
Abstract
Transient cerebral ischemia results in an increase in the tyrosine phosphorylation of proteins associated with postsynaptic densities (PSDs). The authors investigated the possible mechanisms behind this increase by analyzing isolated PSDs for protein tyrosine kinase activity and for the presence of specific tyrosine kinases. Transient (15 minutes) global ischemia was produced in adult rats by four-vessel occlusion, and PSDs were isolated immediately after ischemia or after 20 minutes or 6 hours of reperfusion. Tyrosine phosphorylation of several PSD proteins, including the N-methyl-
One of the early changes which occurs after cerebral ischemia is an increase in tyrosine phosphorylation (Hu and Wieloch, 1994; Ohtsuki et al., 1996; Takagi et al., 1997). A relationship between ischemia, tyrosine phosphorylation, and neuronal cell damage has been suggested (Campos-Gonzalez and Kindy, 1992; Hu and Wieloch, 1994), and tyrosine kinase inhibitors have been found to be neuroprotective against ischemia-induced cell loss (Kindy, 1993; Ohtsuki et al., 1996). Tyrosine phosphorylation has been implicated in the control and modulation of synaptic and neuronal functions (reviewed in Gurd, 1997; Leβmann, 1998; Salter, 1998) and ischemia-induced increases in tyrosine phosphorylation may result in altered synaptic activity.
The postsynaptic density (PSD) is composed of a network of interacting proteins, including neurotransmitter receptors, anchoring proteins such as PSD95/SAP90-family proteins, cytoskeletal proteins such as alpha-actinin and fodrin, and protein kinases, including Ca2+/calmodulin-dependent protein kinase II, protein kinase C, and protein tyrosine kinases (reviewed in Ziff, 1997; Kennedy, 1998). The assembly of these molecules into a macromolecular complex at the PSD suggests that the PSD may be dynamically involved in the regulation of a variety of synaptic functions, including synaptic adhesion, receptor clustering and function, and the primary stages of signal transduction in the postsynaptic cell (Ziff, 1997; Kennedy, 1998). Because many of the initial events (e.g., activation of glutamate receptors, Ca2+ influx) after an ischemic challenge occur at or in the vicinity of the PSD, this seems a likely site for ischemia-induced biochemical cascades leading to neuronal cell death to originate. Consistent with this notion, ischemia-induced changes in the protein composition, morphology, and tyrosine phosphorylation of PSDs have been recently reported (Hu et al., 1998; Martone et al., 1999; Takagi et al., 1999).
To begin to identify mechanisms which may be responsible for the ischemia-induced changes in the tyrosine phosphorylation of PSD proteins, we investigated the effect of transient cerebral ischemia on the tyrosine kinase activity of PSDs and on the association of specific tyrosine kinases with the postsynaptic structure. The findings demonstrate that PSDs from postischemic brains are characterized by enhanced tyrosine kinase activity and increased levels of several tyrosine kinases potentially involved in the tyrosine phosphorylation of PSD proteins.
MATERIALS AND METHODS
Surgical procedures
All procedures using animals were approved by the Animal Care Committee of the University of Toronto and were in accordance with the guidelines established by the Canadian Council on Animal Care. Male Wistar rats, weighing between 200 and 250 g, were given food and water ad libitum before surgery. Transient global ischemia (15 minutes) was induced as described in detail previously (Shinno et al., 1997; Zhang et al., 1997) using a modified version of the method described by Pulsinelli and Brierley (1979). Briefly, male Wistar rats were anesthetized, and both vertebral arteries were permanently occluded by electrocauterization on day 1. On the second day, the common carotid arteries were reexposed and both were occluded with aneurysm clips for 15 minutes. Animals that did not meet the following criteria were excluded from the study: 1) completely flat bitemporal EEG during the duration of the carotid occlusion; 2) maintenance of a diluted pupil and an absence of a cornea reflex attributable to light stimulation; 3) consistent body temperature recordings; and 4) femoral artery blood gas parameters which did not vary from normal values. Morphologic examination revealed that under these conditions less than 6% of the CA1 neurons were damaged at 3 or 24 hours after reperfusion, there was pronounced damage by 3 days, and greater than 90% of the CA1 neurons were injured by 7 days (Shinno et al., 1997). Sham-operated animals received identical treatment except that the vertebral arteries were not cauterized and the carotid arteries were not occluded. Rats were killed by decapitation either at the end of the ischemic episode, after 20 minutes, or after 6 hours of reperfusion. Heads were quickly frozen by immersion in −42°C isopentane to avoid dephosphorylation and translocation of proteins to the PSD (Suzuki et al., 1994) and stored at −72°C until use.
Tissue preparation
Frozen heads were warmed to 0°C and the forebrains from two to five ischemic animals, or from two to five sham-operated animals, were dissected and pooled for the preparation of PSDs according to the method of Cho et al. (1992) using two extractions with Triton X-100 to obtain the final PSD preparation. PSDs were suspended in 0.32 mol/L sucrose and stored at −72°C. Initial experiments indicated that although the yield of PSDs from frozen brains was decreased relative to unfrozen brains, the concentration of several PSD-associated proteins, including NR1, NR2A, NR2B, PSD-95, and Src, were unaffected by the freezing step.
Gel electrophoresis, immunoprecipitation, and immunoblotting
Protein was determined by the method of Lowry (1951) using bovine serum albumin as standard. For gel electrophoresis, PSDs were solubilized by heating at 100°C for 3 minutes in 62.5 mmol/L Tris-HCl, pH 6.8, containing 2% sodium dodecyl sulfate (SDS), 1% β-mercaptoethanol, and 5% glycerol (sample buffer). Proteins (2 μg) were separated on 8% polyacrylamide minigels and detected by silver staining (Pierce GelCode; Rockford, IL, U.S.A.). In some experiments, 100 μg of forebrain homogenate was extracted with 1% sodium deoxycholate (DOC) containing 50 mmol/L Tris-HCl, pH 9.0, 10 μmol/L EDTA, 200 μmol/L phenylmethylsulfonyl fluoride, 1 mmol/L sodium orthovanadate, and 5 μg/mL each of antipain, aprotinin, and leupeptin for 30 minutes at 37°C followed by centrifugation at 100, 000 × gAv for 10 minutes. The supernatant and pellet were solubilized in SDS as above for analysis by gel electrophoresis and immunoblotting.
For immunoprecipitation, antibodies specific to NR2A or NR2B were precoated on 30 μL Protein A/G PLUS-Agarose (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) by incubating 2 μL of immune serum with 30 μL of Protein A/G for 2 hours at 4°C in 500 μL of 50 mmol/L HEPES buffer, pH 7.1, containing 1.5% Triton X-100, 150 mmol/L NaCl, 1.5 mmol/L MgCl2, 10% glycerol, 100 μmol/L each of phenylmethylsulfonyl fluoride and sodium orthovanadate, 1 mmol/L each of NaF, ZnCl2, and EGTA, and 5 μg/mL each of antipain, leupeptin, and aprotinin (binding buffer). PSDs (20 μg) that had been solubilized by boiling in 1% SDS containing 1% β-mercaptoethanol were diluted 10-fold with binding buffer. Diluted samples were incubated with 30 μL of Protein A/G PLUS-Agarose for 1 hour at 4°C, centrifuged, and the supernatant added to the precoated Protein A/G agarose and incubated at 4°C overnight. The immune complexes were isolated by centrifugation, washed three times with 50 mmol/L HEPES buffer, pH 7.1, containing 0.15% Triton X-100, 150 mmol/L NaCl, 100 μmol/L sodium orthovanadate, 10% glycerol, and proteins eluted by heating at 100°C in sample buffer.
For immunoblotting, SDS-solubilized samples (5 to 10 μg) were separated on 8 or 10% polyacrylamide gels, transferred to nitrocellulose as described (Gurd et al., 1992), and protein blots reacted with specific antibodies as below. Bound antibodies were detected by enhanced chemiluminescence (Pierce Super Signal, Rockford, IL, U.S.A.). For quantification, exposed X-ray film was scanned using a BioRad GS 700 Gel scanner as described (Gurd et al., 1992). Individual proteins were detected by Western immunoblotting using the following antibodies: phosphotyrosine (clone 4G10) and c-Src (clone GD11) from Upstate Biotechnology Inc. (Lake Placid, NY, U.S.A.), NR2A (polyclonal antibody produced as described below), NR2B (clone 13), c-Fyn (clone 25), proline-rich tyrosine kinase 2 (PYK2) (clone 11), FAK (clone 77), and TrkB (clone 47) from Transduction Laboratories (Lexington, KY, U.S.A.). NR2A and NR2B antibodies used for immunoprecipitation were generated against polyhistidine fusion proteins containing the C-terminal region of each subunit encompassing amino acid residues 934–1203 for NR2A and 935–1455 for NR2B. Expression vectors containing the appropriate coding regions were kindly provided by Dr. R. J. Wenthold (National Institutes of Health, Bethesda, MD, U.S.A.). The polyclonal NR2A and NR2B antibodies reacted with NR2A and NR2B which had been immunoprecipitated from rat brain with anti-NR2A antibodies from Upstate Biotechnology Inc. or anti-NR2B antibodies from Transduction Laboratories but did not cross-react with immunoprecipitated NR2B or NR2A, respectively.
Endogenous protein tyrosine kinase assays
PSD samples used for these experiments were frozen and thawed only once. PSD (5 μg) from sham-operated or ischemic animals after 6 hours of reperfusion were incubated for various times in 30 μL of 20 mmol/L Tris buffer, pH 7.2, containing 10 mmol/L MgCl2, 2 mmol/L ATP, 100 μmol/L sodium orthovanadate, 100 μmol/L phenylmethylsulfonyl fluoride, and 5 μg/mL each of antipain, aprotinin, and leupeptin. Reactions were stopped by the addition of 10 μL of 4 × sample buffer and heating at 100°C for 5 minutes. For detection of in vitro phosphorylation of NR2A and NR2B, 20 μg of PSDs were incubated with ATP for 30 seconds as above, and the reactions were stopped by the addition of 10 μL of 4% SDS containing 4% β-mercaptoethanol. NR2A and NR2B were immunoprecipitated and analyzed as above.
RESULTS
Increased tyrosine phosphorylation induced by transient ischemia occurs primarily on deoxycholate insoluble proteins
Ischemia followed by reperfusion results in increased tyrosine phosphorylation of proteins in several regions of the brain, including the hippocampus, striatum, and frontal cortex (Kindy, 1993; Hu and Wieloch, 1994; Ohtsuki et al., 1996; Takagi et al., 1997). To further investigate the effects of ischemia on tyrosine phosphorylation we examined the distribution of tyrosine-phosphorylated proteins between DOC-soluble and-insoluble fractions prepared from rat forebrains after ischemia and 6 hours of reperfusion. Extraction with 1% DOC solubilized greater than 85% of the total homogenate protein for both sham-operated and ischemic samples (Fig. 1A). Immunoblotting with antiphosphotyrosine antibodies showed that most of the tyrosine-phosphorylated proteins in sham-operated samples were located in the DOC-soluble fraction (Fig. 1). In contrast, the majority of tyrosine-phosphorylated proteins were present in the DOC-insoluble residue after ischemia and reperfusion (Fig. 1). The DOC-insoluble fraction is enriched in proteins derived from the cytoskeleton and PSD (Matus and Taff-Jones, 1978; Moss, 1983). The relative contribution of each of these to the increase in tyrosine phosphorylation of proteins in the DOC residue was not further investigated, but together with our earlier finding of enhanced tyrosine phosphorylation of PSD proteins after ischemia (Takagi et al., 1999), the results suggest that a substantial proportion of ischemia-induced tyrosine phosphorylation is associated with the postsynaptic structure.
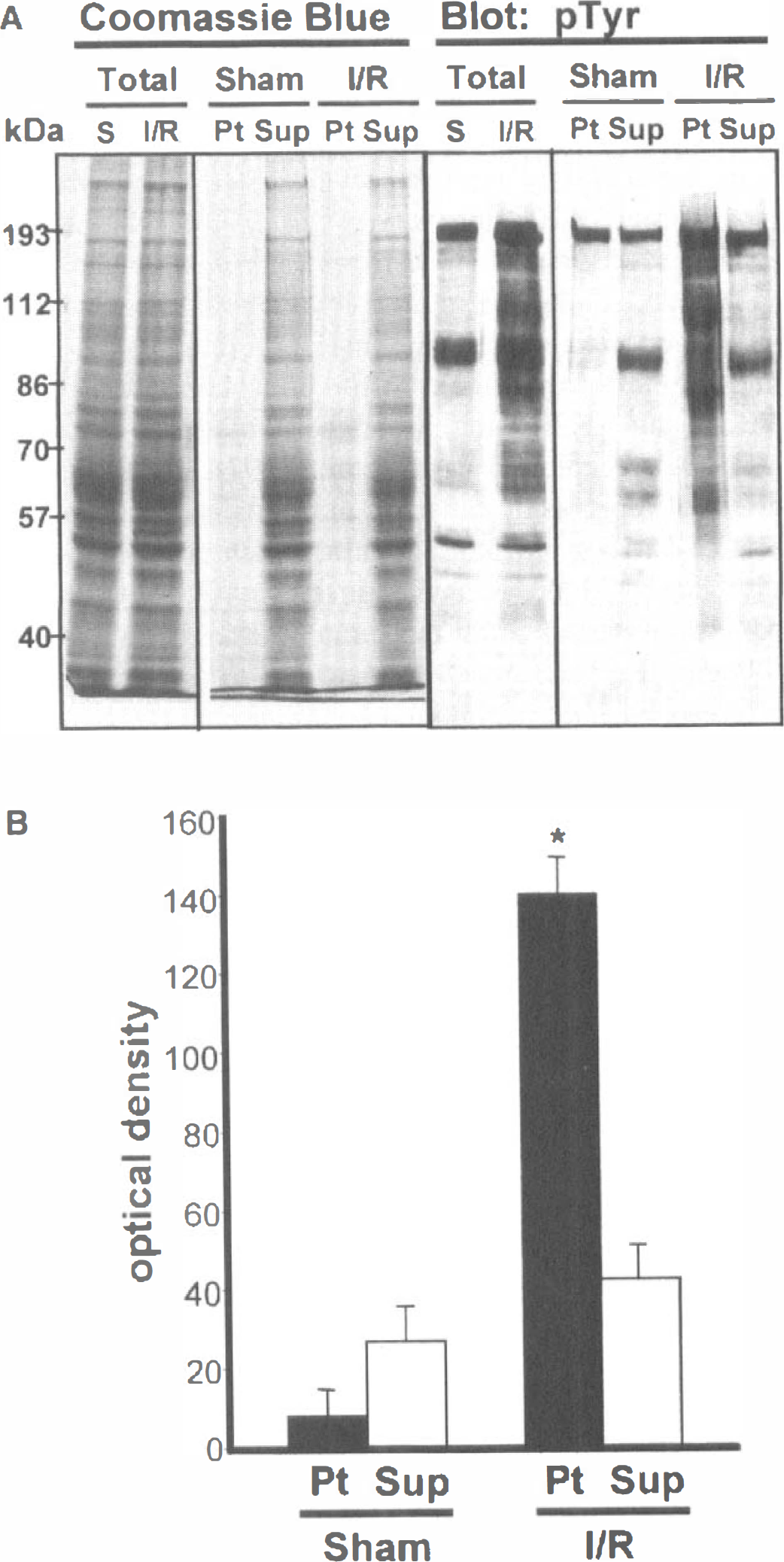
Distribution of tyrosine-phosphorylated proteins between sodium deoxycholate (DOC)-soluble and-insoluble fractions after transient global ischemia. (
Tyrosine phosphorylation of PSD proteins increases rapidly after ischemia
We next examined the relationship between reperfusion time and the tyrosine phosphorylation of PSD proteins. PSDs were prepared from rat forebrains immediately after ischemia and after 20 minutes or 6 hours of reperfusion. The overall protein composition of PSDs from sham-operated and challenged animals was similar at all times examined (Fig. 2A). Immediately after ischemia, the tyrosine phosphorylation of PSD proteins was decreased relative to shams (Fig. 2B, time 0). PSD proteins were rapidly rephosphorylated after the onset of reperfusion; by 20 minutes tyrosine phosphorylation levels exceeded those of PSDs from sham-operated animals and remained elevated after 6 hours (Fig. 2B).
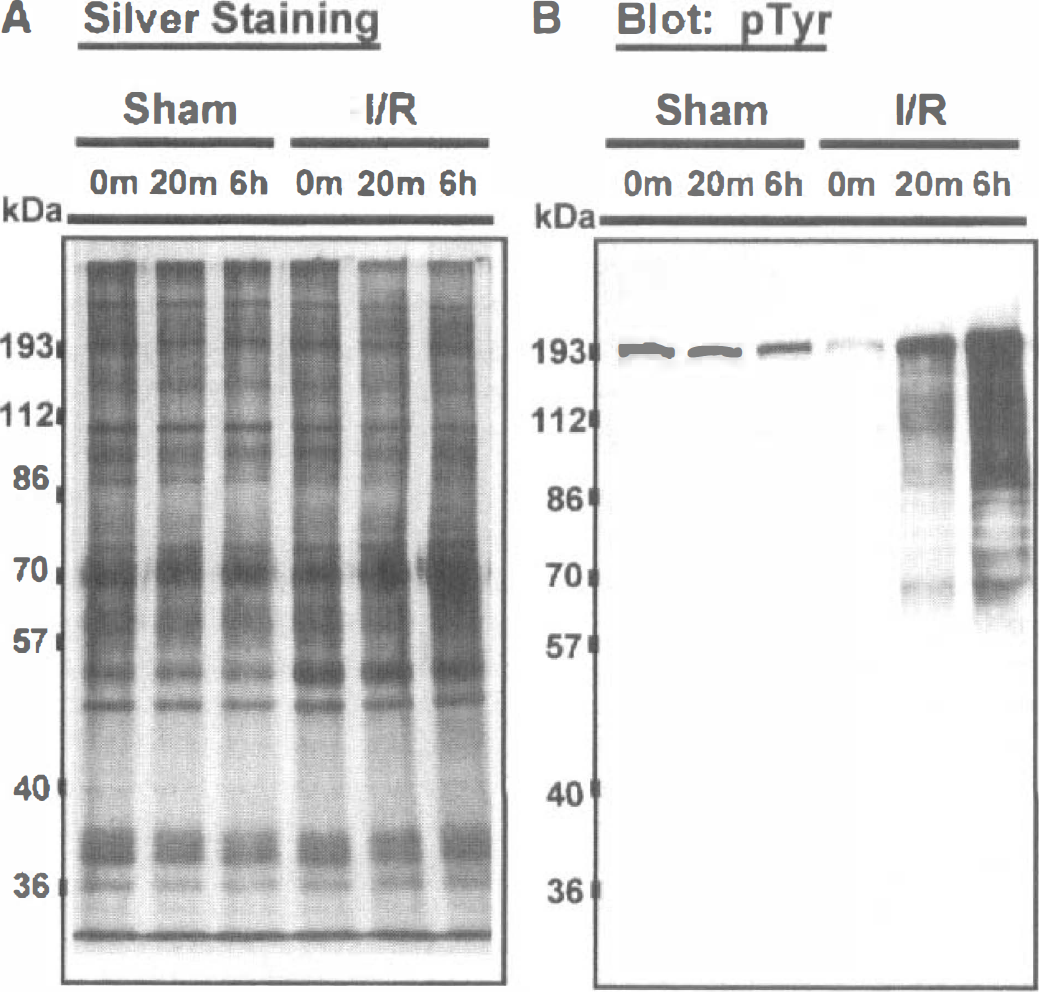
Transient ischemia rapidly increases tyrosine phosphor ylation of postsynaptic density proteins. Postsynaptic *densities were isolated from rat forebrains and proteins detected by (
We reported previously that ischemia results in a marked elevation in the tyrosine phosphorylation of the NR2A and NR2B subunits of the N-methyl-
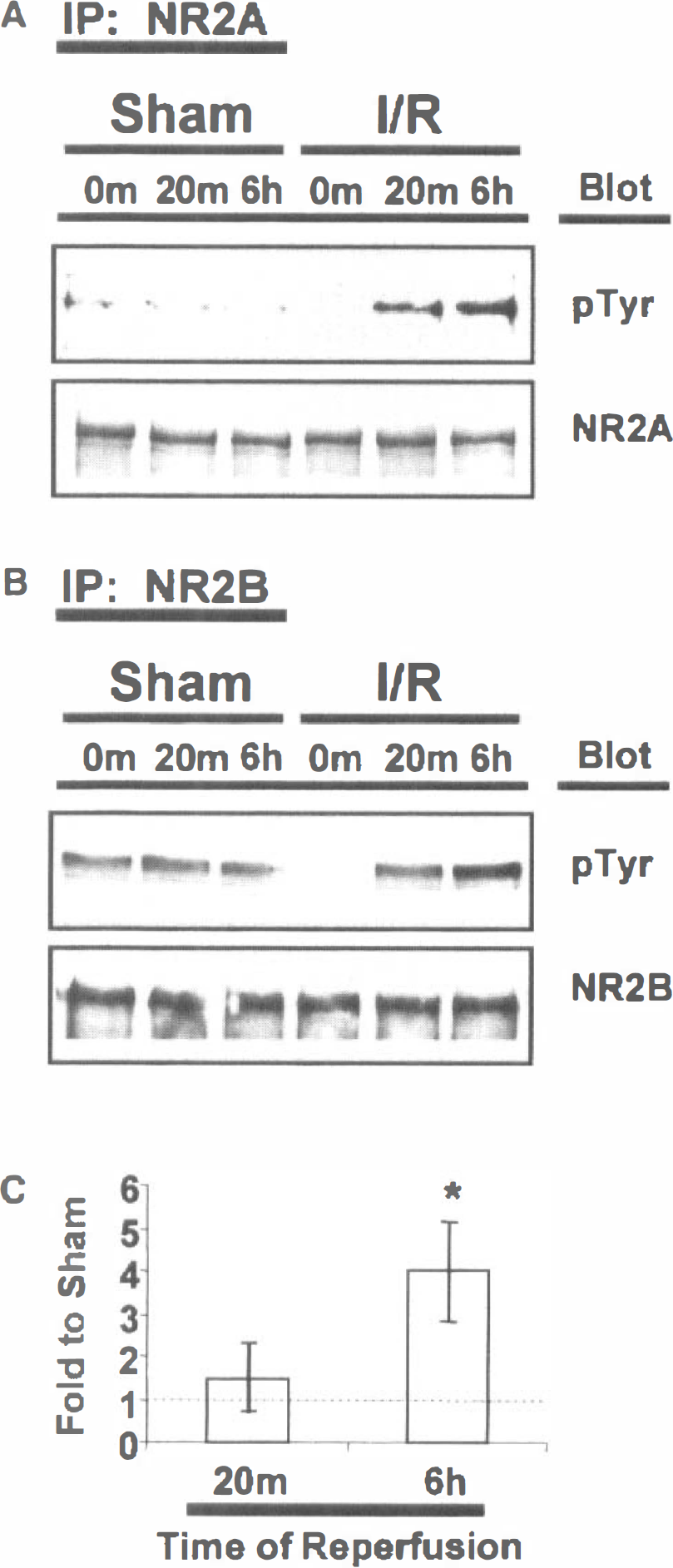
Ischemia-reperfusion results in increased tyrosine phosphorylation of NR2A and NR2B in postsynaptic densities. Postsynaptic densities from sham-operated (Sham) or ischemic (I/R) rat forebrains were immunoprecipitated with (
Ischemia results in enhanced protein tyrosine kinase activity in PSDs
To determine if the ischemia-induced increase in tyrosine phosphorylation of PSD proteins was paralleled by an increase in PSD-associated tyrosine kinase activity, PSDs prepared from forebrains after 6 hours of reperfusion were incubated with ATP and the tyrosine phosphorylation of endogenous proteins determined by immunoblotting with antiphosphotyrosine antibodies. In experiments with three different PSD preparations from sham-operated and ischemic animals, the rate at which PSD proteins were phosphorylated was 2.7 to 3.3-fold greater in PSDs from ischemic than from sham-operated animals (Figs. 4A and 4B). In vitro phosphorylation of NMDA receptor subunits 2A and 2B was also greater in PSDs from ischemic as compared to sham-operated animals (Figs. 4C to 4E).
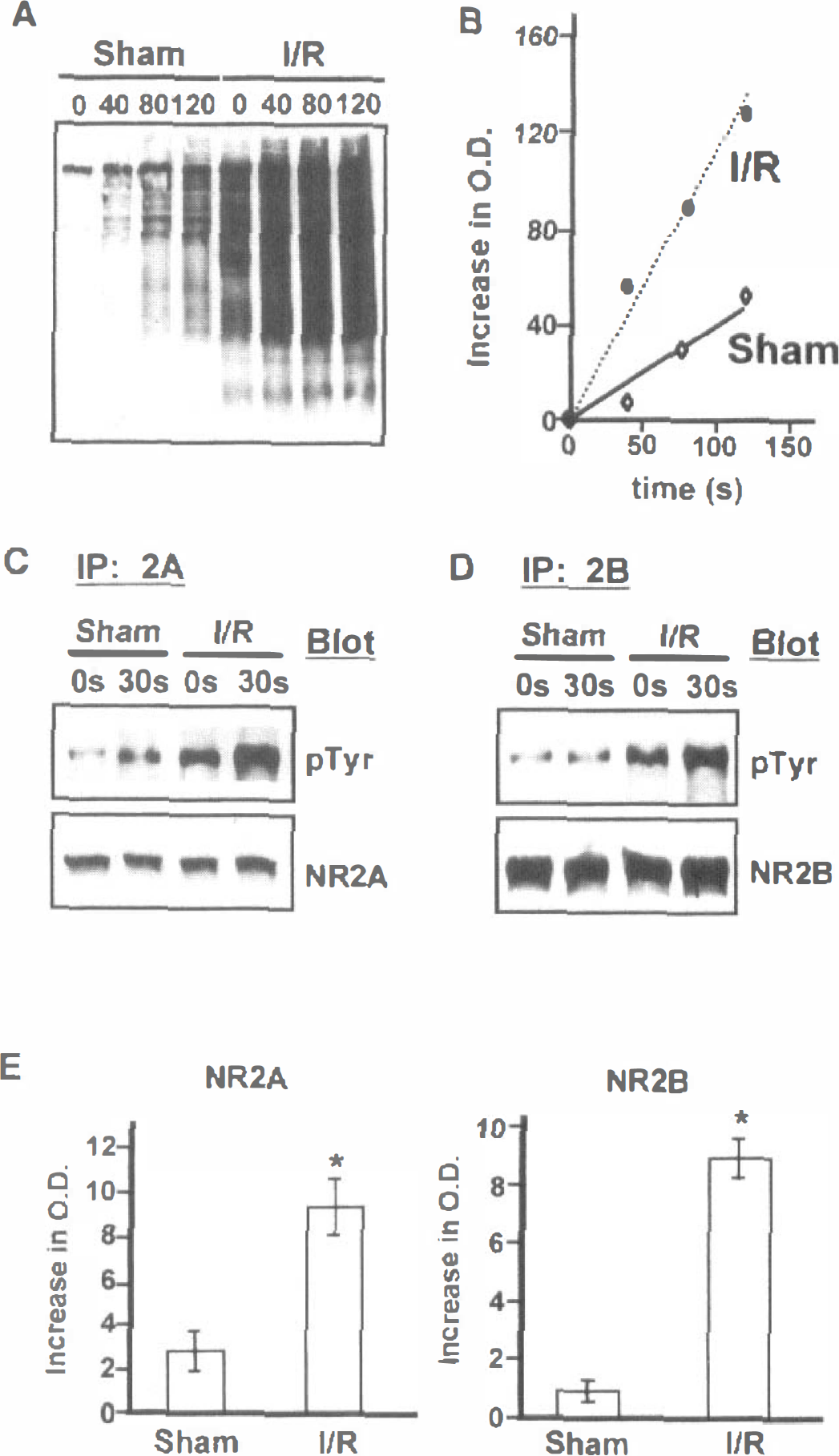
Transient ischemia results in enhanced protein tyrosine kinase activity in postsynaptic densities. (
Ischemia results in increased association of tyrosine kinases with the PSD
The ischemia-induced increase in tyrosine phosphorylation of PSD proteins might have reflected the activation of PSD-associated tyrosine kinases, the recruitment of tyrosine kinases to the PSD, or both. To assess if the enhanced phosphorylation activity reflected changes in the association of specific tyrosine kinases with the PSD, PSDs from sham-operated and ischemic animals were examined for the presence of Src, Fyn, PYK2, FAK, and the brain-derived neurotrophic factor (BDNF) receptor gp145TrkB at increasing times of reperfusion.
Each of the five tyrosine kinases examined was present in PSDs from sham-operated animals (Fig. 5). Individual kinases responded differently to the ischemic challenge. Immediately after ischemia, there was a small increase in PSD-associated PYK2 (1.79 ± 0.05-fold relative to shams, average ± SD, n = 3), and a large (11.7 ± 1.2-fold) increase in gp145TrkB (Fig. 5). In contrast, the levels of Src, Fyn, and pp125FAK in the PSD were not different from shams at this time, although a new 77kDa protein (p77) immunoreactive with the FAK antibody was detected (Fig. 5), suggesting that some proteolysis of FAK had occurred during the ischemic episode. After 20 minutes, PYK2 and gp145TrkB remained elevated relative to shams, but there was no change in the levels of Src, Fyn, or pp125FAK. Between 20 minutes and 6 hours, PYK2 and gp145TrkB underwent a further increase to 3.9 ± 1.0 and 23 ± 2.9 times sham levels, respectively. By 6 hours, Src and Fyn had both increased to 2.5 times the sham levels. In contrast, pp125FAK levels were 30% lower than shams after 6 hours.
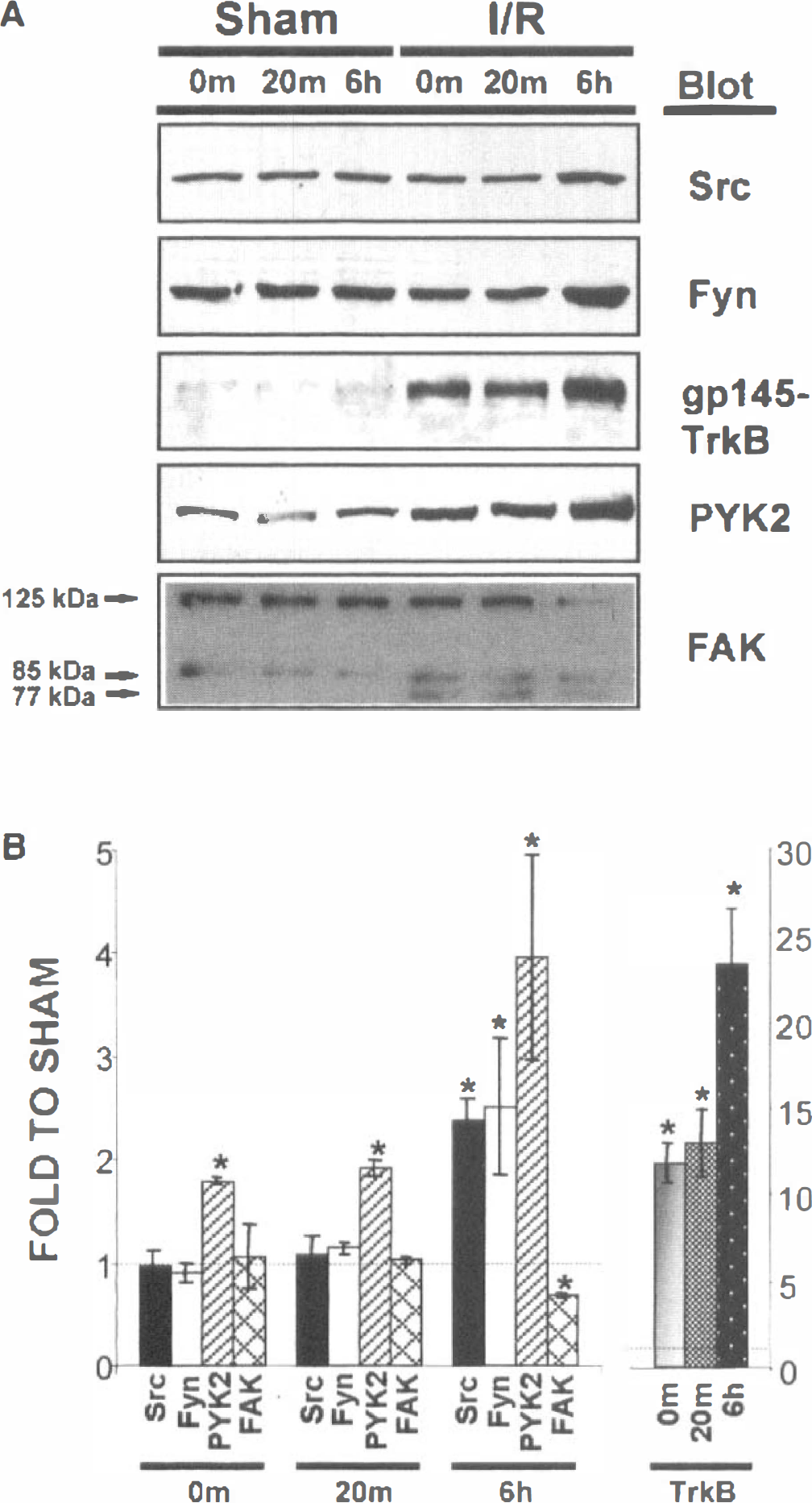
The effect of transient ischemia on the association of protein tyrosine kinases with postsynaptic densities. (
Tyrosine kinase levels in synaptosomes are not altered after ischemia
We also assessed the concentrations of each of the tyrosine kinases in synaptosomes after 6 hours of reperfusion. The synaptosomal levels were unaffected by the ischemic challenge (Fig. 6), indicating that the ischemia-induced changes associated with the PSD were specific for this structure and did not reflect a general increase in enzyme levels after ischemia.
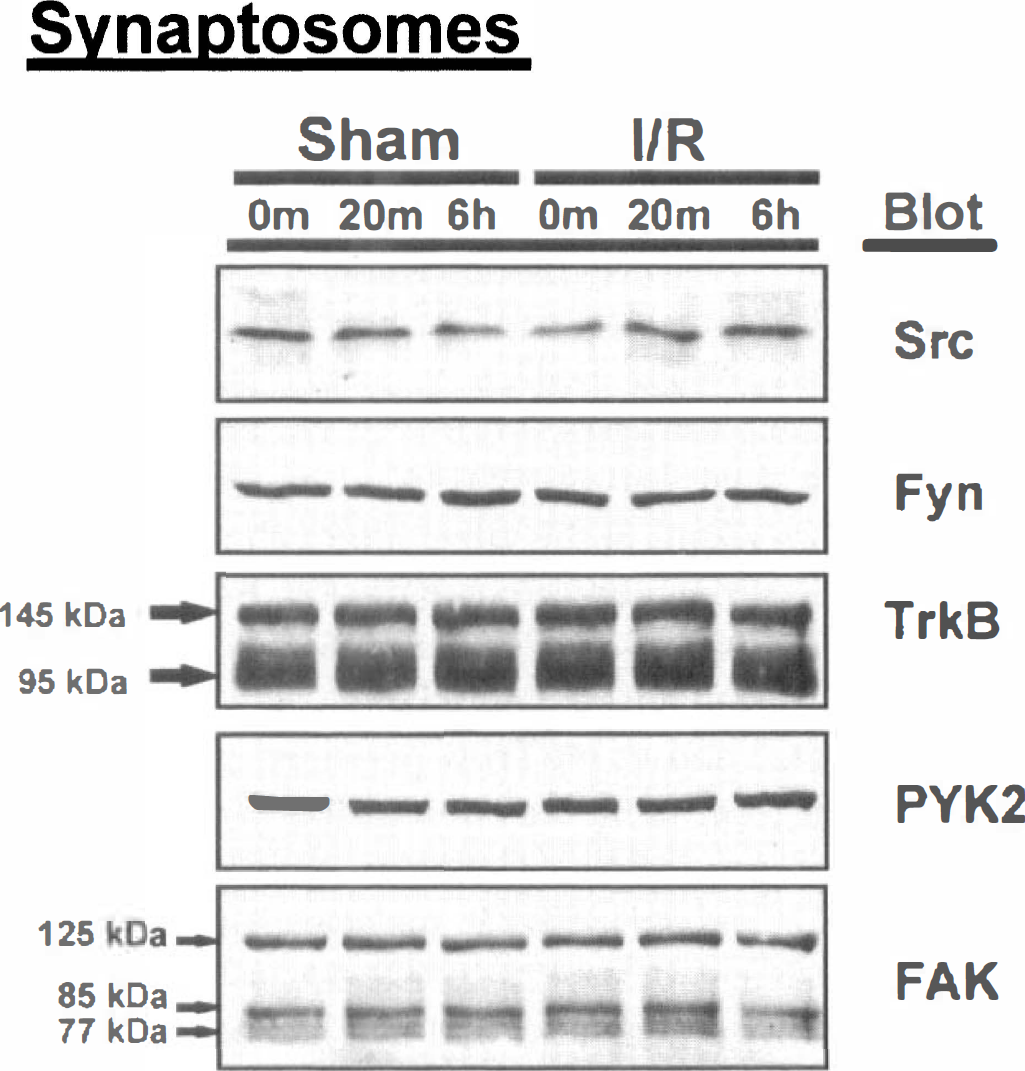
Transient ischemia does not change protein tyrosine kinase levels in the synaptosomal fraction. Synaptosomes were isolated from the forebrains of sham-operated (Sham) or ischemic (I/R) animals after 0 minutes, 20 minutes, or 6 hours of recovery and analyzed for the presence of Src, Fyn, TrkB, PYK2, and FAK. Results are typical of those obtained with at least three separate preparations of synaptosomes for each condition. Immunoreactivity of individual protein tyrosine kinases in ischemia and sham-operated animals was not significantly different.
DISCUSSION
Ischemia induces a rapid, large, and sustained increase in the tyrosine phosphorylation of a number of proteins, including the NMDA receptor, in the rat brain. The magnitude and timing of these changes suggests that they may be critically involved in the initial response of the brain to the ischemic event. In the present study, we focused on ischemia-induced changes in the PSD because of its temporal and spatial proximity to the initial events which occur after an ischemic challenge. The results demonstrate that ischemia results in an increase in the tyrosine phosphorylation of a number of PSD proteins, including the NMDA receptor subunits NR2A and NR2B, and that this increase is accompanied by enhanced intrinsic PSD tyrosine kinase activity and by specific changes in the association of protein tyrosine kinases with the PSD.
Although the hippocampus is more sensitive to ischemia-induced cell damage than the forebrain, we used the latter for the preparation of PSDs because of the difficulty of isolating sufficient amounts of material from the more sensitive hippocampal region. Previous studies have shown that ischemia induces increased tyrosine phosphorylation of proteins in the frontal cortex and striatum, although these changes are smaller than those that occur in the hippocampus (Hu and Wieloch, 1994; Takagi et al., 1997). In addition, changes in the composition and morphology of forebrain PSDs have been reported to occur after an ischemic challenge (Hu et al., 1998; Martone et al., 1999). It now becomes important to determine the relationship between ischemia-induced changes in the tyrosine kinase activity of the PSD and the pathophysiologic response of different brain regions to an ischemic challenge.
The present results demonstrate that ischemia differentially affects the association of individual tyrosine kinases with the PSD: the levels of PSD-associated PYK2 and TrkB increased during the ischemic episode, Src and Fyn levels increased after a delay of at least 20 minutes of reperfusion, and the amount of PSD-associated FAK decreased. Earlier studies reported that Src, Fyn, and TrkB levels were increased in the PSD as a result of transient ischemia followed by 4 to 6 hours of reperfusion (Hu et al., 1998; Takagi et al., 1999). Because the tyrosine phosphorylation of PSD proteins increases rapidly after the onset of reperfusion, it was important to determine the effect of shorter reperfusion times on tyrosine kinase levels. The delayed increase in PSD Src and Fyn protein levels indicates that recruitment of these enzymes to the PSD does not account for the initial rise in tyrosine phosphorylation. In contrast to Src and Fyn, PSD-associated PYK2 was elevated at the start of reperfusion. PYK2 is activated by calcium (Lev et al., 1995). Activation of PYK2 by ischemia-induced calcium influx could therefore result in a rapid increase in the phosphorylation of PSD proteins, either by direct phosphorylation by PYK2 itself or indirectly via PYK2-mediated activation of Src and/or Fyn (Dikic et al., 1996; Li et al., 1996). Interestingly, Src, Fyn, and PYK2 have each been implicated in potentiation of the NMDA receptor ion channel (Köhr and Seeberg, 1996; Huang et al., 1999; Lu et al., 1999). Enhanced activity of one or more of these enzymes at the PSD at the onset of reperfusion could therefore play a critical role in the regulation of calcium fluxes immediately after the ischemic challenge, and we are now investigating the roles of each of these enzymes in the ischemia-induced increase in the phosphorylation of PSD proteins.
The level of gpl45TrkB in PSDs was increased 20 to 25-fold relative to shams after 6 hours of reperfusion, in general accord with the earlier report of elevated PSD-gp145TrkB after 4 hours of reperfusion (Hu et al., 1998). Notably, the concentration of gp145TrkB in the PSD increased during the period of ischemia, so that, like PYK2, elevated levels of gpl45TrkB were present at the start of reperfusion. Gpl45TrkB has been implicated in the phosphorylation of the NMDA receptor (Suen et al., 1997), and BDNF, acting through TrkB receptors, may modulate glutamatergic synaptic transmission (reviewed in Leβmann, 1998). Ischemia results in moderate increases in BDNF in the dentate gyrus (Kokaia et al., 1996), and the elevation of gpl45TrkB in the PSD at early times after the onset of reperfusion may increase the sensitivity of the postsynaptic neuron to small changes in BDNF concentrations. BDNF is neuroprotective against neuronal damage resulting from transient global ischemia (Kiprianova et al., 1999) and was recently reported to mediate the antiapoptotic action of NMDA in cerebellar granule neurons (Bhave et al., 1999). Whether or not the increase in PSD-associated gp145TrkB is involved in the neuroprotective actions of BDNF remains to be determined.
In addition to regulating the phosphorylation and activity of postsynaptic proteins, tyrosine kinases may link the PSD to downstream signaling pathways. Src, Fyn, PYK2, FAK, and gp145TrkB are all potential activators of mitogen-activated protein kinase cascades (Marsh et al., 1993; reviewed in Lopez-Ilasaca, 1998), and tyrosine-phosphorylated NMDA receptor subunits may interact directly with signaling proteins that contain Src Homology 2 domains (Gurd and Bissoon, 1997; Takagi et al., 1999). Thus, changes in the interaction of tyrosine kinases with the PSD and the associated change in the tyrosine phosphorylation of PSD proteins may impact on both upstream (receptors) and downstream (signaling pathways) activities in the postsynaptic neuron.
The specific mechanisms involved in the interactions of individual tyrosine kinases with the PSD, and the effect of ischemia on these interactions, remain to be determined. Whether the ischemia-induced increase in PSD-associated tyrosine kinases reflects the recruitment of new enzymes from the local environment, or the stabilization of preexisting interactions of these enzymes with the network of PSD proteins remains to be determined. Whatever the specific mechanisms involved, the changes now described are likely to have significant implications for the pathophysiologic response of neurons to ischemia.