
Abstract
Pathophysiology of the neurovascular unit (NVU) is commonly seen in neurological diseases. The typical features of NVU pathophysiology include tissue hypoxia, inflammatory and angiogenic activation, as well as initiation of complex molecular interactions between cellular (brain endothelial cells, astroctyes, pericytes, inflammatory cells, and neurons) and acellular (basal lamina) components of the NVU, jointly resulting in increased blood–brain barrier permeability, brain edema, neurovascular uncoupling, and neuronal dysfunction and damage. The evidence of important role of the brain vascular compartment in disease pathogenesis has elicited the debate whether the primary vascular events may be a cause of the neurological disease, as opposed to a mere participant recruited by a primary neuronal origin of pathology? Whereas some hereditary and acquired cerebral angiopathies could be considered a primary cause of neurological symptoms of the disease, the epidemiological studies showing a high degree of comorbidity among vascular disease and dementias, including Alzheimer's disease, as well as migraine and epilepsy, suggested that primary vascular pathology may be etiological factor causing neuronal dysfunction or degeneration in these diseases. This review focuses on recent hypotheses and evidence, suggesting that pathophysiology of the NVU may be initiating trigger for neuronal pathology and subsequent neurological manifestations of the disease.
Keywords
Neurovascular unit anatomy to function: overview of recent progress
Although the concept of the neurovascular unit (NVU) arose from long-known and studied phenomenon of coupling between neuronal activity (energy demand) and local blood flow (energy supply), in the last decade, the term came to symbolize and promote the research into the integrated
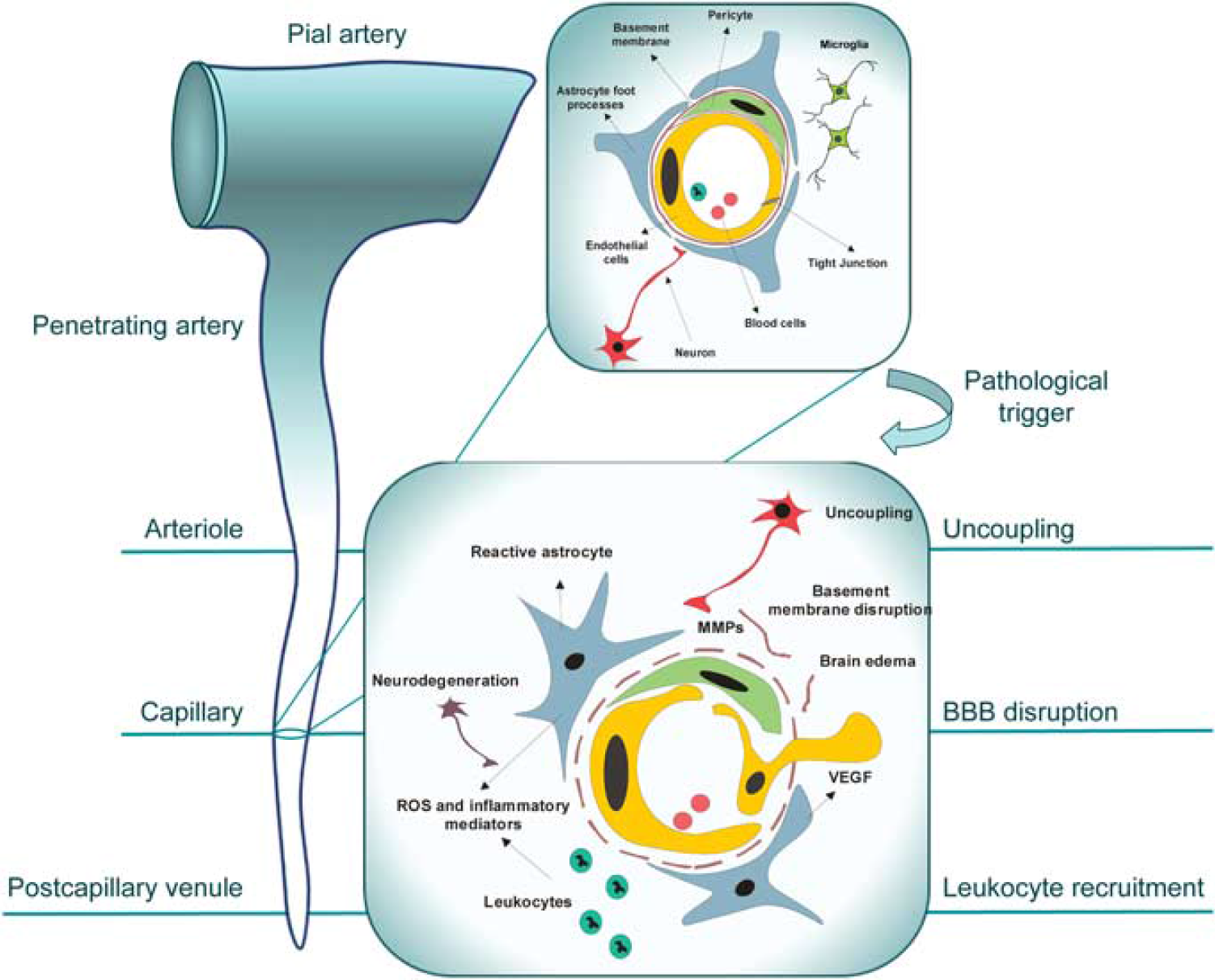
Neurovascular unit (NVU) reorganization in response to pathogenic stimulus. Highly structured multicellular anatomy of the NVU (shown in the upper right inset) undergoes profound changes in response to pathogenic stimulus, such as tissue hypoxia, schematically shown in the lower inset. The subsequent ‘sequence of events’ leading to neuronal injury includes the expression and release of pro-angiogenic factors, such as vascular endothelial growth factor (VEGF), by astrocytes and surrounding cells and upregulation of their receptors in brain endothelial cells, stimulating endothelial proliferation and migration with consequent disruption of tight junctions and increased blood–brain barrier (BBB) permeability. Release of metaloproteases by migrating endothelial cells and pericytes leads to proteolytic disruption of the basement membrane and additional release of pro-angiogenic breakdown products of the extracellular matrix. Accompanying influx of serum proteins and water through disrupted BBB results in vasogenic edema, which further disconnects cellular interactions within the NVU; toxic serum components cause astrocyte activation. Upregulation and secretion of inflammatory mediators from both activated astrocytes and endothelial cells stimulates the expression of adhesion molecules in endothelial cells and the recruitment of inflammatory cells into the brain. Reactive oxygen species and proteases released from leukocytes and activated perivascular cells cause oxidative injury to neurons. Secondary injury to neurons, if prolonged or repeated, could cause dissociation of neuronal projections from the NVU, uncoupling and subsequent retrograde degeneration. Some of the described events and their manifestation occur at specific sites of the brain microcirculatory tree: uncoupling at the level of arterioles, BBB disruption at the level of capillaries, and leukocyte recruitment at the level of postcapillary venules.
In the last decade, a significant progress has been made in the understanding of the role of different cellular constituents of NVU and their intercellular and molecular interactions and signaling during development, physiological, and pathological conditions. During development, the neuroepithelium signals to the vascular endothelium through the canonical Wnt/
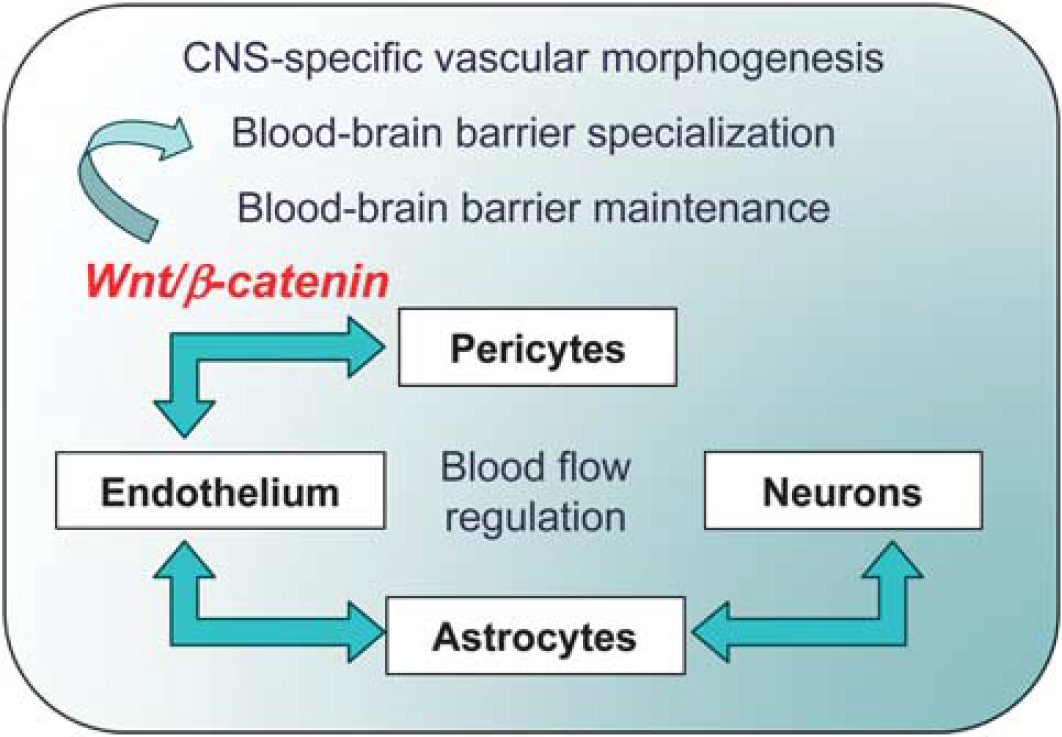
Cellular interactions implicated in the development, maturation and functional responses of the neurovascular unit (NVU). Current understanding places pericyte–endothelial cell interactions, mediated via canonical Wnt/
This accelerated understanding of molecular mechanisms governing NVU functions has aided in understanding the pathophysiology of brain diseases. It has become apparent from these studies that NVU is not simply a ‘passive responder’ but rather an active participant in pathogenesis of virtually all brain pathologies, ranging from chronic neurodegenerative diseases to brain tumors and infections (for extensive list, see Neuwelt et al, 2011). This has recently elicited the debate whether primary vascular events may be a cause of the neurological disease, as opposed to being a mere participant in disease pathogenesis recruited by a principal neuronal origin of pathology? This article aims to further this debate by summarizing current hypotheses, experimental and clinical evidence in support of vascular origin for some CNS pathologies. If indeed specific CNS diseases originate from primary NVU pathology which then causes neuronal dysfunction, then, conceptually, the diagnostic and treatment strategies should be (re)focused to chiefly address/target vascular component of the disease, avoiding often insurmountable problems of drug delivery across the BBB and side effects associated with targeting neuronal compartment(s).
Neurovascular unit pathophysiology: common molecular features
The pathophysiology of the NVU, whether a source or participant in brain disease, could be scoped through the lens of common molecular/functional processes that are observed across the neurological disease spectrum. Perhaps, the most prevalent characteristics of the NVU pathophysiology detected at various degrees of interplay in many neurological diseases, where they combine with disease-specific pathologies (e.g., A
Tissue hypoxia is often an initial trigger of a cascade of pathophysiological changes in the NVU. Tissue hypoxia could be caused by nonvascular mechanisms (e.g., altitude disease) or by acute or chronic processes affecting brain vasculature, including common cardiovascular risk factors such as atherosclerosis and hypertension. In hypoxic tissues, immediate early genes that encode many functionally different products including secreted pro-inflammatory cytokines and chemokines, cytoplasmic enzymes such as cyclo-oxygenase-2, and inducible transcription factors, are promptly induced in a protein synthesis-independent manner. Hypoxia-inducible factor-1 (HIF-1) is a transcription factor that is specifically activated by hypoxia by both modifications in protein processing and by transcriptional induction (Semenza, 2011). Hypoxia-inducible factor-1 is a heterodimeric protein composed of HIF-1
Hypoxia-inducible factor-1
Angiogenic NVU remodeling initiated by the secretion of VEGF and modulated by inflammatory milieu involves dynamic interactions of proliferating cells with the extracellular matrix mediated by specific adhesion molecules, integrins, transmembrane receptors composed of different heterodimeric combinations of
In summary, the process of NVU reorganization in response to tissue hypoxia and inflammatory activation (schematically shown in the lower inset of Figure 1) includes the disruption of interendothelial tight junctions, retraction of pericytes from the abluminal surface of the capillary, breakdown of the basal lamina with transudation of plasma, infiltration of inflammatory cells, endothelial cell proliferation and migration, and in some cases formation of new vessels through angiogenesis and remodeling. At the molecular level, this reorganization is accompanied by increased expression of endothelial cell-leukocyte adhesion receptors, loss of endothelial cell and astrocyte integrin receptors, loss of their matrix ligands, expression of members of several matrix-degrading protease families, and the appearance of receptors associated with angiogenesis and neovascularization (del Zoppo, 2010). This remodeling causes profound functional changes in the NVU unit, including BBB dysfunction, impaired neurovascular coupling, leukocyte adhesion and infiltration, pro-thrombotic conversion, angiogenesis and vasculogenesis. Although initiated as adaptive response aiming to limit injury and promote recovery, in the environment of neurological disease, this adaptive process can be either interrupted or pathologically perpetuated, leading to amplification of initial pathology.
The relationship of the NVU reorganization initiated by the noxious stimulus to subsequent acute or chronic neuronal injury will be further examined in this article using examples from experimental and clinical studies. The issue in focus will remain: can primary vascular pathophysiology elicit and sustain neuronal pathophysiology that produces neurological manifestations often identified as ‘disease,’ such as for example Alzheimer's disease?
Vasculopathies and neurological phenotype
Epidemiological studies have recently provided strong evidence of cooccurrence of neurological manifestations such as migraine, dementia, and mood disorders with primary genetic vasculopathies, most notably cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL), as well as acquired small vessel disease.
CADASIL is the hereditary disease of small cerebral arteries that affects middle-aged adults and leads to disability and dementia, which accounts for 11% of lacunar stroke cases with leukoaraiosis in patients younger than 50 years (Chabriat et al, 2009; Herve and Chabriat, 2010). The disease is essentially characterized by five main symptoms: migraine with aura, subcortical ischemic events, mood disturbances, apathy, and cognitive impairment. The most frequent manifestations of CADASIL, occurring in 60% to 85% of patients, are transient ischemic attacks and subcortical ischemic strokes, in most cases in the absence of conventional vascular risk factors (Chabriat et al, 2009; Herve and Chabriat, 2010). Microscopic ultrastructural studies show a specific arteriopathy affecting mainly the small penetrating cerebral and leptomeningeal arteries characterized by a thickening of the arterial wall leading to luminal stenosis, accumulation of a nonamyloid granular osmiophilic material in the BM, and prominent morphological alterations of smooth muscle cells with their eventual disappearance (Chabriat et al, 2009; Herve and Chabriat, 2010). Clinical and neuroimaging features resemble those of sporadic small artery disease (Jouvent et al, 2008). CADASIL is autosomal dominant disease caused by mutations in NOTCH3 (Notch homolog 3), which encodes a transmembrane receptor primarily expressed in systemic arterial smooth muscle cells (Monet-Leprêtre et al, 2009). Pathogenic mutations alter the number of cysteine residues in the extracellular domain of NOTCH3, which accumulates and forms aggregates around smooth muscle cells and pericytes of brain arteries and capillaries (Monet-Leprêtre et al, 2009). Recent studies suggest that CADASIL mutations produce gain of novel function(s) of mutated protein arising from novel protein–protein interactions rather than a loss of its canonical function (Yamamoto et al, 2011). On the basis of genetic, clinical, and imaging studies, the proposed pathogenic mechanism of the disease arising from both structural and functional changes in brain arteries centers on an early decrease in the CBF with chronic subcortical ischemia. Functional studies have indicated a blunted increase in CBF response to carbon dioxide inhalation in patients with CADASIL (Pfefferkorn et al, 2001). This chronic subcortical ischemia can cause recurrent lacunar infarcts and microstructural alterations that correlate with cognitive decline (Chabriat et al, 2009; Herve and Chabriat, 2010).
By contrast, the pathophysiology of another common symptom of CADASIL, migraine with aura, is mostly unknown. Migraine with aura generally starts long before the first ischemic attacks and before changes on magnetic resonance imaging are detectable (Stam et al, 2009; Chabriat et al, 2009). Furthermore, migraine with aura is not seen in chronic, hypertension-related small artery diseases of the brain, which suggests specific mechanisms for migraine with aura in CADASIL.
Cortical spreading depolarization, a slowly propagating wave of neuronal and glial depolarization that can be evoked in the cortex, cerebellum, basal ganglia, thalamus, and hippocampus, is a putative biologic substrate for migraine aura (Charles and Brennan, 2009). Indeed, migraine visual aura and spreading depolarization share several characteristic functional magnetic resonance imaging findings including that both are associated with an initial hyperemia lasting 3 to 4.5 minutes, which is followed by mild hypoperfusion lasting 60 to 120 minutes (Charles and Brennan, 2009). The hypothesis that
Experimental and epidemiological evidence suggested that hypoperfusion disorders, stroke, and microemboli reduce the threshold for the induction of spreading depolarization (Dalkara et al, 2010). Whereas migraine aura and ischemic stroke share spreading depolarization as a common element, they differ fundamentally in that the ischemic stroke is typically associated with nonspreading or persistent depression, with sudden multiple neurological deficits, whereas in migraine aura slowly spreading depression of spontaneous activity correlates with clinical presentation of slowly creeping neurological deficits that successively affect various functions such as vision, language, somatosensory, or motor function (Dreier, 2011).
Spreading depolarization, induced by acute neuronal hyperexcitability, normally stimulates the NVU to respond with marked vasodilatation and spreading hyperemia (normal neurovascular coupling) to match the increased neuronal energy demand. In the healthy tissue, spreading depolarization does not cause neuronal damage (Nedergaard and Hansen, 1988), because it is coupled with reactive hyperemia. In contrast, under pathological conditions, NVU may respond to spreading depolarization with marked vasoconstriction and spreading ischemia (inverse neurovascular coupling), leading to widespread secondary neuronal injury (Dreier et al, 1998, 2001; Shin et al, 2006). Direct recordings from the surface of the brain from patients with subarachnoid hemorrhage, traumatic or ischemic brain injuries support the high frequency of spreading depolarization waves in these patients, and indicate that they may be associated with ‘inverse’ neurovascular coupling increasing the likelihood of cortical damage (Lauritzen et al, 2011; Dreier et al, 2009). Inverse neurovascular coupling and spreading ischemia may explain the clinical syndrome of migraine stroke (Dreier, 2011).
Whereas pathogenesis of the third common symptom of hereditary and acquired vasculopathies, mood disorders, is far from being clear, a recent hypothesis (Shalev et al, 2009) proposed that a focal BBB breakdown triggers events within the NVU associated with dysfunction of brain astrocytes and local inflammatory response, leading to pathological synaptic plasticity, increased network connectivity with manifestations of psychiatric illness (see below). Observations of increased markers of the BBB breakdown, CSF albumin and serum S100
Neurovascular hypothesis of Alzheimer's disease: or is it vasculoneuronal?
Acquired cerebrovascular disease is common in several cognitive disorders, including multiinfarct dementia, poststroke dementia, mild cognitive impairment, degenerative dementias such as Alzheimer's disease (AD) and others (Iadecola et al, 2010; Iadecola, 2010; Humpel and Marksteiner, 2005). Dementia is more likely to be present when vascular and AD lesions coexist, and association between stroke and AD in elderly individuals is especially strong in patients with vascular risk factors (Jellinger, 2008; Iadecola, 2010). Pathologically, cerebral amyloid angiopathy is found in both AD and cerebral atherosclerosis and is associated with the development of cognitive deficits (Iadecola, 2010; Jellinger, 2008; Humpel and Marksteiner, 2005). These epidemiological observations and experimental studies have started challenging the common view that ‘the earliest manifestation of AD is synaptic failure,’ (Selkoe, 2002) and that, consequently, the disease is inherently neuronal, and have brought about an alternative, vascular hypothesis of AD.
The vascular hypothesis of AD proposes that vascular risk factors including hypercholesterolemia, hypoglycemia, hypertension, etc. damage the NVU during the process of aging, leading to chronic hypoperfusion, BBB dysfunction, and common NVU pathophysiological responses described in ‘Neurovascular unit pathophysiology: common molecular features’ (Figure 2) (Zlokovic, 2010; Iadecola, 2010). Dysregulation and uncoupling of NVU due to disassembly of cellular architecture and dissociation of cholinergic nerve terminals establishes vicious circle, leading to degeneration of nerve endings and retrograde death of cholinergic neurons. These processes also impair the BBB functions, diminishing its ability to clear soluble brain
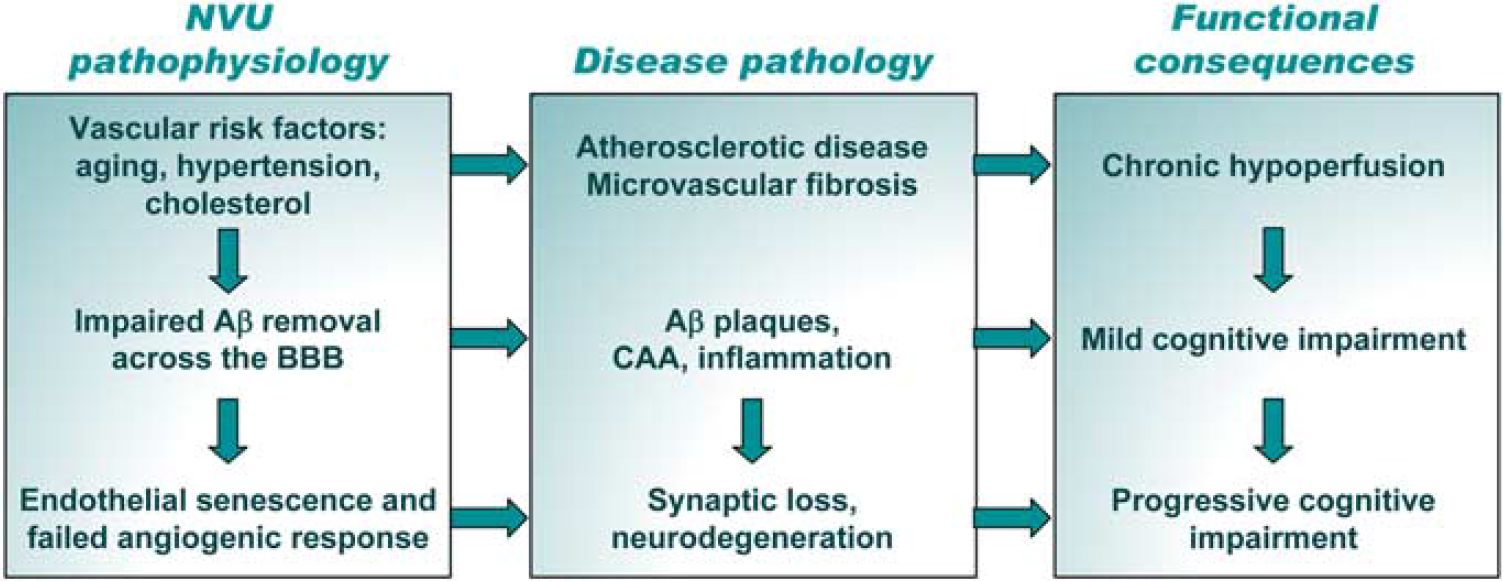
Vascular hypothesis of neurodegeneration in Alzheimer's disease. Early triggers of homeostatic misbalance are common vascular risk factors, including age, hypertension, and cholesterol, leading to atherosclerotic disease and microvascular fibrosis. Functional consequence of these changes is chronic hypoperfusion that initiates neurovascular remodeling cascade. While the aberrant clearance of amyloid-
Several elements of the vasculoneuronal hypothesis of AD are supported by strong experimental evidence obtained in animal models. Morphological and architectural analyses of cerebral vasculature using scanning electron microscopy of brain vascular corrosion casts (Meyer et al, 2008) showed that significant microvascular alterations, often accompanied by small amyloid deposits attached to the vessels, could be detected in APP23 transgenic mice at young ages before the appearance of parenchymal amyloid plaques. In older animals, vasculature abruptly ended at amyloid plaques, resulting in holes in microvascular and capillary network; between such holes, the surrounding vascular array appeared more dense and showed features typical of angiogenesis (Meyer et al, 2008).
Vascular insufficiency with underlying tissue hypoxia can accelerate A
Mechanistic studies have generated a substantial body of evidence that brain accumulation of A
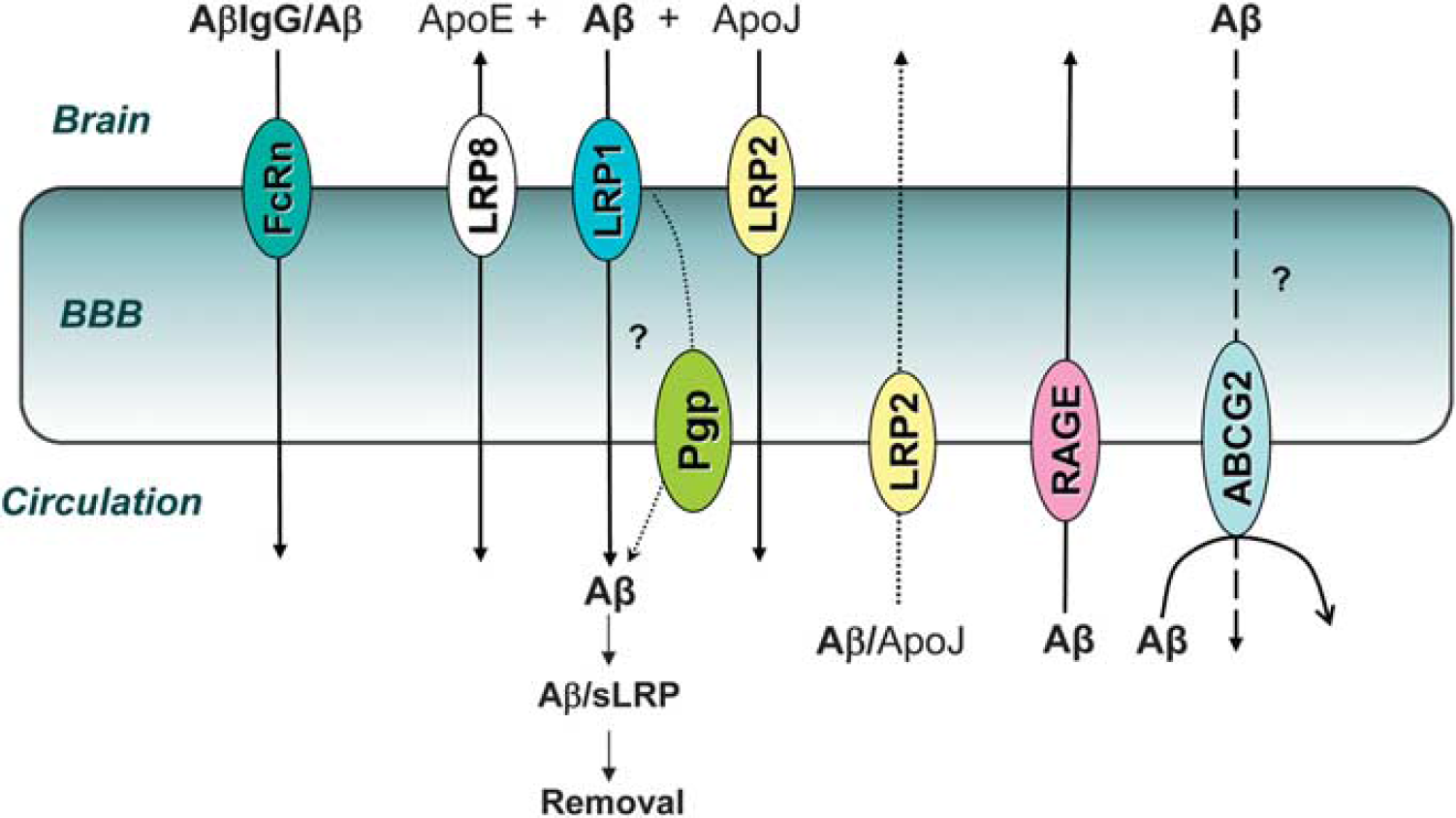
Current understanding of amyloid-
Passive and active immunotherapy against A
Nevertheless, the question whether AD is principally a vascular disease remains difficult to answer based solely on pathological and clinical evidence of association. Recent comparative studies in two animal models of AD-like pathology attempted to dissect whether cerebrovascular insufficiency/chronic brain hypoperfusion is causally linked to functional manifestations of the disease, most notably cognitive impairment (Nicolakakis et al, 2011; Nicolakakis and Hamel, 2011). In aged mice transgenically overexpressing TGF
Evidence for blood–brain barrier dysfunction as trigger of disease
In addition to generalized or localized cerebrovascular insufficiency and hypoperfusion, recent focus has also been directed to a transient or chronic dysfunction of the BBB as a potential trigger of acute or chronic neurological symptoms. Perhaps, the two most striking examples of the BBB breakdown causing or transforming clinical presentation of neurological disease are the hemorrhagic transformation after thrombolysis in stroke patients, and iatrogenic seizures caused by the osmotic BBB disruption procedure in neuro-oncological patients (Neumann-Haefelin et al, 2002; Marchi et al, 2007). Occasional clinical reports also indicated a role for BBB breakdown in familial hemiplegic migraine-associated spreading depolarization (Dreier et al, 2005) and in pathogenesis of seizures in the cerebral hyperperfusion syndrome (Ivens et al, 2010). Whereas notion that the massive, overt BBB opening will lead to brain influx of circulating proteins, water and cells, thus causing acute neurological manifestations is not questioned, whether more subtle, transient and presumed innocuous changes in the BBB integrity could lead to evolution of neurological syndromes requires further scrutiny.
Indeed, accumulating experimental evidence support the hypothesis that primary vascular lesions and, specifically an opening of the BBB, could trigger a chain of events leading to neuronal dysfunction and damage, as well as to specific clinical syndromes, including epilepsy and dementia (Friedman et al, 2009; Marchi et al, 2007; van Vliet et al, 2007). A direct evidence that localized and transient BBB breakdown could trigger both acute epileptiform activity and chronic seizures has been recently provided by a series of studies showing that focal application of bile salts onto the rat neocortex, which opened the BBB in a spatially and temporally restricted manner (Greenwood et al, 1991), resulted in delayed appearance of a hypersynchronous epileptiform activity (Seiffert et al, 2004; Ivens et al, 2007: Tomkins et al, 2007). More recently, Marchi et al (2011) demonstrated that glucocorticoids attenuated both pilocarpine-induced seizures in animals and in pediatric drug-resistant epileptic subjects by protecting BBB integrity. The authors further suggest that because BBB dysfunction plays an etiological role in seizure disorders, that combination therapies utilizing an antiepileptic drug in conjunction with a ‘cerebrovascular’ drug should be used to control seizures more effectively (Marchi et al, 2011). Interestingly, the loss of BBB integrity may also be considered as causative for specific neurological manifestations in AD in light of data showing frequent subclinical seizures, as well as BBB leakiness, in animal models of the disease (Noebels, 2011).
What are the mechanisms by which such BBB dysfunction leads to neuro-glial changes? The accumulating data suggest that acute increase in BBB permeability changes brain extracellular ionic environment (e.g., increase [K+]o and decrease [Ca++]o and [Mg++]o) to promote increased synchronicity and excitability of the neuronal network (Ivens et al, 2007; Marchi et al, 2007). Furthermore, when the BBB is disrupted for large-size molecules such are serum proteins, their accumulation in the brain, apart from driving water influx and edema, may affect glial and neuronal cells by triggering specific signaling pathways. For example, serum albumin has been shown to potently induce calcium signaling and DNA synthesis in astrocytes in culture (Nadal et al, 1995) and in brain slices (Nadal et al, 1998).
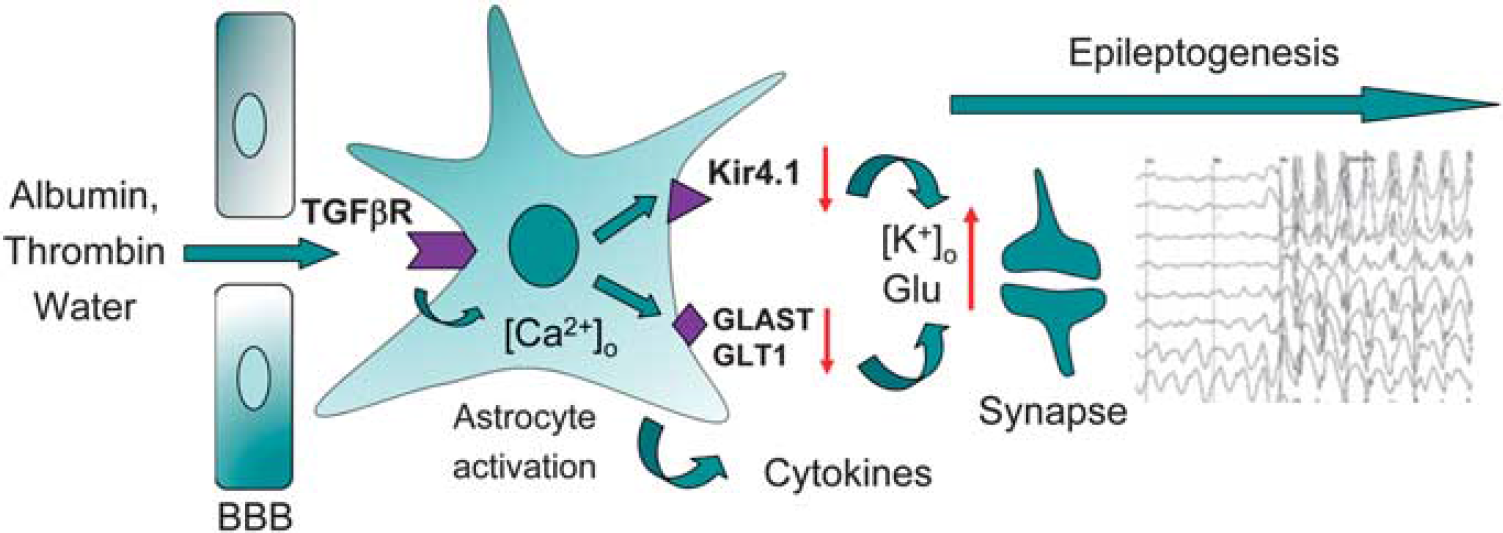
Mechanisms involved in epileptogenesis triggered by the blood–brain barrier (BBB) permeability changes. Brain influx of serum proteins at sites of the BBB disruption, in particular that of albumin and thrombin, trigger profound astrocytic responses. Astrocytes internalize albumin via transforming growth factor (TGF)
In summary, accumulating experimental data suggest that BBB dysfunction is not simply an ‘epiphenomenon’ or ‘biomarker’ for injury, but by itself contributes to glial immune response within the exposed brain, which directly affects the function of the neuronal network and may be sufficient to cause clinical manifestation of epilepsy.
Diagnostic and therapeutic targeting of the neurovascular unit
The recognition of the importance of the NVU in etiology and pathogenesis of a variety of neurological diseases, including several that were not specifically discussed in this review such as multiple sclerosis, Parkinson's disease, and amyotrophic lateral sclerosis, led to efforts to identify informative biomarkers of brain vessel disease and to develop therapeutic strategies that target ‘vascular’ component of the disease. Since brain-specific biomarkers, compartmentalized by the BBB, are not readily accessible for noninvasive detection and targeting, brain vascular compartment is particularly attractive. Vessel-specific molecular biomarkers of disease are often expressed in early stages of the disease and can be detected
For example, specific and selective biomarkers of angiogenic brain tumor (Pen et al, 2007) identified by microarray screening have been exploited for the detection of pathological vasculature by targeted molecular imaging agents (Iqbal et al, 2010). Particularly thick (∼90 nm compared with ∼40 nm in cardiac EC) brain endothelial glycocalyx, the surface coating comprised of oligosaccharide moieties of plasmalemmal glycoproteins and glycolipids (Figure 6), has been an exceptionally rich source of vascular-specific circulating biomarkers, as it readily sheds in response to inflammatory or ischemic conditions. Not surprisingly, several identified blood biomarkers that associated to various degrees with the risk or progression of AD are ‘shed’ or secreted molecules of vascular origin, including endothelial adhesion molecules, adrenomedulin, and endothelin-1 (Ewers et al, 2010). A key future challenge in multiparametric disease evaluation and staging remains meaningful integration of disease-specific cerebrovascular biomarker profiles with functional (neuro)vascular assessments using advanced imaging techniques.
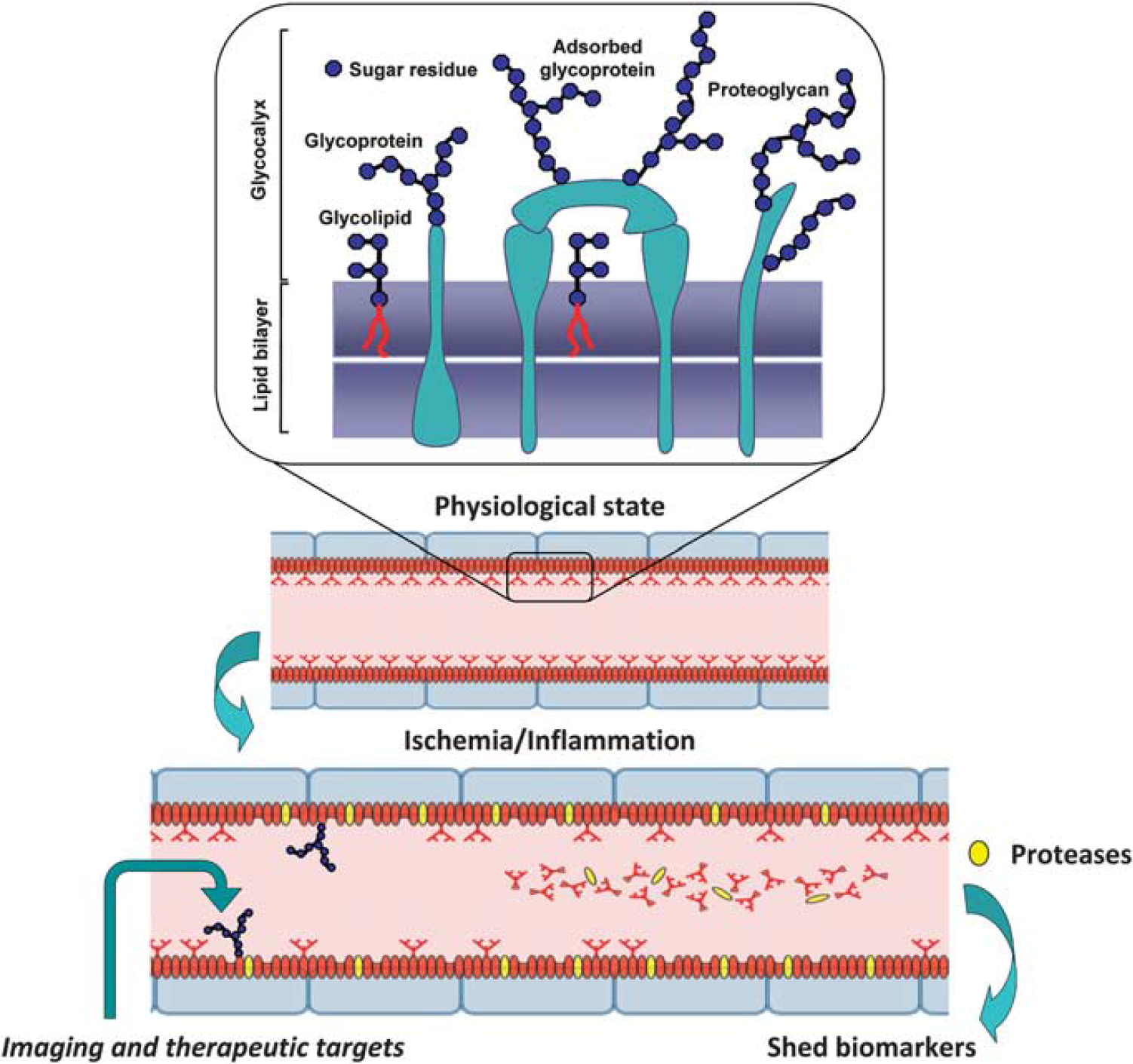
Brain endothelial glycocalyx as a source of circulating biomarkers and therapeutic targets for neurovascular injury. Brain endothelial cell (BEC) glycocalyx is an exceptionally thick layer composed of sugar residues decorating glycolipids, membrane and adsorbed glycoproteins and proteoglycans that cover luminal lining of BECs and participate in essential functions of the neurovascular unit (NVU) (i.e., blood–brain barrier permeability, blood flow control, interactions with inflammatory and immune cells, as a source of adsorbed growth factors, thrombogenesis, and angiogenesis). Through the activation of membrane proteases by hypoxic or inflammatory stimuli, BEC glycocalyx components (proteins, glycosylated fragments of proteins, glycosylated lipids, oligosaccharides, etc.) are promptly ‘shed’ into circulatory compartment, creating a pool of unique endothelial-derived biomarkers that could be used to assess NVU and brain pathology. Luminal BEC (glyco)proteins, such are for example adhesion molecules and transporters, are systemically accessible imaging and therapeutic targets for assessing and modifying functions of the NVU in disease.
Along with advances in molecular understanding of the NVU in disease, some new targets and strategies to protect and repair damaged NVU have started to emerge. For example, nuclear receptor PPAR
In neuroinflammatory diseases, such as multiple sclerosis, endothelial-expressed molecules are targeted to inhibit brain infiltration of inflammatory cells. While intercellular adhesion molecule-1 and vascular cell adhesion molecule 1 are ‘prototypical’ of such brain endothelial targets, novel targets compartmentalized in endothelial membrane subdomains such as lipid rafts (Cayrol et al, 2008) have been implicated in recruitment of specific T-cell subsets. Antibodies against some of these targets, such as activated leukocyte cell adhesion molecule 1, have shown efficacy in suppressing neurological symptoms in experimental allergic encephalomyelitis model(s) of neuroimmune demyelination (Cayrol et al, 2008). Notwithstanding a long history of vascular therapeutic targeting in the area of tumor angiogenesis, these examples demonstrate an emerging spectrum of therapeutic targets and experimental therapeutics identified specifically to protect or modify functions of the NVU. It remains to be seen whether this emerging arsenal of experimental strategies will translate into efficacious clinical therapeutics confirming that NVU, if not origin of neurological disease, is targetable participant that is inextricably linked to disease pathogenesis.
After-Word
The title of this review would have us debate whether it is the blood vessel or the neuron that determines
Footnotes
Disclosure/conflict of interest
The authors declare no conflict of interest.