoward effects such as an increased risk of infection (Becker et al, 2011) and a higher incidence of hemorrhagic stroke (Collins et al, 2004; Goldstein et al, 2008; Vergouwen et al, 2008), stressing the need for further clinical evaluation of HMG-CoA-reductase inhibitors in the context of acute ischemic injury.
Rho Kinase (ROCK)—Inhibitors
There is ample evidence that the neuroprotective effect of statins/statin-induced upregulation of eNOS activity is at least in part mediated by inhibition of rho kinase (Laufs and Liao, 1998; Laufs et al, 1999, 2000a). Rho kinase (ROCK) is a serine–threonine kinase whose activity is modulated by isoprenoid intermediate geranygeranylpyrophosphate (GGPP). Rho kinase is implicated in the pathophysiology of atherosclerosis (Miyata et al, 2000), myocardial infarction, and hypertension (Uehata et al, 1997). Activation of Rho kinase in endothelial cells under hypoxic conditions (Wolfrum et al, 2004) has been related to the mechanism responsible for the downregulation of eNOS during and after ischemia (Takemoto et al, 2002; Ming et al, 2002; Wolfrum et al, 2004; Jin et al, 2006).
Rho kinase inhibitors such as fasudil or hydroxyfasudil rapidly lead to increased eNOS activity by Akt kinase-dependent phosphorylation of eNOS at S1179 (Fulton et al, 1999). This results in a significant reduction of ischemic brain damage when given before (Rikitake et al, 2005; Shin et al, 2007) but also when given after (Shin et al, 2007) experimental cerebral ischemia. The observed neuroprotective mechanism was proven to be eNOS dependent, based on the observation that ROCK inhibitors failed to improve CBF or reduce infarct volumes in eNOS null mice (Rikitake et al, 2005; Shin et al, 2007).
Apart from their well-documented neuroprotective effect in experimental stroke models, Rho kinase inhibitors lower systemic blood pressure potently, which may limit their use for the treatment of acute stroke (Uehata et al, 1997; Shimokawa, 2002; Takahara et al, 2003). Interestingly, however, the ROCK inhibitor hydroxyfasudil, which is neuroprotective, caused hypotension in wild type as well as in eNOS null mice (Shin et al, 2007), suggesting that the blood pressure lowering effect may be mediated by actions independent of eNOS activity. Therefore, the development of ROCK inhibitors with selectivity toward the cerebral circulation or toward ROCK2 (ROCK
Phosphodiesterase Inhibitors
Emerging evidence suggests that phosphodiesterase (PDE) inhibitors such as dipyramidol exhibit neuroprotective effects beyond their well-known antiplatelet properties. It was shown that dipyramidol augmented the neuroprotective effects of statins in an eNOS-dependent way (Kim et al, 2008). The putative mechanism involves an increase in eNOS S1177 phosphorylation, as demonstrated in spontaneously hypertensive rats (Oyama et al, 2011) based on the reduction in postischemic brain damage following pretreatment with PDE inhibitor cilostazol. While these are the first encouraging data suggesting that PDE inhibitors can also increase eNOS activity in stroke patients (Serebruany et al, 2011), clinical use of PDE inhibitors is so far restricted to secondary prevention of ischemic stroke: Two large clinical trials (ESPRIT: European/Australasian Stroke Prevention in Reversible Ischaemia Trial (Halkes et al, 2007), ESPS-2: European Stroke Prevention Study 2 (Diener et al, 1996)) proved that dipyramidole in combination with acetylsalicylic acid (ASA) is more effective in preventing secondary ischemic insults than ASA alone. Use of PDE inhibitors in the early postischemic phase is currently not recommended due to a possibly increased bleeding risk by PDE-induced platelet inhibition. However, a recently completed clinical trial with cilostazole (CAIST: Cilostazol in acute ischemic stroke treatment (Lee et al, 2011)) showed that cilostazol is comparable to aspirine in terms of safety and bleeding complications so further clinical evaluation of cilostazol mono- or combination-therapy in acute stroke is ongoing (Nakamura et al, 2011).
Other Neuroprotective Strategies Influencing the Endothelial Nitric Oxide Synthases Pathway
PI3 K/Akt pathway—S1177 phosphorylation—drugs
Niacin (vitamin D3) was shown to increase the levels of high-density lipoprotein (HDL), thus increasing levels of HDL-cholesterol (HDL-C) and lowering levels of serum triglycerides (Guyton, 1998; Elam et al, 2000). High-density lipoprotein promotes reendothelialization and stimulates migration of endothelial cells (Seetharam et al, 2006) by increasing NO concentration by activation of eNOS activity via the PI3/Akt pathway (Mineo et al, 2003; Drew et al, 2004).
After experimental ischemic stroke niacin (vitamin D3) and niacin derivative niaspan significantly improved functional outcome independently of HDL-C levels due to improved angiogenesis (Chen et al, 2007; Shehadah et al, 2010); again, this is thought to be conferred mainly by eNOS via the PI3 K/Akt pathway.
Niacin and niacin derivative niaspan are potent drugs to increase levels of HDL-cholesterin and thus help to restore vascular function and reduce occurrence of subsequent cardiovascular events without obvious side effect (Guyton et al, 2000; Grundy et al, 2002). However, a recently completed study (AIM-HIGH (Boden et al, 2011)) could not detect significant benefit of niacin treatment in addition to statins compared with statins alone in primary prevention of cardiovascular events; ischemic stroke was recorded as a secondary parameter.
Currently, a randomized double-blinded phase II study is investigating whether niaspan can improve recovery after ischemic stroke (Randomized, Controlled Trial of Extended-Release Niacin (Niaspan, Abbott Laboratories, Abbott Park, IL, USA) to Augment Subacute Ischemic Stroke Recovery, start date: April 2009, NCT00796887).
Another group of compounds known to increase levels of HDL-cholesterol are liver X receptor agonists (Brunham et al, 2006). TO901317, a synthetic liver X receptor agonist, was shown to improve neurological function after MCA occlusion in healthy but not in eNOS-deficient mice (Chen et al, 2009). The suggested neuroprotective mechanism is promotion of angiogensis via increased eNOS phosphorylation, but also antiinflammatory effects are discussed (Morales et al, 2008). Further evaluation of the mechanisms involved are necessary before clinical evaluation.
PI3 K/Akt pathway—S1177 phosphorylation—hormones
Steroids hormones when used in pharmacological doses increase CBF and improve outcome after experimental ischemic stroke (de Court et al, 1994; Bertorelli et al, 1998). This effect is due to a rapid onset of transcription-independent eNOS activity via the PI3/Akt pathway (Limbourg et al, 2002; Hafezi-Moghadam et al, 2002). In an animal model of transient cerebral ischemia, dexamethasone increased CBF and reduced infarct volume for up to 3 days after MCAo in wild type but not in eNOS knockout mice (Limbourg et al, 2002). Despite these promising data, clinical trials yielded mixed results (Patten et al, 1972; Bauer and Tellez, 1973); corticosteroids are therefore not recommended for the treatment of acute stroke (Adams et al, 2007).
Other hormones are proposed to exert their neuroprotective effect by an increase in eNOS activity: Thyroid hormone-mediated neuroprotection is mediated by the PI3/Akt pathway and eNOS phosphorylation after acute stroke (Hiroi et al, 2006). Furthermore, estrogen confers increased eNOS activity via the same mechanism (Haynes et al, 2000; Simoncini et al, 2000). While these results might add to our understanding of gender related differences in stroke incidence and outcome, it remains unclear whether hormone replacement or treatment might be a tool to acutely increase CBF after ischemic stroke. Adiponectin, an adipokine generated by visceral fat cells was shown to increase eNOS phosphorylation. Following MCAO, infarct size was larger in adiponectin-deficient mice, an effect reversed by increasing adiponectin expression in its knockout mouse (Nishimura et al, 2008). Low plasma levels of adiponectin have been implicated in obesity, hypertension, and diabetes.
Although there is ample evidence that eNOS phosphorylation is an important mechanism of action for the hormones mentioned, one has to keep in mind that given the multifaceted and sometimes contrasting effects of hormones eNOS modulation might not be the sole mechanism of action.
Other Therapeutic Strategies Conferring Neuroprotection by Endothelial Nitric Oxide Synthases Activation
There are a variety of other pharmacological and nonpharmacological strategies that seem to exert neuroprotection mainly via modulation of eNOS activity.
Experimental studies proved that exercise increases eNOS activity, thus improving infarct volume and neurological outcome (Endres et al, 2003; Gertz et al, 2006). The mechanism may relate to increased shear stress. In line with these findings, the protective effect of prestroke exercise was absent in eNOS null mice (Gertz et al, 2006). However, there is no conclusive evidence that physical activity can be neuroprotective when started after stroke (Johansson, 2003; Gertz et al, 2006). Nevertheless, the importance of exercise in primary and secondary prevention of ischemic stroke is undoubted.
Among other drugs thought to confer stroke protection via the eNOS/cGMP pathway are the phytoalexine resveratrol, the neuro- and cardio-protectant found in red wine (Tsai et al, 2007), peroxisome proliferator-activated receptor-
Inhaled Nitric Oxide Donors
As noted above, many promising strategies that increase the availability of NO to the vessel wall have distinct problems in the context of acute cerebral ischemia. Nevertheless, a rapidly acting, easily accessible treatment is actively being sought that increases the availability of NO to acutely improve CBF—especially within the penumbra.
Nitric oxide inhalation (inhaled nitric oxide, iNO) has been used to treat diseases characterized by pathological pulmonary vasoconstriction, for example, pulmonary hypertension since the early 1990s. Initially, it was hypothesized that NO was rapidly inactivated in pulmonary vessels once in contact with oxyhemoglobin, thus restricting the vasodilatory effect to the lung. This was consistent with the findings that iNO therapy at moderate doses did not alter systemic vascular resistance and—thus—systemic blood pressure in experimental and clinical studies. Based on this notion and the fact that other significant adverse effects could be excluded, iNO was approved for the treatment of persistent pulmonary hypertension in neonates and later used for other pulmonary conditions (Griffiths and Evans, 2005; Kinsella and Abman, 2005). Over the ensuing decade, evidence was presented that strongly suggested that inhaled NO led to distinct extrapulmonary effects, for example, in mesenteric, renal, and cardiac vessels. Recently, there is growing evidence that iNO might be protective in reducing ischemia–reperfusion injury within mesentery (Fox-Robichaud et al, 1998), and myocardium (Nagasaka et al, 2008), thus reducing tissue damage. While experimental (Rosenberg et al, 1995; Lopes Cardozo et al, 1996; Kusuda et al, 1999) and clinical (Vavilala et al, 2001) studies could not detect changes in CBF during iNO therapy, one experimental study revealed that while it did not alter cerebral perfusion, iNO was shown to possess a cerebrovascular effect (Kuebler et al, 2003). Nitric oxide inhalation is a clinically approved treatment that has few side effects that might positively influence the outcome following ischemic stroke. Terpolilli et al (2011) recently demonstrated an effect of iNO on the cerebrovasculature using
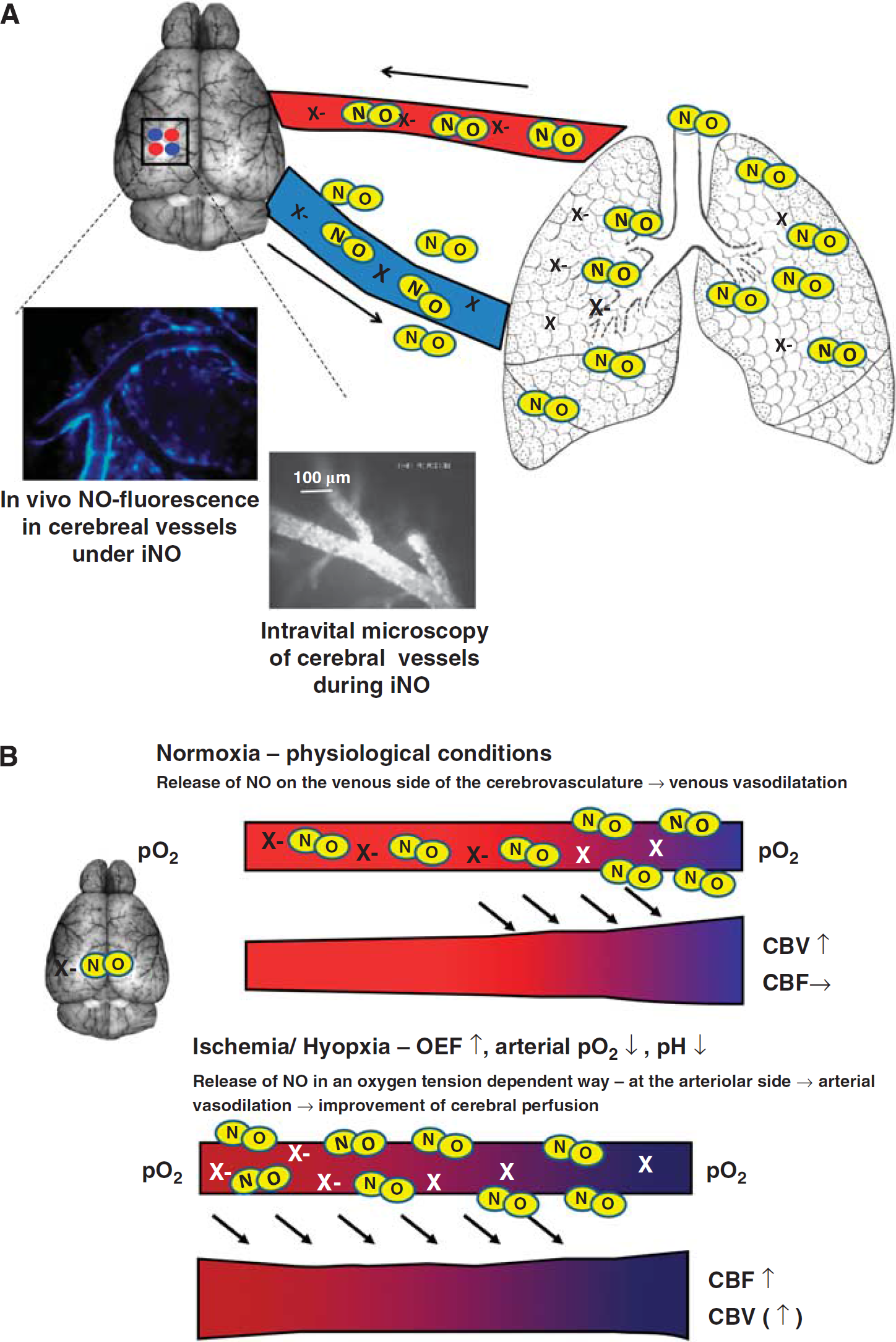
Nitric oxide (NO) inhalation (iNO). (
The extrapulmonary effects of other inhaled NO donating agents such as nebulized sodium nitroprusside (Fattouch et al, 2005), inhaled ethyl nitrite (Auten et al, 2007), and inhaled nitrite (Blood et al, 2011) are being investigated in the context of pulmonary pathologies because the observed effects seem at least in part to be mediated by the same pathways as iNO. In the case of inhaled ethyl nitrite, neuroprotection after experimental subarachnoid hemorrhage has recently been demonstrated (Sheng et al, 2011), so that this might be another promising inhalative treatment strategy for cerebral ischemia.
Conclusion
Reduction in NO signaling pathways have been well documented after ischemic stroke, especially within the blood vessel wall. Many strategies have been used to restore these pathways in order to improve CBF and improve histopathological and functional outcome after cerebral ischemia. While some NO-based therapeutical approaches have been implemented into clinical guidelines for stroke prevention, most strategies for acute stroke treatment are still in the experimental stage and more research is needed to explore novel, side effect-free and specific NO-based therapeutic approaches for cerebral ischemia.
Footnotes
Disclosure/conflict of interest
The authors declare no conflict of interest.