
Abstract
The blood–brain barrier (BBB) plays critical roles in the maintenance of central nervous system (CNS) homeostasis. Dysfunction of the BBB occurs in a number of CNS diseases, including Alzheimer's disease (AD). A prevailing hypothesis in the AD field is the amyloid cascade hypothesis that states that amyloid-β (Aβ) deposition in the CNS initiates a cascade of molecular events that cause neurodegeneration, leading to AD onset and progression. In this review, the participation of the BBB in the amyloid cascade and in other mechanisms of AD neurodegeneration will be discussed. We will specifically focus on three aspects of BBB dysfunction: disruption, perturbation of transporters, and secretion of neurotoxic substances by the BBB. We will also discuss the interaction of the BBB with components of the neurovascular unit in relation to AD and the potential contribution of AD risk factors to aspects of BBB dysfunction. From the results discussed herein, we conclude that BBB dysfunction contributes to AD through a number of mechanisms that could be initiated in the presence or absence of Aβ pathology.
Keywords
INTRODUCTION
Alzheimer's disease (AD) is the most common type of dementia, and affects ~36 million individuals worldwide (http://www.alz.co.uk/research/world-report). The majority of individuals afflicted with AD initially present with symptoms of memory loss and cognitive impairment late in life (≥65 years old). These symptoms increase in severity as AD progresses, often become so debilitating that institutionalization is necessary, and are eventually fatal. Therefore, AD is a disease with tremendous socioeconomic cost. Because of the growing aged population and lack of treatments that can prevent or slow disease progression, future AD prevalence is expected to increase to an extent that it may threaten the sustainability of healthcare systems worldwide. This necessitates a global effort to better understand AD, with the goal of developing treatments that can prevent or slow AD progression.
Much of the focus in AD research has been on amyloid-β peptide (Aβ) and tau, which are the protein constituents of the hallmark senile plaques and neurofibrillary tangles that pathologically define AD. The amyloid cascade hypothesis, initially proposed by Hardy and Higgins in 1992, 1 updated by Hardy and Selkoe in 2002, 2 and still a prevalent focus of AD research today, states that deposition of Aβ in the brain is the initiating event in AD. Although the hypothesis remains generally accepted, it is also increasingly evident that Aβ plays a complex, multifaceted role in AD progression. In rare, familial forms of AD, mutations in the Aβ precursor protein (APP) or in presenilin proteins, the enzymes that cleave Aβ from APP, cause increased Aβ deposition. In the remainder of AD cases, it is thought that Aβ deposition results from deficient clearance. 2 Multiple routes of clearance have been elucidated for Aβ, including enzymatic degradation, 3 bulk flow of cerebrospinal fluid (CSF), 4 and transport across central nervous system (CNS)–blood barriers. 5 These pathways are illustrated in Figure 1, and will be described in detail in later sections. In this review, we will discuss the participation of the blood–brain barrier (BBB) in AD with special focus on its potential roles both upstream and downstream of the amyloid cascade.
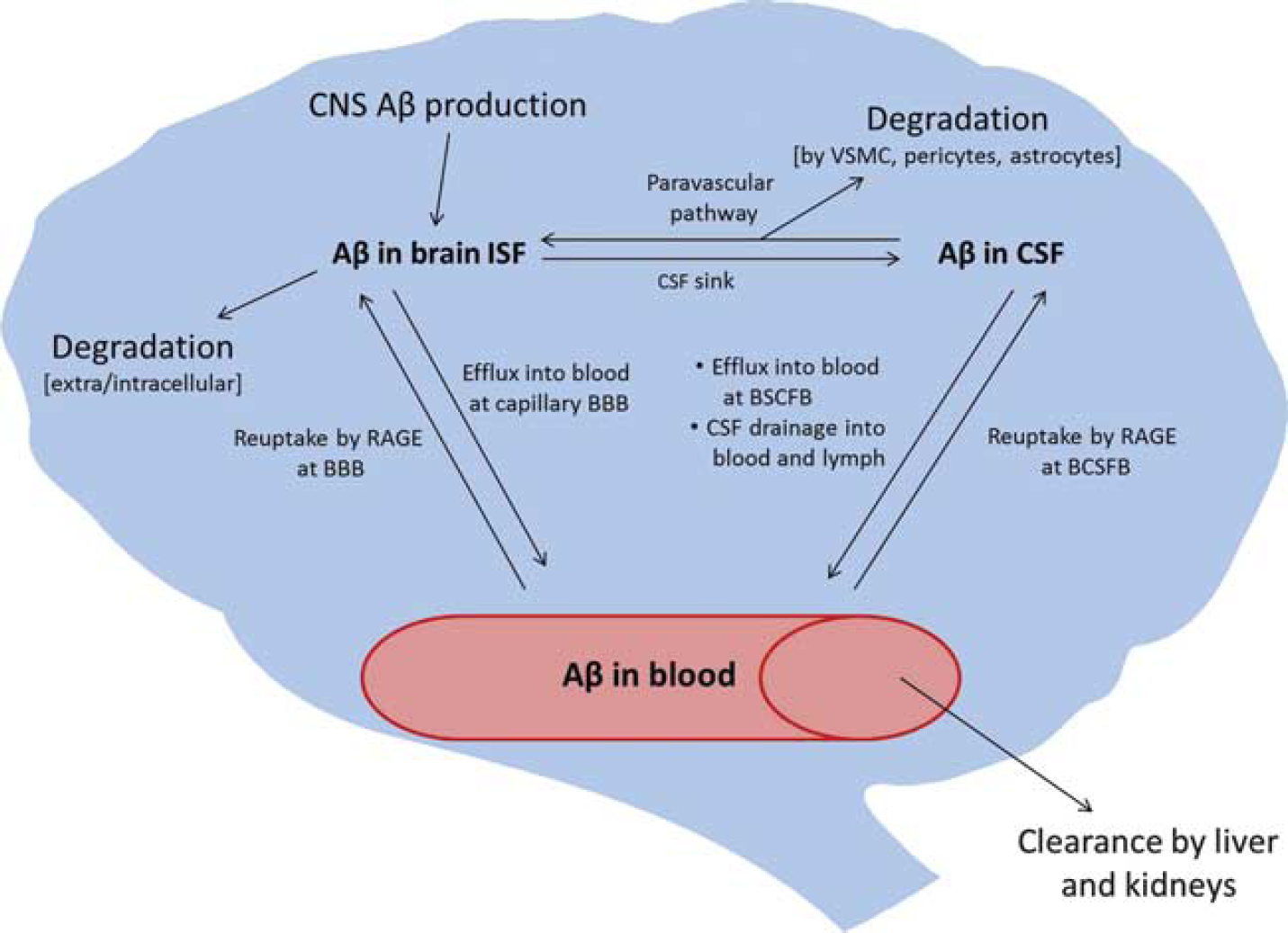
Clearance pathways of amyloid-β (Aβ) from the brain. BBB, blood–brain barrier; CNS, central nervous system; BCSFB, blood–cerebrospinal fluid barrier; CSF, cerebrospinal fluid; ISF, interstitial fluid; RAGE, receptor for advanced glycation endproducts; VSMC, vascular smooth muscle cell.
OVERVIEW OF THE blood–brain BARRIER
Endothelial cells of brain capillaries are the primary anatomic units of the BBB. 6 These cells are highly specialized to support the essential functions of the BBB that include: (1) acting as a diffusion barrier, (2) transporting substances in and out of the brain, and (3) serving as an interface for communication between the CNS and periphery. 7 Barrier properties of brain endothelial cells are conferred through expression of tight junctions at cell-cell contacts, absent fenestrae, degradative enzymes, reduced pinocytosis, and efflux transporters.6,8 Tight junctions also restrict lateral diffusion of membrane proteins, and therefore support transporter function by maintaining their polarity. 6 The BBB transporters play many roles in supporting proper CNS function by allowing selective passage of molecules in and out of the brain. Supportive cells such as astrocytes and pericytes are closely apposed to the capillary endothelium and play essential roles in BBB induction and maintenance.9–13 Neurons and microglia also communicate with BBB endothelial cells. 6 Collectively, these cell types are referred to as the neurovascular unit (NVU). Because the BBB plays critical roles in maintaining CNS homeostasis, its dysfunction contributes to multiple diseases. The dysfunction of BBB includes (1) BBB disruption, which results in leakage of circulating substances into the CNS that can be neurotoxic; (2) transporter dysfunction, which has consequences such as inadequate nutrient supply, buildup of toxic substances in the CNS, and increased entry of compounds that are normally extruded; and (3) altered protein expression and secretions by endothelial cells and other cell types of the NVU that can result in inflammatory activation, oxidative stress, and neuronal damage. All three effects have been reported in AD, and are described in the remainder of this review.
DISRUPTION OF THE blood–brain BARRIER IN ALZHEIMER'S DISEASE
The possibility that the BBB is leaky in AD, that is, it does not prevent the uncontrolled entry into the brain of blood proteins and other molecules, has been investigated for ~30 years. This is clearly an important question as disruption of even a transient or localized nature could have devastating consequences for brain function, inducing a cascade of events involving neurotoxicity, neuroinflammation, and oxidative stress that eventually could produce the AD phenotype. Indeed, some, but not all, animal models of AD exhibit BBB disruption. But to determine whether BBB disruption is actually a feature of AD, one can only consider clinical material.
Disruption of BBB can be readily shown in conditions such as stroke and multiple sclerosis, but showing a disrupted BBB in AD has been more controversial. Many papers have examined the question of BBB disruption in AD, some concluding that disruption occurs and some concluding that it does not. Here, we review selected papers on this topic to give a sense of the problems and surrounding controversies. In general, investigations fall into three categories: comparison of CSF/serum ratios of substances of peripheral origin, usually of albumin; histologic examination for evidence of blood proteins in the CNS; and imaging studies. These will each be considered below.
Albumin Cerebrospinal Fluid/Serum Ratios
The rationale as crystallized by Tibbling
Substances other than albumin have also been used to assess BBB function in AD. Immunoglobulin G levels, either as the CSF/serum ratios or as the IgG index of Tibbling
Not all studies have found increases in CSF/serum albumin ratios. Mecocci
Both Frolich
Histologic Studies
Most of these studies have relied on immunocytochemical techniques to detect the presence of plasma or serum proteins in the CNS. Assuming the proteins are not produced in the CNS, they can be used to indirectly indicate BBB disruption. Wisniewski and Kozlowski
32
examined brains from 7 patients with AD aged 63 to 85 years and 5 nondemented controls aged 29 to 77 years for albumin and immunoglobulin staining. They found light staining for both albumin and immunoglobulins in controls and in the regions of brain with single or no amyloid plaques. In areas with heavy plaque burden, they found intensive staining of the plaques and the neuropile, with the heaviest staining being in the corona of the amyloid plaques. They also found perivascular staining in AD brains that was indicative of BBB disruption. In a later study, Wisniewski
Substances other than albumin and immunoglobulins have been used to suggest BBB disruption. Peptides related or derived from hemoglobin are found in increased concentration in cerebellums from AD patients. 34 These peptides may have arisen from an increased leakage of blood in AD as suggested by the work of De Reuck 35 or by decreased clearance of these peptides by the AD brain. Prothrombin is not produced by the normal brain but shows increased levels in AD brains consistent with leakage across a disrupted BBB. 36 Prothrombin levels in brain were elevated and it was found to be surrounding microvessels. Levels were highest in patients with the highest Braak stage and with at least one ApoE4 allele, but did not correlate with cerebral amyloid angiopathy. However, prothrombin mRNA is expressed by brain under some conditions.37,38 If prothrombin is expressed by the AD brain, these results could be because of defective clearance mechanisms for prothrombin. Claudio 39 found increased vesicularization of brain endothelial cells from AD patients, consistent with increased transcytotic disruption of the BBB.
Others have failed to find evidence for BBB disruption in AD. Alafuzoff
Using fibrinogen and immunoglobulins as markers, Tomimoto
Others have examined histologic material for cerebrovascular lesions in dementias, including AD. One clear finding of these studies is that patients with evidence for only a single cause of dementia such as AD, vascular dementia, or Lewy body disease are a minority. De Reuck
35
examined the occurrence of small cerebral bleeds in various neurodegenerative diseases. He found that ~50% of age-matched nondemented controls had microscopic bleeds. The percent of microscopic bleeds in AD patients was not reported, but in a severity scale of microscopic bleeds, those with AD had a worse score than age-matched controls and those with AD plus cerebral amyloid angiopathy still worse. Deramecourt
Overall, the clearest conclusions from histologic studies are that those with vascular dementia clearly have evidence of small vessel disease including that consistent with BBB disruption, that such evidence is more common in vascular dementia than in AD, that there are cases of AD with little or no evidence of BBB disruption, and that the patients with only a single disease, such as AD or vascular dementia, are in the minority of any study. The evidence also strongly suggests that vascular lesions occur to some extent in nondemented age-matched controls and that studies that rely on immunostaining as evidence of leakage must be particularly careful regarding issues such as time from death to fixation and fixation techniques. Those studies relying on immunostaining should also consider whether the increased detection of the index substance in the CNS is only dependent on its blood-to-brain leakage and not also affected by its induction of CNS production, alteration in clearance from the CNS, or increased CNS retention. Given these considerations, it is difficult to conclude that histologic studies have shown that AD patients without a cerebrovascular component have increased BBB disruption when compared with age-matched controls. However, as the histologic studies show that the majority of AD patients also have evidence of infarct (and the majority of vascular dementia patients have evidence of neurodegenerative disease), the studies suggest that the most relevant clinical question may not be whether the BBB is disrupted in AD, but whether or how neurodegenerative diseases and cerebrovascular disease interact to promote the onset and progression of dementia. Such interactions might not only induce BBB disruption, but contribute to the many other altered BBB functions found in AD.
Imaging
Several studies using various modalities of imaging have addressed the question of whether the BBB is disrupted in AD. Two computed tomography (CT) studies45,46 and a positron emission tomography (PET) study with [68Ga]EDTA
47
did not find increased BBB permeability in AD. A magnetic resonance imaging (MRI) study by Starr
Summary of Results for blood–brain Barrier Disruption The CSF/serum ratios for serum proteins, immunohistochemical evaluations, and imaging modalities have all been useful in showing BBB disruption in a host of clinical conditions, including dementia resulting from infarcts. Application of these techniques to AD has led some to conclude that AD patients, or at least a subset of AD patients, have disruptions of the BBB. Others have not found such evidence, whereas some others have pointed out the difficulties in applying CSF/serum ratios and immunohistochemical approaches to AD. Several MRI, CT, and PET studies have failed to show convincing evidence of BBB disruption in AD. Any method will have a lower limit of detection and any disruption of the BBB, no matter how transient or limited in scope, could be relevant, especially in a susceptible population. To date, however, it is difficult to claim that BBB disruption is a major driver of pathology in AD patients who do not also have a vascular component to their dementia.
DEFECTS IN blood–brain BARRIER TRANSPORTERS THAT MAY CONTRIBUTE TO ALZHEIMER'S DISEASE
Transport of Amyloid-β Efflux
The first observation of Aβ efflux from the brain was by Ghersi-Egea
More recently, it has become evident that other transporters in the BBB facilitating Aβ efflux also become impaired in AD. One example is the multidrug transporter P-glycoprotein (Pgp). Evidence supporting this role for Pgp includes: (1) observations of Pgp-dependent efflux of Aβ
Based on present results, it is conceivable that deficient BBB efflux of Aβ could both initiate and be initiated by the amyloid cascade. Pathologic states such as inflammation, obesity, diabetes, stroke, and others are known to alter the function of many transporters in the BBB. Interestingly, many of these conditions are also considered risk factors for AD. There is evidence that such risk factors alter the function of Aβ transporters in the BBB, and details of this will be described in a later section. More severe defects in Aβ transport are also likely to occur as a result of Aβ accumulation in the CNS. Amyloid-β has a much higher propensity to transition to β-sheet conformation and aggregate as its concentration increases. 70 Optimal BBB efflux occurs for Aβ in its monomeric state, and transporter affinity decreases with increased aggregation/β-sheet content.71,72 Therefore, increased Aβ accumulation in the CNS would preclude its efflux. Amyloid-β also decreases microvascular expression of its transporters. This is evident for LRP-1 and Pgp in transgenic mouse models of AD,63,71 and in nontransgenic mice treated with Aβ. 73 Impaired Aβ efflux is also observed in the SAMP8 mouse model of AD, which derives from a spontaneous mutation found to cause accelerated senescence. 56 These mice show a modest increase in APP and Aβ compared with transgenic models, yet have marked age-associated cognitive impairment.74,75 Evidence supports that the Aβ efflux deficit occurs as a result of Aβ accumulation in this model as well, as antisense oligonucleotide that reduces APP expression and Aβ accumulation also restores Aβ efflux to normal levels. 76 Together, these results suggest that deficits in efflux of Aβ across the BBB feedforward as AD progresses, and unique therapeutic interventions may be necessary at each stage of disease.
Transport of Amyloid-β Influx and Systemic Clearance The amyloid precursor protein is expressed in a variety of tissues other than brain, and low levels of Aβ are detectable in the circulation. 77 Before the realization of an efflux system for Aβ, it was tested whether circulating Aβ could contribute to Aβ deposition in the brain. Studies in rodents showed that a 28-amino-acid fragment, as well as the 40- and 42-amino-acid forms of Aβ, were taken up by capillaries and could completely cross the BBB from the circulation.78–82 Later, the transporter that mediates luminal-to-abluminal transcytosis of Aβ was identified as the receptor for advanced glycation endproducts (RAGE). 83 In transgenic rodent AD models, influx was shown to be a substantial contributor to Aβ deposition; blocking the Aβ/RAGE interaction significantly reduced the appearance of plaques in these models.83,84 Furthermore, RAGE upregulation is observed in the CNS microvasculature of humans with AD, as analyzed by histochemical methods in post-mortem tissue.85,86
Systemic clearance of Aβ is facilitated by the liver and, to a lesser extent, the kidneys and spleen.55,87 Lipoprotein receptor-related protein-1 was shown to be the predominant transporter that mediates clearance by the liver. The short half-life of circulating Aβ (on the scale of minutes),55,87 and the capacity of the liver to clear Aβ at levels far exceeding its physiologic concentration in blood suggest that there is a biologic necessity to protect against elevations in circulating Aβ. 87 This also suggests that rapid systemic clearance of Aβ prevents reuptake by RAGE after efflux. Liver ligation was shown to be an effective method to maintain high levels of Aβ in the circulation up to 1 hour after intravenous injection. In the same study, liver-ligated rats with sustained increased levels of Aβ in blood showed markedly reduced efflux of Aβ from the brain. 88 Despite the results of these studies in rodents, data on plasma Aβ changes in human AD have been contradictory. Increased,89,90 decreased,91,92 or unchanged93,94 circulating levels of Aβ have been reported in clinical studies. Furthermore, it remains unclear whether changes that are detected have any predictive value for cognitive decline.91,92,94
Lack of human evidence showing changes in circulating levels of Aβ in AD suggests that an alternative mechanism is relevant for influx. An early study showed that Aβ entering the brain from the circulation is unbound. 82 Amyloid-β in the circulation is bound to a number of proteins naturally occurring in blood such as albumin, 95 ApoE and ApoJ,96,97 and a soluble form of LRP-1 (sLRP-1). 98 The latter was shown to bind the majority of circulating Aβ, and Aβ bound to sLRP-1 prevents RAGE-dependent influx but enhances systemic clearance. 98 This evidence supports a peripheral sink hypothesis, 99 where binding proteins in serum facilitate systemic clearance of Aβ and prevent its uptake by RAGE. Peripheral sink mechanisms may therefore at least in part explain how therapeutics such as antibodies against Aβ 100 and other binding proteins such as gelsolin 101 and sLRP-1 98 lower Aβ levels in the CNS. Furthermore, individuals with AD have slightly lower circulating levels of sLRP-1, as well as increased oxidative damage to sLRP-1, which markedly lowers its binding affinity for Aβ. 98 This would increase the pool of free circulating Aβ that is available for RAGE-dependent influx. Future studies are necessary to investigate the utility of sLRP-1, both as a plasma biomarker of AD and as an Aβ-lowering therapeutic.
GLUT-1 Transporter, Alzheimer's Disease, and the blood–brain Barrier
Glucose use by brain is decreased in AD, with low glucose metabolism predating clinical symptoms, predicting subsequent decline in cognitive function, and with further decreases in glucose use correlating with those cognitive declines. 102 Glucose uptake from blood to brain is dependent on GLUT-1 expression by the BBB, with GLUT-1 increasing the transport rate of D-glucose across the BBB by ~30- to 100-fold. 103 With the capillary bed comprising ~0.1% of brain weight and the brain using ~20% of the body's glucose, it is not surprising that GLUT-1 is highly expressed by the BBB or that a reduction in GLUT-1 expression is associated with seizures and mental retardation. 104 The brain microvessels in AD, including those from the hippocampus, have a reduction in the expression of GLUT-1 protein,105–107 but not a decrease in GLUT-1 mRNA expression. 108
Decreased expression of GLUT-1 in the BBB raises the question of whether this is because of decreased demand by a dysfunctional brain or whether the brain is dysfunctional in AD because the BBB is not delivering adequate amounts of glucose, essentially starving the brain. The answer to this question is vital: if GLUT-1 activity is a primary defect in AD, then providing more energy to the brain would be the major step to preventing and treating AD. Harik
109
postulated that the decrease in GLUT-1 in the BBB was secondary because no corresponding decrease in GLUT-1 was found in the erythrocytes of the AD patients, with erythrocytes being a readily accessible tissue that contains GLUT-1. This contrasts with De Vivo's disease, an inherited condition in which GLUT-1 is underexpressed in both the BBB and erythrocytes. The brain capillaries of the aged APP/PS1 transgenic mouse have both an absolute decrease in GLUT-1 expression and a decreased density of GLUT-1 that seems to occur after a critical level of Aβ peptide accumulates in brain.
110
In imaging studies of humans, however, glucose use by brain correlates with ApoE genotype but not with fibrillated Aβ peptide load in brain as assessed
In conclusion, GLUT-1 expression and activity is decreased in the BBB of AD patients and in transgenic animal models. Although the reason for this decrease is not known, current thinking seems to lean toward it being in response to the decreased metabolic demand by brain.
CEREBROSPINAL FLUID/INTERSTITIAL FLUID BULK FLOW, BRAIN BARRIERS, AND ALZHEIMER'S DISEASE
The Role of Cerebrospinal Fluid/Interstitial Fluid Bulk Flow in Alzheimer's Disease, and Participation of the blood–brain Barrier
The CSF has many critical functions in the CNS, including physical protection, regulation of intracranial pressure, waste removal, and providing a supportive milieu (for detailed review, see Johanson
The ability of CSF to supply nutrients to and clear waste from the brain ISF depends on its ability to mix with ISF. No barrier exists between ISF and CSF, and therefore solute transfer can occur between both fluid compartments. It has been proposed that a predominant mechanism driving solute transfer is bulk flow of ISF into CSF compartments. In this model, CSF acts as a ‘sink’ for solutes in ISF.117,118 A distinct paravascular anatomic pathway for CSF volume transfer has also been shown,119,120 and a recent study has elucidated a role for aquaporin 4 (AQP4), a water channel that is localized to astrocytic endfeet of the microvasculature, in this pathway.
121
Iliff
Blood-Cerebrospinal Fluid Barrier Transport in Aging and Alzheimer's Disease
The BCSFB, like the BBB, is an active transport interface for a number of peptides between the blood and CSF compartments. Impaired BCSFB function could therefore cause CNS disturbances that would promote AD. This is evident with respect to reduced CSF production in AD, 31 whose consequences on CSF turnover were discussed in the previous section. Evidence also supports, however, that CP epithelial cells play a direct role in regulating levels of Aβ in CSF. Choroid plexus epithelial cells show saturable uptake and complete transcytosis of Aβ that favors efflux in the CSF-to-blood direction. 135 The BCSFB, like the BBB, expresses transporters for Aβ that include LRP-1, 136 LRP-2/megalin, 137 Pgp, 138 and RAGE. 139 The efflux transporters LRP-1 and Pgp in the CP increase with aging, 139 which is opposite to expression patterns observed with aging in the BBB. Similarly, the bidirectional transporter LRP-2 and influx transporter RAGE were decreased and unchanged with age, respectively. 139 These changes were associated with decreased concentrations of Aβ42 in the CP, suggesting that the BCSFB compensates for age-dependent transporter dysfunctions in the BBB. 139 In humans with AD, increased Aβ deposition in the CP is observed. 140 Furthermore, Aβ deposition in CP epithelial cells causes oxidative damage, disruption of the BCSFB, and cell death. 140 This supports that transporter dysfunction and Aβ deposition in the BCSFB is an important pathologic distinction from normal aging and AD, and likely contributes mechanistically to AD pathogenesis.
ALZHEIMER'S DISEASE AND THE NEUROVASCULAR UNIT
Many cell types in addition to brain endothelial cells contribute to the essential function of the BBB, including pericytes, microglia, astrocytes, and neurons. These cell types and the extracellular matrix are collectively referred to as the NVU. 141 This section will discuss how pathologic changes in the NVU in AD could cause BBB dysfunction, and potentially lead to subsequent neurodegeneration.
Pericytes
Among all cell types of the NVU, pericytes have the closest anatomic association with the capillary endothelium. They are located beneath a layer of the basement membrane, and direct communication between pericytes and endothelial cells is possible through gap and peg-and-socket junctions. The ratio of pericytes to endothelial cells in the brain ranges from 1:5 in rats to 1:3 in humans in the CNS, which is much higher than in peripheral tissues.
142
Pericytes are important for induction and maintenance of BBB functions as supported by (1) findings of higher transendothelial electrical resistance and reduced permeability to fluorescein when pericytes are cocultured with brain endothelial cells
Although anatomically distinct, pericytes have many similarities to vascular smooth muscle cells (VSMCs). For example, like VSMCs, pericytes have receptors for vasoactive substances and can regulate capillary diameter and local blood flow. 150 Vascular smooth muscle cells play an important role in Aβ uptake and clearance by degradation, and their ability to clear Aβ is impaired in AD. 122 This function in VSMCs is dependent on Aβ endocytosis by LRP-1 or scavenger receptors,122,123,151 followed by lysosomal degradation.123,151 Whether pericytes have a similar Aβ clearing function is presently unknown, but they do express LRP-1.152,153
Astrocytes
Astrocytes are the most abundant cell type in the brain, and have important functions in the BBB. Their endfeet surround the endothelium of capillaries, arterioles, and venules.
154
At capillaries, the astrocytic endfeet are closely apposed to the endothelium and surrounding basement membrane such that interstitial space is absent.
155
This proximity allows for cross-talk between astrocytes and capillary endothelial cells that promotes the phenotypic specialization of both cell types.
Astrocytes can internalize and degrade Aβ, 159 resulting in reduced Aβ accumulation in the CNS. 160 In AD, astrocytes are found in proximity to Aβ plaques, 161 and perivascular astrocytes accumulate Aβ. 154 Furthermore, in response to Aβ-ApoE complexes, astrocytes from nondemented human brain tissue upregulate their expression of scavenger receptor B1 and neprilysin, which participate in the internalization and degradation of Aβ, respectively. 162 This function is lost in astrocytes derived from AD brains, 162 suggesting that the Aβ clearance function of astrocytes is impaired in AD. Exposure of astrocytes to Aβ also results in astrogliosis, oxidative stress, and impaired glutamate uptake. 163 This has clear negative consequences for neuronal survival, 164 but the contribution of Aβ-induced astrocytic changes to BBB function is less clear. It has been shown that astrocyte changes precede widespread amyloid pathology and BBB disruption in a mouse model that develops plaques and CAA. 165 This suggests that Aβ-induced astrocyte changes in AD may contribute to BBB dysfunction, but the mechanisms by which this occurs have yet to be determined.
Microglia/Perivascular Macrophages
The resident immune cells of the brain are microglia that derive from monocyte populations during embryonic development, but may also be replenished by circulating monocytes that enter the brain during adulthood. 166 Microglia constantly survey the CNS, 167 and transition to an activated phenotype on contact with an immune stimulus. Activated microglia interact with the BBB through secretions of cytokines and vasoactive substances, and physically shield blood vessels after injury. 167 They are therefore an important constituent of the neurovascular unit. In AD, activated microglia are found surrounding amyloid plaques, 168 and microglia activation correlates with cognitive impairment in living AD patients. 169 Microglia activation has somewhat conflicting roles in AD. Proinflammatory molecules secreted from activated microglia could result in direct or indirect neurotoxicity. The BBB may contribute to this indirect neurotoxicity through disruption, secretions, and/or aberrant transport in response to microglial activation. The details of AD-related BBB changes in response to inflammation will be discussed in a later section. Conversely, activated microglia can promote Aβ clearance through phagocytosis.168,170 Phagocytosis of Aβ was shown to be a property of a specialized subset of brain macrophages that are recruited from the circulation, migrate to parenchymal plaques, and inhibit plaque deposition.170,171 Perivascular macrophages also phagocytose Aβ, and protect against vascular deposition of Aβ in a mouse model of cerebral amyloid angiopathy. 172 Furthermore, peripheral blood mononuclear cells from individuals with AD show inefficient phagocytosis of Aβ compared with healthy controls. 173 Together, these data support that deficient phagocytosis of Aβ by brain macrophages could contribute to Aβ deposition in AD. Although participation of the BBB in recruitment of these cells into the brain is implicit, the mechanistic details of this process are presently not well understood.
Neurons
The brain is an organ with an exceptionally high metabolic demand, and is highly vascularized to meet such a demand. This allows for neurons to regulate their own blood supply according to their metabolic needs through communication with the endothelium. Dysregulation of this communication (neurovascular uncoupling) is thought to promote AD, and this topic has been extensively reviewed in a previous issue of this journal.
174
Therefore, this section will discuss neurovascular cross-talk in the context of how soluble factors produced in the BBB in AD influence neurodegeneration. A substantial amount of information on factors whose expression and/or secretion is altered in the BBB in AD has been elucidated by Grammas
ALZHEIMER'S DISEASE RISK FACTORS AND THE blood–brain BARRIER
Despite the continued focus on Aβ and tau in AD, it is increasingly evident that other pathologic events and conditions such as inflammation, oxidative stress, ApoE4 genotype, diabetes/metabolic syndrome, head injury, and vascular pathologies are associated with AD. This section will consider these changes in the context of BBB participation, and their potential contribution to AD neurodegeneration.
Inflammation/Oxidative Stress
Evidence supports that both inflammation and oxidative stress are early events in AD184,185 and activation of inflammatory and oxidative stress pathways in the BBB recapitulate many of the AD-associated deficiencies described in previous sections. The BBB endothelial cells respond to inflammatory stimuli such as cytokines,
186
LPS,
187
,
188
Aβ,189–191 and a truncated tau fragment
192
by activating signaling pathways that result in the upregulation of proinflammatory second messengers and reactive oxygen/nitrogen species, ultimately causing BBB disruption and paracrine activation of surrounding cells that are responsive to such stimuli, such as astrocytes
193
and pericytes153,194 of the NVU. Inflammation impairs systemic clearance of Aβ by the liver and kidneys, as well as bulk flow of CSF.
195
Furthermore, BBB transporter changes occur in response to inflammation and oxidative stress that can promote the accumulation of Aβ in the brain. Increased oxidative damage to LRP-1 in the CNS and circulation has been observed in human AD.98,196 In a mouse lacking the antioxidant vitamin E, LRP-1-dependent brain efflux and systemic clearance of Aβ are impaired.
197
Both LRP-1 and Pgp are functionally downregulated in the BBB in mice with systemic inflammation induced by LPS,66,198 and this corresponds with impaired Aβ efflux.66,198 In contrast to AD, decreased function of these transporters after LPS was not attributed to reduced protein levels or oxidative damage.
66
Despite this, the antioxidant
Apolipoprotein E Genotype
Apolipoprotein E is an apolipoprotein that facilitates lipid transport. Humans have a polymorphic APOE allele that results in three isoforms: APOE2, APOE3, and APOE4. The predominant APOE allele in human populations is APOE3, with mean frequency at 77.9%, whereas APOE4 and APOE2 are substantially less abundant, at 13.7% and 8.4%, respectively.
199
The APOE4 allele is the strongest genetic risk factor for nonfamilial AD.
200
The influences of ApoE4 on CNS function and AD pathogenesis have been described in a recent review,
201
and this section will primarily consider functional differences of ApoE isoforms in the context of their influence on the BBB that are relevant to AD. The ApoE production occurs in both the CNS and peripheral compartments, where it is made primarily by astrocytes and the liver, respectively.
201
An early pharmacokinetic study in guinea pigs showed that circulating ApoE does not cross the BBB,
202
and all three ApoE isoforms limit the influx of circulating Aβ.202,203 Unlike Aβ in complex with ApoE2 and 3, circulating ApoE4-Aβ complexes can cross the BBB.
202
All isoforms of ApoE show significant efflux across the BBB, although at slower rates than both Aβ40 and Aβ42.
204
Furthermore, the efflux rate of ApoE4 across the BBB is slower than that for ApoE2 and 3.
204
Efflux of ApoE2 and 3 is dependent on LRP-1 and very-low-density-lipoprotein receptor (VLDLR), whereas ApoE4 efflux is primarily through VLDLR and is LRP-1 independent.
204
When in complex with Aβ, all three isoforms of ApoE reduce the efflux rate of Aβ.72,204 This reduction is most pronounced for ApoE4-Aβ complexes, which was attributed to the slower endocytosis rate of VLDLR versus LRP-1.
204
Despite these findings for ApoE complexed to Aβ
Apolipoprotein E also plays a role in maintenance of BBB integrity. This was first shown
Diabetes/Metabolic Syndrome
Diabetes mellitus is hyperglycemia that results from insufficient insulin action, the insufficiency caused by either decreased insulin production by pancreatic β-cells (type 1 diabetes) or by insulin receptor resistance (type 2 diabetes). Both types of diabetes are associated with cognitive decline and an increased risk of developing AD.209,210 One probable factor linking these diseases is insulin resistance in the CNS. 211 Under physiologic conditions, CNS insulin signaling regulates feeding and cognition. 212 Therefore, some have proposed that AD should be considered ‘type 3 diabetes.’ 213
Because of a lack of detectable insulin production in the CNS with the exception of a small group of cells in the olfactory mucosa, 214 CNS insulin is derived primarily from the circulation through BBB transport. 215 Pharmacokinetic studies of insulin uptake by the brain confirm that insulin crosses the BBB by a saturable transport system.216–218 Insulin transport across the BBB increases in a rodent model of diabetes 219 and associated pathologic factors such as inflammation 220 and triglycerides. 221 However, insulin transport is decreased in obese dogs and rodents.221–223 Together, these results indicate that multiple conditions of the metabolic syndrome influence insulin transport across the BBB. The BBB therefore plays a likely role in mediating CNS insulin resistance, but the precise mechanisms of this are presently unclear.
It is also evident that altered BBB functions in the diabetic state recapitulate many of those observed in AD. Insulin transport in the BBB is increased in a transgenic mouse model of AD. In a rodent model of diabetes, LRP-1-dependent BBB efflux of Aβ is impaired, 224 whereas RAGE-dependent influx of Aβ increases. 225 However, Pgp function is unaltered. 226 Disruption of BBB occurs in a rodent model of diabetes, 227 and has also been reported in diabetic humans. 228 One factor contributing to BBB disruption is pericyte loss 229 that occurs from hyperglycemia-induced mitochondrial oxidative stress in a rodent model of diabetes. 149 Pericyte loss in this model is blocked by the carbonic anhydrase inhibitor topiramate that shifts glucose metabolism to aerobic glycolysis and prevents generation of superoxide. 149 Together, these results suggest that diabetes causes BBB dysfunction that could contribute to AD.
Traumatic Brain Injury
Traumatic brain injury (TBI) increases the risk for developing AD by 2- to 4.5-fold, depending on severity of the injury.
230
Individuals who die from TBI have elevated Aβ plaques
231
and tau pathology is evident in those who have experienced repeated head injuries such as boxers, football players, and combat veterans.
230
Disruption of BBB occurs as an early response to TBI, but resolves by 1 week after injury.
232
Endothelial cell activation, tight junction disruption, and migration of pericytes away from brain capillaries occur within this time period.148,232 Recently, Pop
Cardiovascular Disease
Cardiovascular disease is a risk factor for AD, and is associated with other AD risk factors such as diabetes and obesity. 230 Furthermore, cerebrovascular disease is often evident in AD. 235 Ischemia caused by cerebrovascular disease can cause Aβ accumulation, inflammation, and oxidative stress in the CNS that trigger BBB dysfunction. 236 In a rodent model of brain ischemia induced by microsphere embolism, microvascular accumulation of Aβ occurs and coincides with parenchymal Aβ deposition and tau hyperphosphorylation. 237 This raises the question of whether vascular pathologies themselves contribute to AD. 238
CONCLUSION
From the information presented in this review (summarized in Table 1), it is evident that Aβ accumulation in the CNS can be both a cause and consequence of BBB dysfunction in AD. However, other Aβ-independent pathologies can phenotypically mimic BBB dysfunction observed in AD, and many changes in the BBB cause neurotoxicity independently of Aβ. This highlights the complexity of AD, and the likelihood that AD has diverse etiologies that converge on Aβ and tau. A lack of clearcut evidence of BBB disruption in AD necessitates the consideration of other critical functions of the BBB that may become impaired in AD, such as altered transport and communication within the neurovascular unit. We conclude that the BBB plays a multifaceted role in AD both upstream and downstream of the amyloid cascade, and is therefore important to consider in future efforts towards understanding this devastating and widespread disease.
Potential causes and consequences of proposed BBB dysfunction in AD
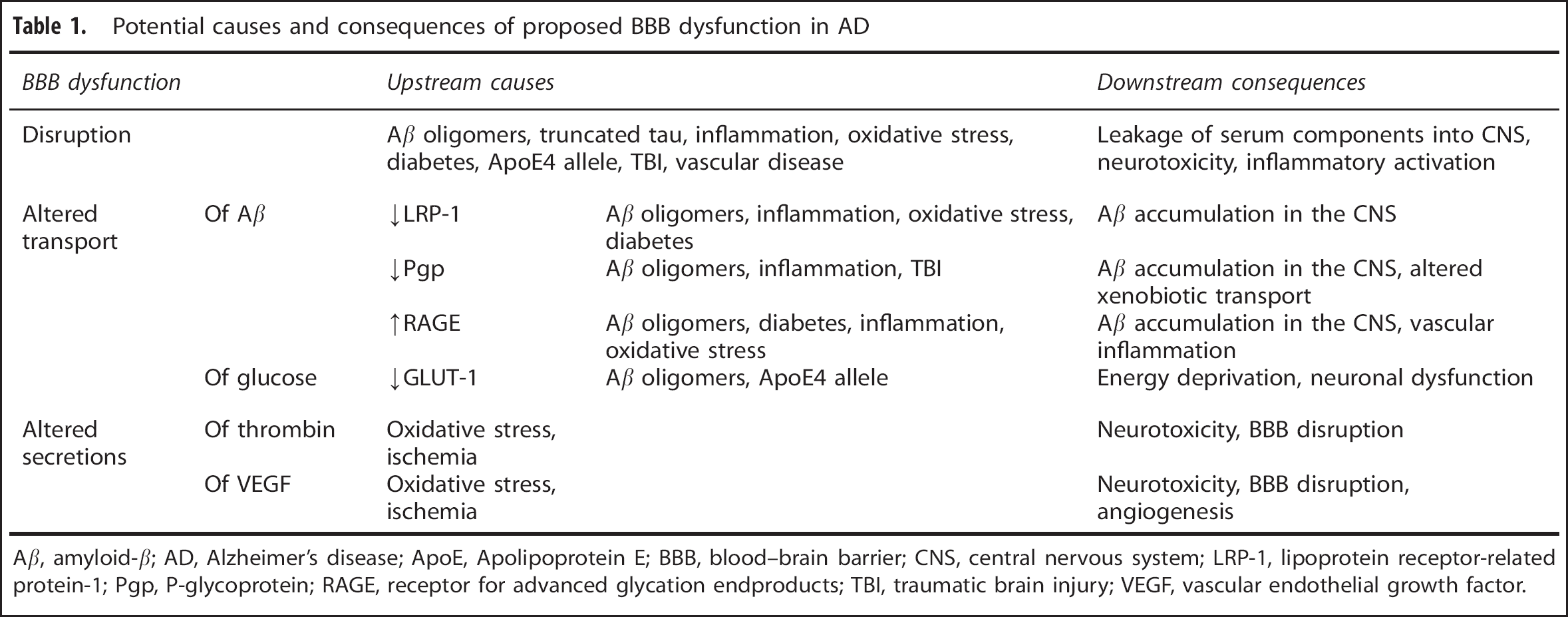
Aβ, amyloid-β; AD, Alzheimer's disease; ApoE, Apolipoprotein E; BBB, blood–brain barrier; CNS, central nervous system; LRP-1, lipoprotein receptor-related protein-1; Pgp, P-glycoprotein; RAGE, receptor for advanced glycation endproducts; TBI, traumatic brain injury; VEGF, vascular endothelial growth factor.
Footnotes
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
We thank Ms Emily Wing for assistance with figure preparation.