
Abstract
The sulfonylurea receptor 1 (Sur1)-regulated NCCa-ATP channel is a nonselective cation channel that is regulated by intracellular calcium and adenosine triphosphate. The channel is not constitutively expressed, but is transcriptionally upregulated
Keywords
Introduction
The adenosine triphosphate (ATP) binding cassette (ABC) transporters constitute a large superfamily of integral membrane proteins, with >48 genes encoding human ABC transporters (Aguilar-Bryan et al, 1998; Shi et al, 2005; Aittoniemi et al, 2008; Lefer et al, 2009). The subfamily encoded by the
Arguably, the most atypical function thus far identified for members of the ABC superfamily involves the sulfonylurea receptors, Sur1/
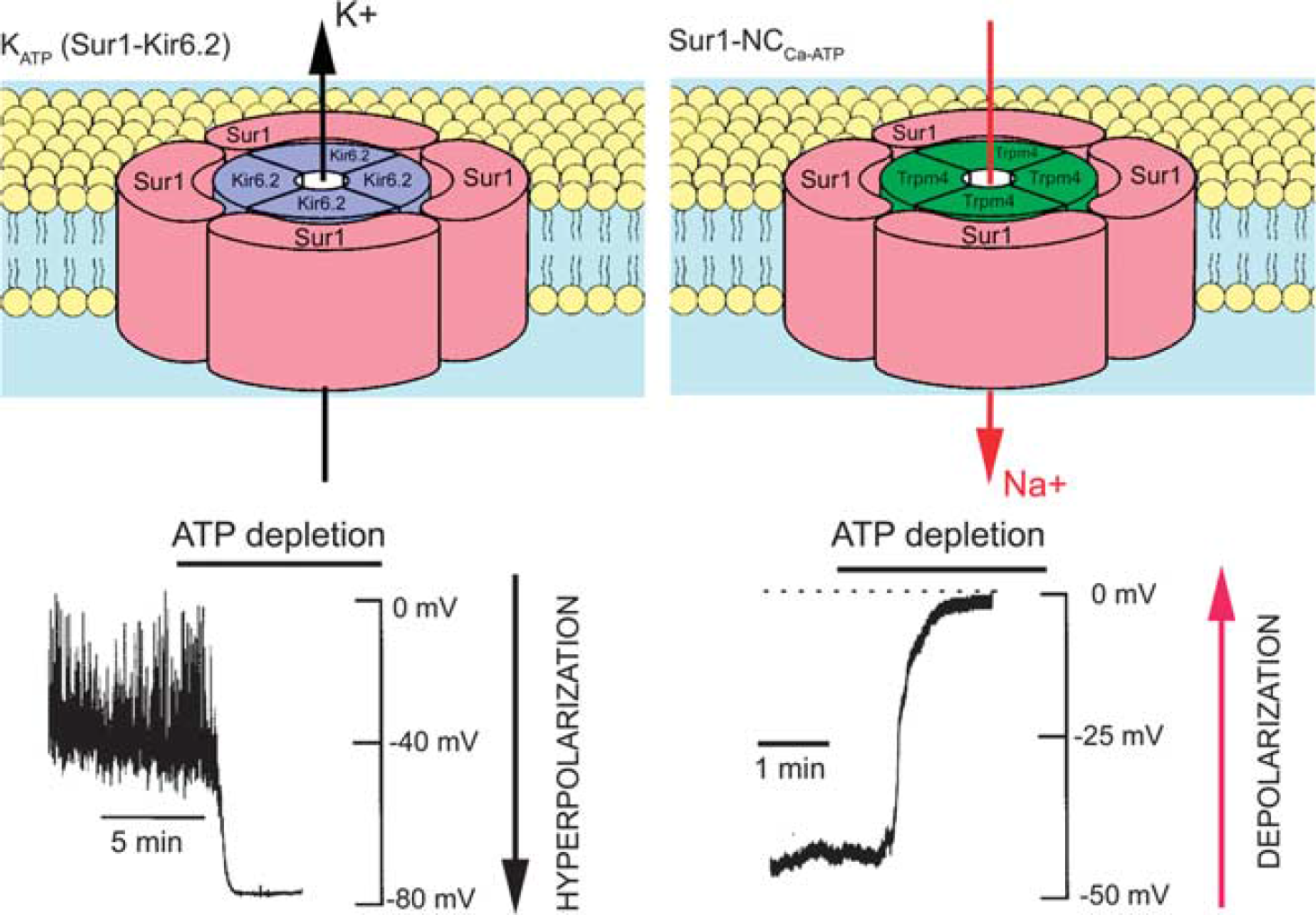
Schematic diagrams of the KATP (Sur1-Kir6.2) and the Sur1-NCCa-ATP channels. The hetero-octameric structure comprising four Sur1 subunits and four Kir6.2 subunits depicted for KATP is widely accepted. The structure depicted for the Sur1-NCCa-ATP channel is hypothesized by analogy. Also shown are the principal physiological actions of the two channels when they are activated by ATP depletion: (1) outward flux of K+ via the K+-selective pore-forming subunit, Kir6.2, resulting in hyperpolarization with the KATP channel; (2) inward flux of Na+ via the nonselective monovalent cation pore-forming subunit, resulting in depolarization with the Sur1-NCCa-ATP channel. The channel schematic was adapted from Seino (1999); the recoding depicting hyperpolarization for KATP was adapted from Harvey et al (1999); the recoding depicting depolarization for NCCa-ATP was adapted from Chen and Simard (2001). ATP, adenosine triphosphate, Sur1, sulfonylurea receptor 1.
Sur1 contains two nucleotide-binding domains as well as high affinity binding sites for therapeutic sulfonylurea drugs and related compounds. Drugs such as glibenclamide (US adopted name, glyburide) and repaglinide bind with nanomolar or subnanomolar affinity, and are potent inhibitors of Sur1-regulated channel activity. Sur1 is the target of sulfonylurea drugs used to treat diabetes mellitus type 2, neonatal diabetes, and some forms of congenital hyperinsulinism.
Sur1 also associates with an ATP- and calcium-sensitive nonselective cation channel to form Sur1-regulated NCCa-ATP (henceforth, Sur1-NCCa-ATP) channels (Figure 1; Chen et al, 2003; Simard et al, 2007b). Although both KATP and Sur1-NCCa-ATP channels are regulated by Sur1, the two have opposite functional effects in central nervous system (CNS) injury—opening of KATP channels hyperpolarizes the cell and may be neuroprotective (Yamada and Inagaki, 2005), whereas opening of Sur1-NCCa-ATP channels depolarizes the cell and, if unchecked, is associated with oncotic (necrotic) cell death (Chen and Simard, 2001; Chen et al, 2003; Simard et al, 2006).
Here, we examine emerging evidence for the role of Sur1 and Sur1-NCCa-ATP channels in acute pathological processes associated with ischemic, traumatic, and inflammatory injury to the CNS.
Biophysical and Pharmacological Properties of the Sur1-NCCa-ATP Channel
The original patch-clamp experiments characterizing the Sur1-NCCa-ATP channel were performed on reactive astrocytes freshly isolated from the hypoxic gliotic capsule
Properties of the Sur1-NCCa-ATP and Trpm4 channels
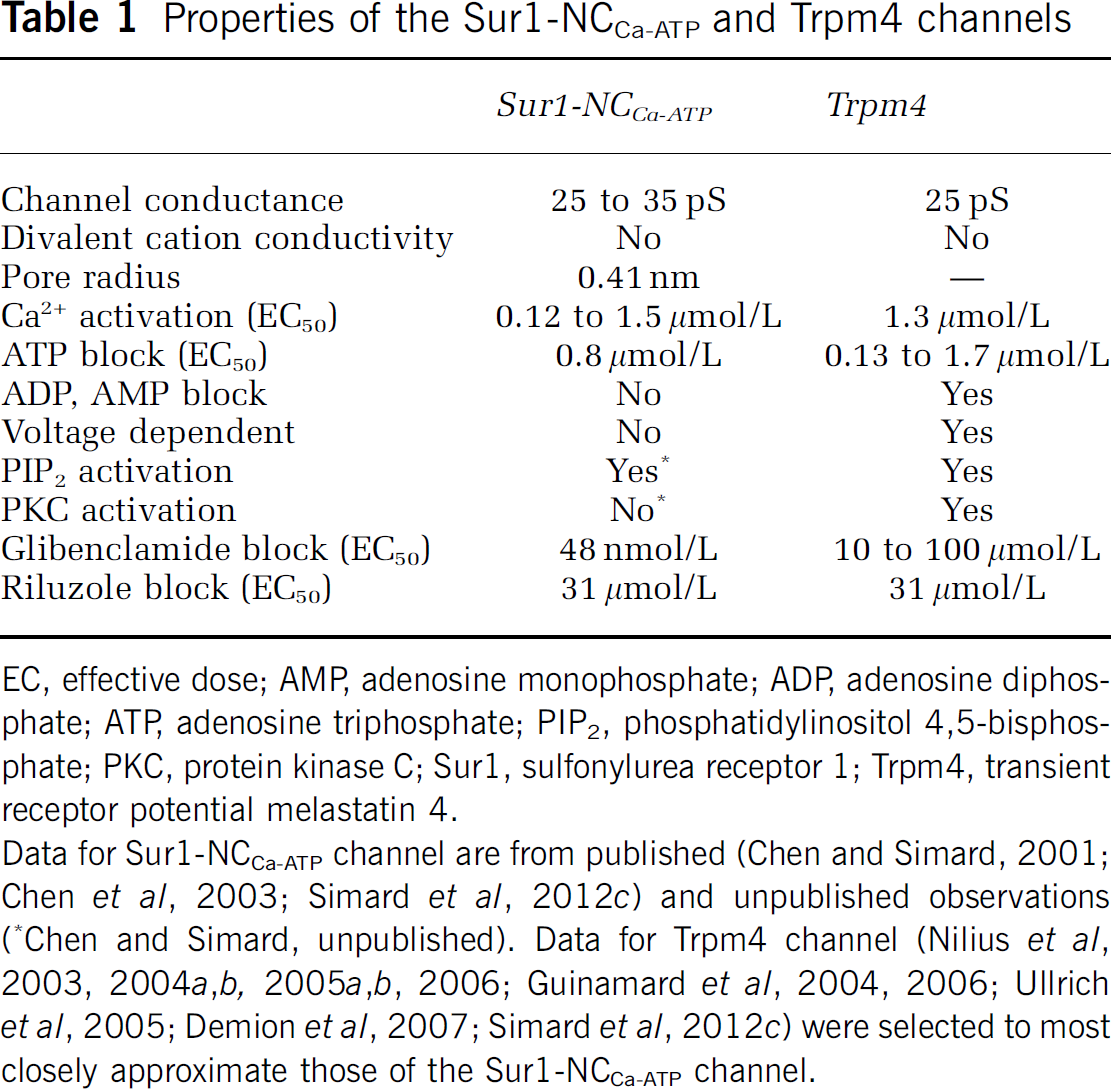
EC, effective dose; AMP, adenosine monophosphate; ADP, adenosine diphosphate; ATP, adenosine triphosphate; PIP2, phosphatidylinositol 4,5-bisphosphate; PKC, protein kinase C; Sur1, sulfonylurea receptor 1; Trpm4, transient receptor potential melastatin 4.
Data for Sur1-NCCa-ATP channel are from published (Chen and Simard, 2001; Chen et al, 2003; Simard et al, 2012c) and unpublished observations (∗Chen and Simard, unpublished). Data for Trpm4 channel (Nilius et al, 2003, 2004a,2004b, 2005a,2005b, 2006; Guinamard et al, 2004, 2006; Ullrich et al, 2005; Demion et al, 2007; Simard et al, 2012c) were selected to most closely approximate those of the Sur1-NCCa-ATP channel.
Certain pharmacological properties of the Sur1-NCCa-ATP channel are determined by the regulatory subunit, Sur1, and therefore approximate closely the pharmacological properties of KATP (Sur1-Kir6.2) channels (Chen et al, 2003). Studies on gliotic capsule astrocytes showed that the channel is blocked by first- and second-generation sulfonylureas, tolbutamide (EC50, 16.1
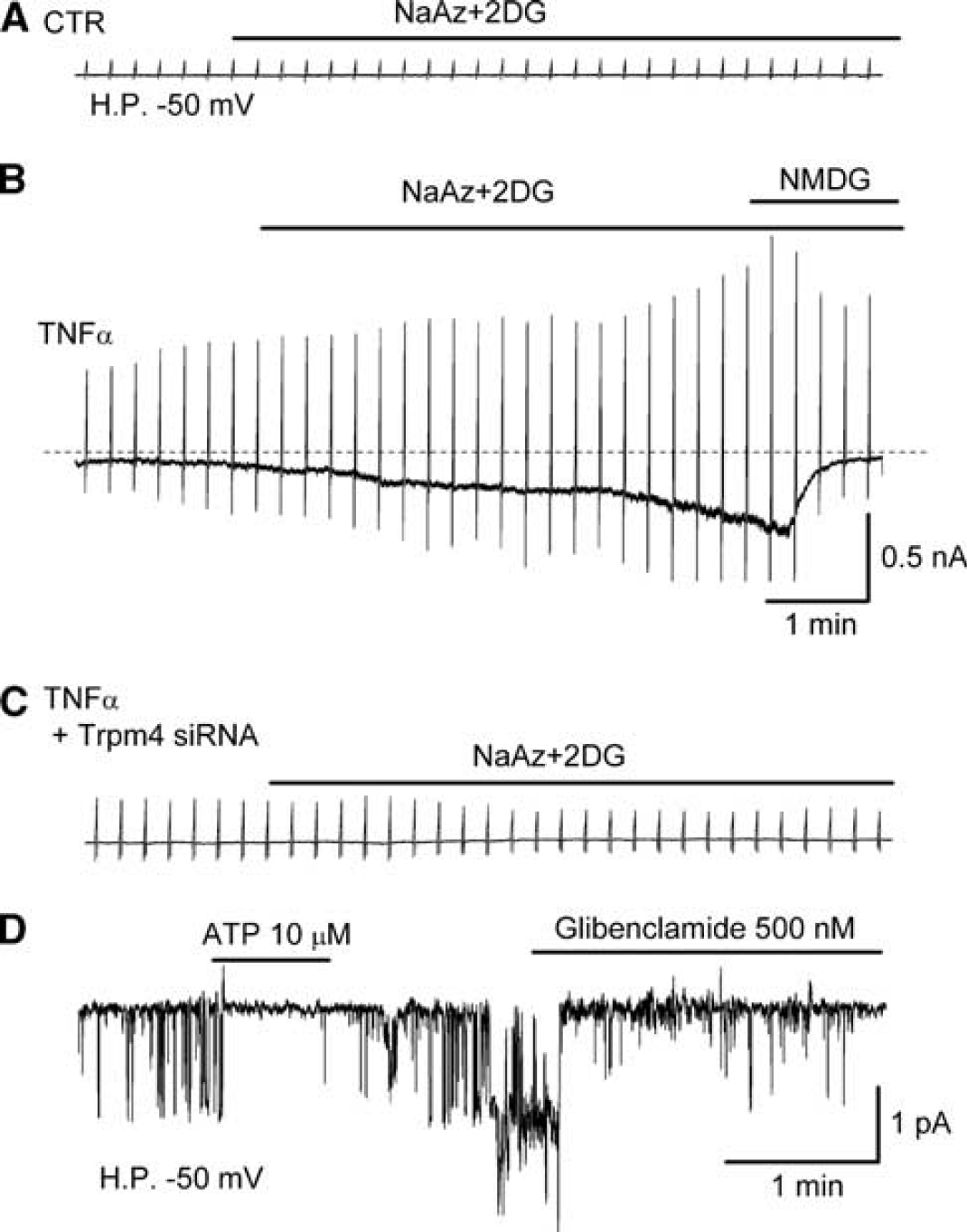
Sur1-NCCa-ATP channel currents in activated brain microvascular endothelial (bEnd.3) cells. (
Other pharmacological properties of the channel are determined by the pore-forming subunit. When the channel is induced in bEnd.3 cells by exposure to TNF
The Pore-Forming Subunit of the Sur1-NCCa-ATP Channel
Like the KATP channel, the Sur1-NCCa-ATP channel is composed of pore-forming and regulatory subunits (Figure 1). The molecular identity of the regulatory subunit, Sur1, has been known for some time (Chen et al, 2003; Simard et al, 2006). It has been postulated that the pore-forming subunit is Trpm4 (Simard et al, 2007b). As noted above, both Trpm4 and Sur1-NCCa-ATP channels exhibit similar biophysical properties (Table 1). Exposure of bEnd.3 cells to TNF
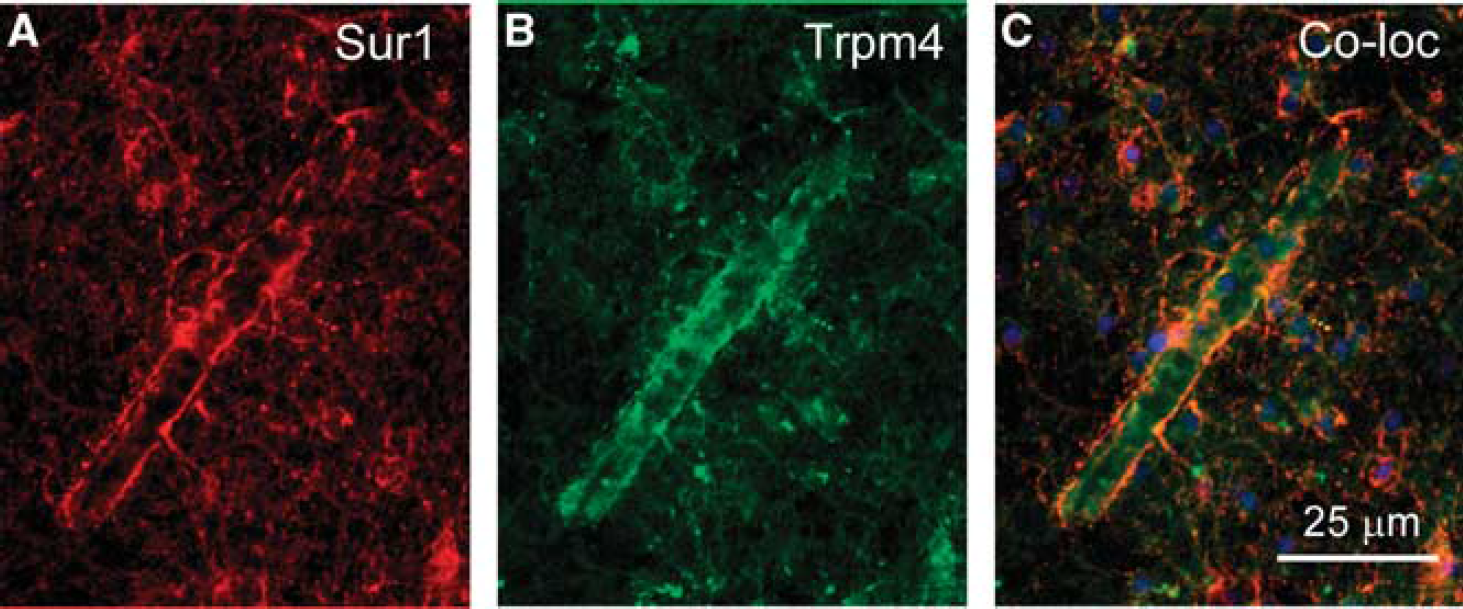
Sulfonylurea receptor 1 (Sur1) and transient receptor potential melastatin (Trpm4) colocalize after central nervous system (CNS) injury in the human. (
To date, it has been difficult to show the simple coassociation and cofunction of Sur1 and Trpm4 in a heterologous expression system (Sala-Rabanal et al, 2012). This is a key experiment that is required for molecular characterization of the channel, and one that is readily performed with Sur1 and Kir6.2 (Aguilar-Bryan et al, 1998). One important difference between Trpm4 and Kir6.2 is that the latter requires assembly with Sur1 before it can be transported to the cell membrane, whereas Trpm4 does not. This property of Trpm4 makes it easy, in a heterologous expression system, to end up with Trpm4 dominating at the cell membrane. The difficulty in showing cofunction in a heterologous expression system may suggest that expression conditions as yet unidentified may need to be developed.
Function of the Sur1-NCCa-ATP Channel
The main function of Trpm4 is the regulation of calcium influx (Vennekens and Nilius, 2007; Guinamard et al, 2011). The entry of calcium into a cell is governed by the electrochemical gradient for calcium, and the electrical portion of that gradient is determined by the cell membrane potential. Depolarization reduces the inward driving force for calcium, thereby decreasing its influx. Two properties of Trpm4 make it ideal for regulating calcium entry: (1) as a nonselective cation channel, its activation results in cell depolarization and (2) because it is activated by calcium, the cell membrane potential becomes directly linked to the intracellular calcium concentration. The same properties that make Trpm4 ideal for regulating calcium influx are shared by the Sur1-NCCa-ATP channel (Figure 4).
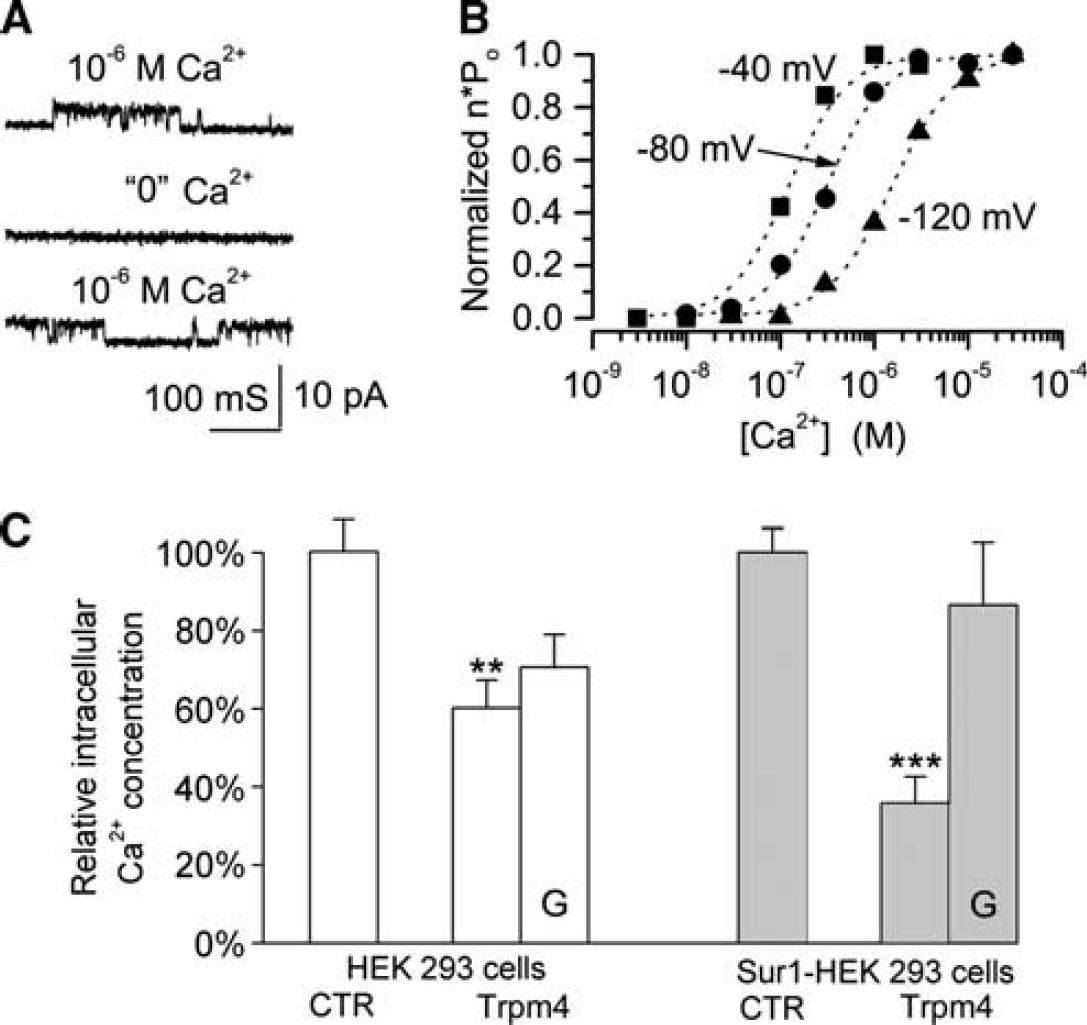
The Sur1-NCCa-ATP channel is activated by intracellular Ca2+ and regulates Ca2+ influx. (
These concepts are illustrated by an experiment in which cells were challenged with the calcium ionophore, A23187, which promotes a rise in intracellular calcium (Simard and colleagues, unpublished). Compared with control cells, cells that expressed Trpm4 accumulated significantly less intracellular calcium at steady state (Figure 4). When the foregoing experiment was repeated with cells that stably overexpress Sur1, again, transfection with Trpm4 resulted in less accumulation of intracellular calcium, compared with controls (Figure 4). Notably, the effect of Trpm4 transfection was greater in cells that also expressed Sur1, and in these cells, the effect was blocked by glibenclamide, consistent with a functional role for Sur1. These observations are consistent with the hypothesis that the Sur1-NCCa-ATP channel normally acts to protect against an excess influx of calcium during pathological conditions.
The Sur1-NCCa-ATP Channel and ‘Accidental Necrotic Cell Death’
During cell death, two types of blebs appear: dynamic blebs and larger stationary blebs (Charras, 2008). Dynamic blebbing is associated with the execution phase of apoptosis and appears closely related to blebbing in ‘healthy’ cells; larger stationary blebs appear during cell necrosis and are a common feature of cells exposed to noxious stimuli such as hypoxia, oxidants, or ATP depletion.
In gliotic capsule astrocytes, depletion of ATP using the cytochrome oxidase inhibitor, sodium azide, causes activation Sur1-NCCa-ATP channels, resulting in rapid cell depolarization to 0 mV that is accompanied by progressive formation of large stationary blebs (Chen and Simard, 2001; Chen et al, 2003). This pathological process is referred to as ‘oncotic cell swelling’ or ‘cytotoxic edema’. Oncotic cell swelling results in part from unchecked Na+ influx via Sur1-NCCa-ATP channels that is presumed to be accompanied by influx of Cl− and water. The formation of large stationary blebs, i.e., oncotic cell swelling, induced by sodium azide is blocked by glibenclamide and is reproduced without ATP depletion by opening the channel with the Sur1-activator, diazoxide (Chen et al, 2003).
Bleb formation of the same sort has been observed
In cells that express the Sur1-NCCa-ATP channel, ATP depletion leads not only to oncotic cell swelling, but eventually to cell death. Cell demise is due predominantly to nonapoptotic, propidium iodide-positive, oncotic (necrotic) cell death (Simard et al, 2006). Oncotic cell death induced by ATP depletion is largely arrested by block of the Sur1-NCCa-ATP channel with glibenclamide (Simard et al, 2006) or by gene silencing of Sur1 (Simard et al, 2010c).
In cells that heterologously express Trpm4, ATP depletion with sodium azide also leads to the formation of large stationary blebs, oncotic cell swelling and propidium iodide-positive, oncotic (necrotic) cell death (Gerzanich et al, 2009), consistent with Trpm4, not Sur1, conveying sensitivity to ATP depletion.
The Sur1-NCCa-ATP channel is a major molecular mechanism of ‘accidental necrotic cell death’ in the CNS. The only other identified mechanism, NMDA receptor-mediated excitotoxicity, is specific to neurons, whereas the mechanism mediated by the Sur1-NCCa-ATP channel involves all members of the neurovascular unit, including neurons, astrocytes, oligodendrocytes, and endothelial cells. Conceptually, ‘accidental’ cell death is substantially different from ‘programmed’ cell death (apoptosis, autophagy, and necroptosis). The designation ‘programmed’ implies that a specific molecular sequence is initiated by an identified stimulus, with the explicit purpose of causing cell death. By contrast, ‘accidental’ implies that molecular machinery that normally serves a role unrelated to death is inadvertently transformed into a cell executioner. In most types of CNS injury, including stroke and traumatic brain injury (TBI), the overwhelming form of cell death is not programmed, but is simply accidental necrosis, due to factors external to the cell or tissue.
The Sur1-NCCa-ATP channel is transcriptionally upregulated (see below), but its upregulation is not targeted specifically to inducing cell death. As reviewed above, evidence suggests that the normal function of Sur1-NCCa-ATP channels is to protect against a pathological rise in intracellular calcium, one of the hallmarks of CNS injury (Arundine and Tymianski, 2003; Bano and Nicotera, 2007). However, because Sur1-NCCa-ATP channels are also sensitive to the intracellular concentration of ATP, extreme depletion of ATP, as occurs in stroke and TBI, can result in persistent activation of the channels. Unchecked channel opening, in turn, leads to persistent sodium influx, resulting in oncotic cell swelling, formation of large stationary blebs, and eventual membrane rupture—oncotic (necrotic) cell death, or ‘accidental necrotic cell death’.
Upregulation of Sulfonylurea Receptor 1 Protein and mRNA in Central Nervous System Injury
The Sur1-NCCa-ATP channel has not been identified in normal tissues or cells, but is upregulated
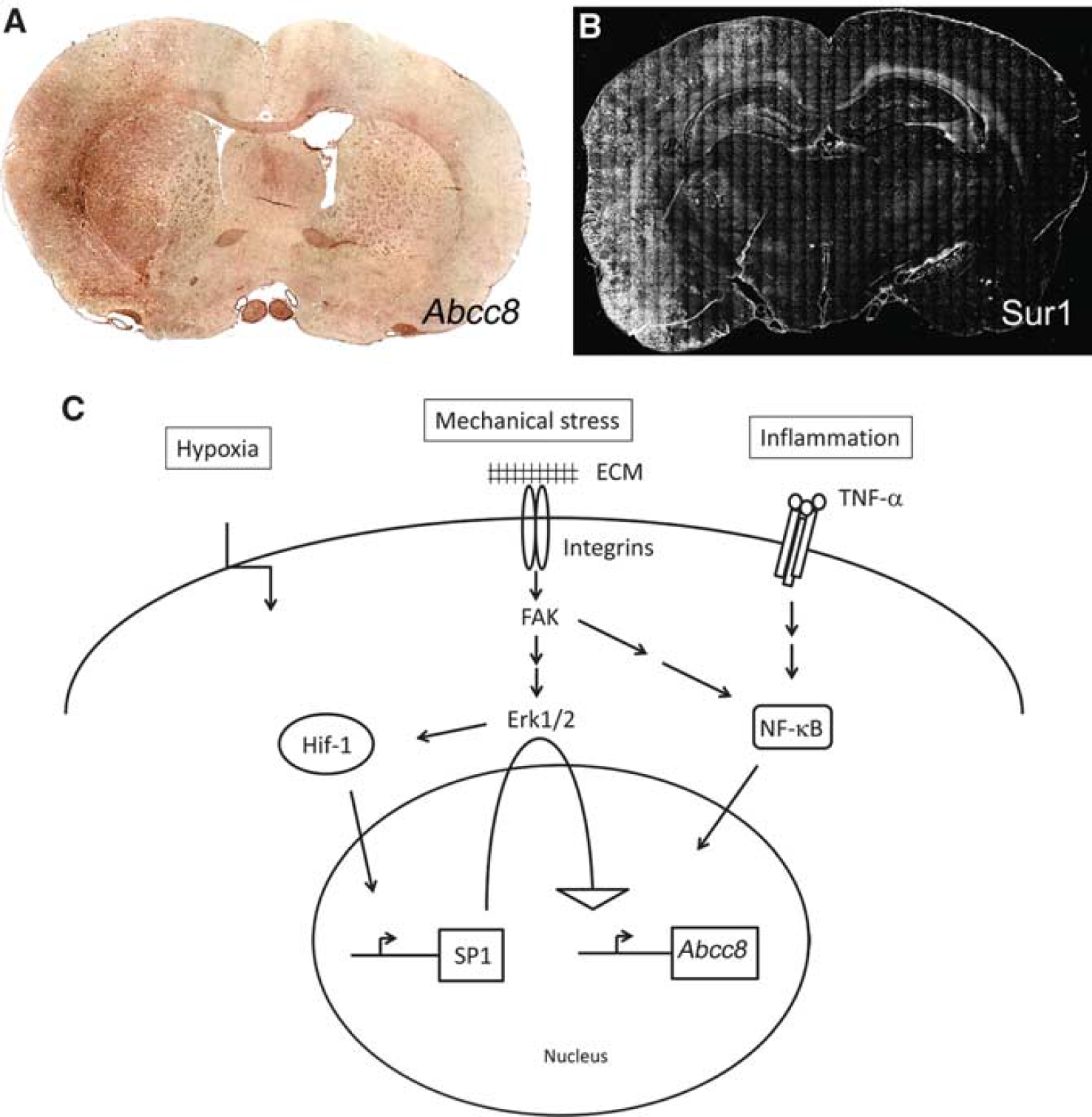
Summary of
Animal models
The original work describing the channel identified Sur1 protein and
Work with multiple models of cerebral ischemia has revealed that Sur1 is upregulated progressively during several hours after the onset of ischemia. After permanent mechanical middle cerebral artery occlusion (MCAo) in the rat, 3 hours are required before
In mechanical trauma to the CNS, upregulation of Sur1 is prominent in microvessels and neurons in both rats and mice. In TBI, upregulation of Sur1 was confirmed using immunohistochemistry, immunoblot analysis, and
In SCI, upregulation of Sur1 in microvessels and neurons has been shown using immunohistochemistry, immunoblot analysis, and
In subarachnoid hemorrhage (SAH), upregulation of Sur1 in microvessels and neurons has been shown using immunohistochemistry, immunoisolation followed by immunoblot analysis, and
Sur1 also is upregulated in cultured cells under conditions that simulate injury. Exposing cultures of brain microvascular endothelial cells, both primary cultures and bEnd.3 cells, to hypoxia or TNF
Human diseases
Two studies have analyzed systematically human tissues for Sur1 expression in the context of injury. Premature infants who die shortly after birth may be found to have a germinal matrix hemorrhage, either unrelated to or as the cause of death. Twelve such cases were examined for expression of Sur1 (Simard et al, 2008b). Regionally-specific upregulation of Sur1 protein and
Seven cases of patients who died within 3 days of SCI were studied using immunohistochemistry and
Sporadic cases have been studied showing prominent expression of Sur1 after injury. Fresh biopsy specimens were obtained from patients undergoing brain surgery for resection of a metastatic tumor surrounded by a gliotic capsule, or for removal of an intracerebral hematoma secondary to rupture of an arteriovenous malformation (see Figure 2 of Simard et al (2008b)). Immunolabeling for Sur1 was prominent in these cases. In the gliotic capsule, astrocytes were labeled exclusively, whereas in tissues adjacent to the intracerebral hemorrhage, immunolabeling for Sur1 as well as
Transcriptional Regulation of Sulfonylurea Receptor 1 Expression
Recent investigations have begun to identify the transcriptional mechanisms involved in
In the context of ischemia/hypoxia, Sur1/
Sequential transcriptional activation is a process that amplifies and prolongs the original signal from a short-lived transcription factor such as Hif, and also delays transcription of the end-target gene, here
Consistent with the role of Hif1 in upregulation of Sur1 in focal ischemia, pharmacological inhibition as well as gene suppression of Hif is protective in cerebral ischemia/hypoxia (Helton et al, 2005; Chen et al, 2007, 2010). Similarly, in the Rice-Vannucci model of neonatal hypoxia-ischemia, inhibiting Hif1 is protective (Chen et al, 2008a,2008b).
In the context of neuroinflammation, as occurs following SAH, Sur1/
In the context of trauma, Sur1/
The mechanism for activation of Sp1 and NF-
Sulfonylurea Block and Gene Suppression of Sulfonylurea Receptor 1 in Spinal Cord Injury
Spinal cord injury caused by a blunt impact is associated with development of the autodestructive process termed as ‘progressive hemorrhagic necrosis’ (PHN). It is characterized by progressive enlargement of the initial (hemorrhagic) contusion, often to twice its original volume, during the first 12 to 18 hours after injury. Microscopically, PHN appears as an advancing front of hemorrhagic necrosis that is due to the progressive catastrophic structural failure of microvessels in the penumbra surrounding the initial contusion. A unique model of impact SCI in the rat was developed specifically to permit study of this phenomenon (Simard et al, 2007c, 2012a). An impact is delivered to the cervical spinal cord of the rat in such a manner as to produce unilateral, but not bilateral, primary hemorrhagic injury. During the hours that follow, as PHN ensues, the hemorrhagic lesion expands to involve more and more of the contralateral spinal cord, converting a unilateral hemorrhagic injury into a bilateral hemorrhagic injury. It is evident that, if PHN could be halted, the hemorrhagic injury would be confined to the site of primary injury, sparing the contralateral spinal cord.
The progressive catastrophic structural failure of microvessels that characterizes PHN is associated with fragmentation of capillaries. Microvessels in the penumbra appear foreshortened, as small segments of nearly the same length and width (Simard et al, 2007c, 2010a). As noted above, microvessels in the penumbra upregulate Sur1 following impact.
Pharmacological block of Sur1 using glibenclamide or repaglinide, administered as late as 3 hours after impact, protects the capillaries, prevents their fragmentation, halts PHN, and stops the spread of the hemorrhagic contusion to the contralateral side (Figure 6; Table 2; Simard et al, 2007c, 2010c, 2012a,2012c). To date, four separate series of rats treated with glibenclamide have been reported, all with similar results, including significant improvements in neurologic function, compared with vehicle-treated rats (
Effects of Sur1 block or

SCI, spinal cord injury; TBI, traumatic brain injury; BBB, Basso, Beattie, Bresnahan rat scale for locomotion; BMS, Basso Mouse Scale for locomotion; Sur1, sulfonylurea receptor 1; CNS, central nervous system.
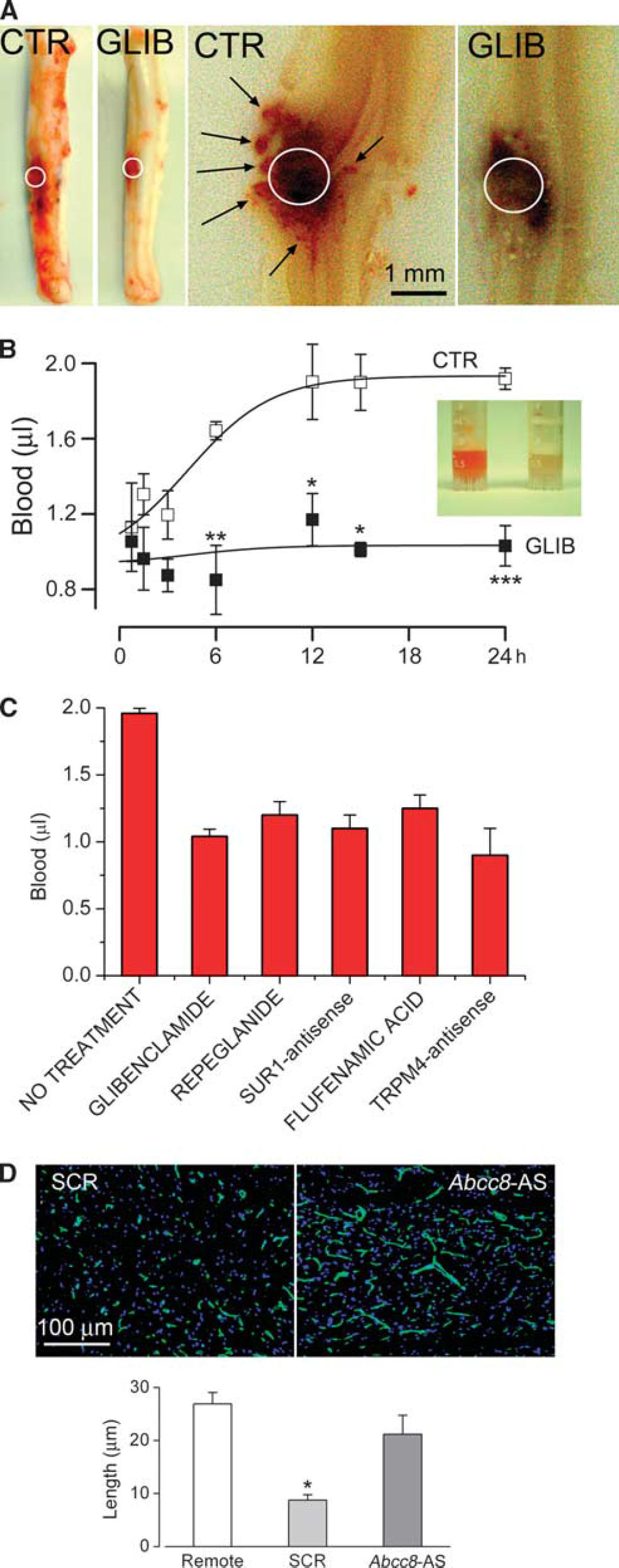
Inhibition of Sur1 or Trpm4, or gene suppression of
Pharmacological block of Trpm4 using flufenamic acid or riluzole, administered as late as 3 hours after impact, also protects the capillaries, prevents their fragmentation, halts PHN, stops the spread of the hemorrhagic contusion to the contralateral side, and is associated with significant improvements in neurologic function, compared with vehicle-treated rats (Gerzanich et al, 2009; Simard et al, 2012c). The findings with riluzole are particularly important, since this agent currently is being considered for a clinical trial in SCI. As noted above, riluzole blocks heterologously expressed Trpm4 channels as well as Sur1-NCCa-ATP channels induced in endothelial cells. Although riluzole may exert multiple biological effects, including inhibition of excitotoxicity, it is likely that block of Sur1-NCCa-ATP channels is a central mechanism for its beneficial effect in SCI (Simard et al, 2012c).
Gene suppression of
Gene suppression of
Sulfonylurea Block and Gene Suppression of Sulfonylurea Receptor 1 in Traumatic Brain Injury
As with SCI, TBI also may be associated with progressive expansion of a hemorrhagic contusion, but in this context a different term is used—hemorrhagic progression of a contusion (Kurland et al, 2012). As in SCI, the progressive catastrophic structural failure of microvessels that characterizes hemorrhagic progression of a contusion is associated with fragmentation of capillaries. Microvessels in the penumbra appear foreshortened, as small segments of nearly the same length and width (Simard et al, 2009b). As noted above, microvessels in the penumbra upregulate Sur1 following impact.
In a cortical impact model of severe injury, the hemorrhagic contusion visibly enlarges in all directions, and the amount of blood that accumulates in the tissues doubles during the first 12 hours after injury (Simard et al, 2009b). Pharmacological block of Sur1 using glibenclamide prevents capillary fragmentation, halts hemorrhagic progression of a contusion, and prevents the further accumulation of blood in the tissues (Table 2; Simard et al, 2009b). Preventing expansion of the hemorrhagic lesion is associated with significant improvements in neurologic function.
As in SCI, administration of AS-ODN directed against
In a cortical impact model in rats that was calibrated to avoid primary contusion injury to the hippocampus, neuronal degeneration and apoptosis in the hippocampus were prominent and were associated with significant deficits in rapid spatial learning, a hippocampus-specific task (Patel et al, 2010). Pharmacological block of Sur1 using glibenclamide significantly reduced neuronal degeneration and apoptosis, and was highly effective in sparing rapid spatial learning.
At present, a prospective, multicenter, placebo-controlled, double-blind, Phase IIa trial of RP-1127 (glyburide for injection; Remedy Pharmaceuticals, Inc., New York, NY, USA) is underway to test the effect of RP-1127 in patients with moderate-to-severe TBI (ClinicalTrials.gov Identifier: NCT01454154).
Sulfonylurea Block of Sulfonylurea Receptor 1 in Cerebral Ischemia
Focal ischemic injury involving the brain is associated with progressive microvascular dysfunction that is manifested initially by the formation of ionic edema, which may be followed by formation of vasogenic edema and, if severe enough, may be followed by the catastrophic structural failure of capillaries—so-called, ‘hemorrhagic transformation’ of an ischemic stroke (Simard et al, 2007a). Ionic and vasogenic edema, as well as hemorrhagic transformation, add mass to the brain that causes tissue swelling. Swollen tissues can compromise adjacent tissues and worsen the original insult. If severe, as in malignant cerebral edema after a large stroke, then death of the organism may ensue. The molecular events resulting in these various forms of microvascular dysfunction are complex, but they include the upregulation of Sur1 and subsequent Sur1-mediated microvascular dysfunction, as suggested by the salutary effects of inhibiting Sur1.
Animal models
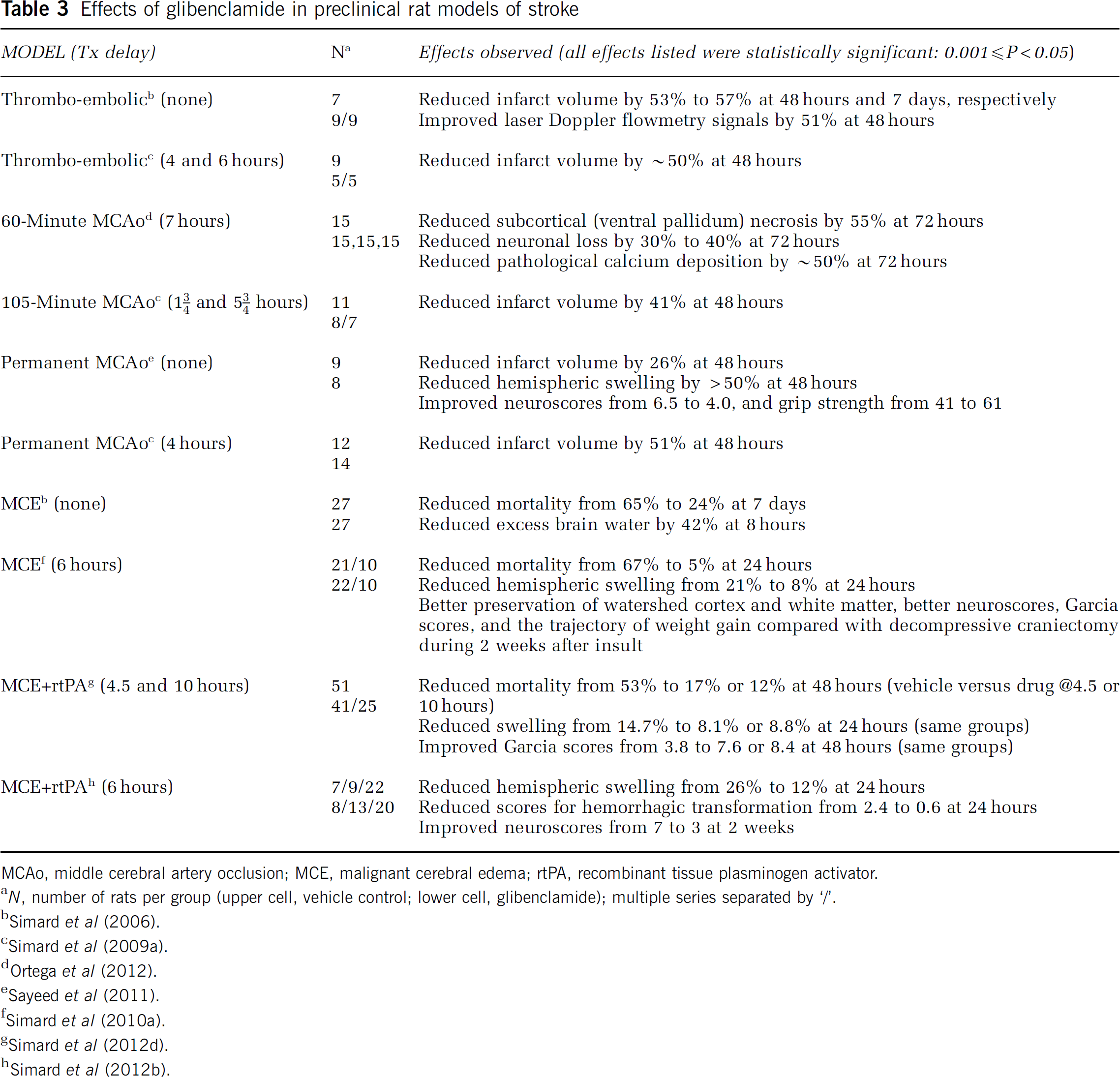
The effect of glibenclamide has been studied in various rat models of stroke (Figure 7; Table 3). The experiments with mechanical MCAo reviewed below were performed using laser Doppler flowmetry to assure a reduction of >75% during monitoring for the initial 30 minutes after occlusion. Unless otherwise noted, a loading dose of 10
Effects of glibenclamide in preclinical rat models of stroke
MCAo, middle cerebral artery occlusion; MCE, malignant cerebral edema; rtPA, recombinant tissue plasminogen activator.
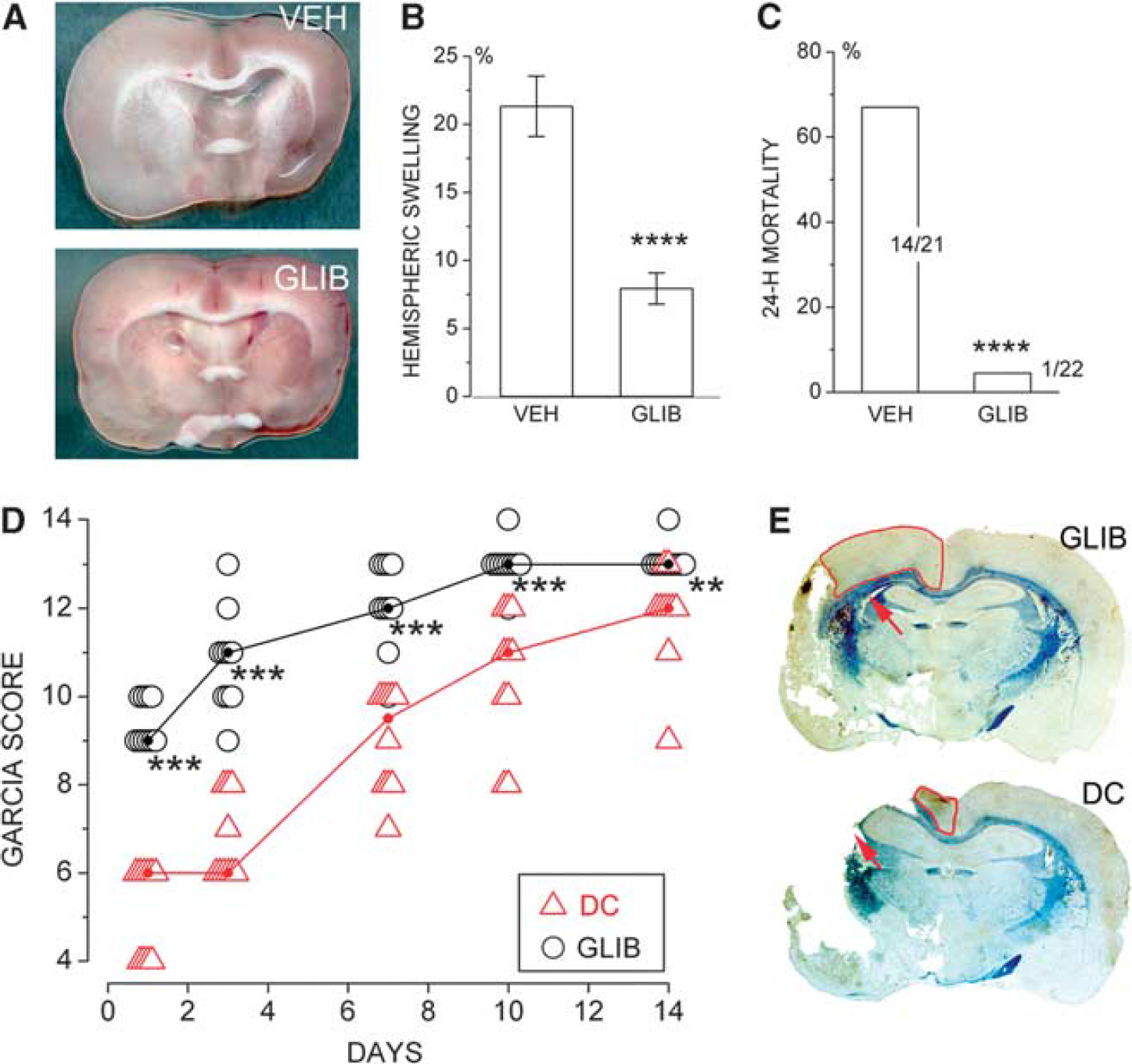
Inhibition of sulfonylurea receptor 1 (Sur1) prevents brain swelling and death after malignant cerebral infarction. (
The effect of glibenclamide was studied in three nonlethal rat models of ischemic stroke, including a thromboembolic model, a 105-minute temporary MCAo model, and a permanent MCAo model (Simard et al, 2006, 2009c). In these experiments, the administration of glibenclamide beginning as late as 5.75 hours after onset of ischemia was associated with 40% to 50% reduction in lesion volumes. In one study with thromboemboli, the use of glibenclamide was associated with cortical sparing that was attributed to improved leptomeningeal collateral blood flow due to reduced mass effect from edema (Simard et al, 2006). In the study with temporary MCAo, smaller lesions were associated with less hemispheric swelling (Simard et al, 2009c).
Independent experiments from other laboratories also have shown beneficial effects of glibenclamide in nonlethal rat models of stroke. One group studied a model with 60-minute MCAo and, beginning 6 hours after the onset of ischemia, administered three intravenous injections of glibenclamide during the first 24 hours (Ortega et al, 2012). Beneficial effects on neurologic outcome and neuronal preservation were observed at 3 days, including reduced subcortical (ventral pallidum) necrosis, reduced neuronal loss, and reduced pathological calcium deposition. A recent study in a model of permanent MCAo replicated the same dosing regimen of glibenclamide detailed in the report by Simard et al (2009c), and found that glibenclamide significantly reduced hemispheric swelling and lesion size, and significantly improved functional outcomes (Sayeed et al, 2011).
The effect of glibenclamide was examined in a rat model of malignant cerebral edema that used particle embolization (Simard et al, 2006). Malignant edema was manifested as an increase in tissue water from a normal value of 78% to 83% at 8 hours and was associated with 65% mortality at 7 days. Administration of glibenclamide (75 ng/h with no loading dose), beginning shortly after onset of ischemia, decreased tissue water to 80%, and reduced mortality at 7 days to 24%.
The effect of glibenclamide was examined in a rat model of malignant cerebral edema involving 6-hour temporary MCAo (Simard et al, 2010b). This insult was associated with thrombosis of proximal and distal middle cerebral artery branches. In some animals, spontaneous thrombolysis required 2 to 3 hours before laser Doppler flowmetry signals returned to normal. Malignant edema was manifested as 22% increase in hemisphere volume and was associated with 67% mortality at 24 hours. Administration of glibenclamide at 6 hours, at the time of recanalization, decreases hemispheric swelling to 7.5% and reduced mortality to 5%. Using this model, the effect of glibenclamide was compared with the effect of a large decompressive hemicraniectomy. Both treatments, administered at 6 hours, were equally effective in essentially eliminating mortality. However, neurologic function during the subsequent 2 weeks showed that glibenclamide was superior to decompressive craniectomy (Figure 7). The superior effect of glibenclamide on neurologic function was associated with better tissue preservation of the corpus callosum and of the watershed cortex of the anterior and middle cerebral arteries.
An important, albeit poorly studied outcome in animal models of stroke is the general health of the organism. Temporary MCAo in rats is associated with severe weight loss of unknown etiology that is not explained by dehydration, stress, or altered hormonal secretions (Petullo et al, 1999; Virtanen et al, 2003, 2004). In the foregoing study (Simard et al, 2010b), weight gain quickly resumed its normal trajectory in the rats treated with glibenclamide, but not in those treated with decompressive craniectomy. Several reasons potentially could account for this, including better sparing of cortico-striatal and hypothalamic tissues in the rats treated with glibenclamide.
At present, the only treatment for stroke that is approved by national medical regulatory agencies worldwide is rtPA, but this treatment, especially if used late and with large strokes, is associated with increased morbidity due to edema and hemorrhagic transformation (Lansberg et al, 2007). The effect of glibenclamide was examined in a rat model of malignant cerebral edema involving 4.5-hour MCAo plus administration of rtPA (0.9 mg/kg intravenously) at the time of occluder withdrawal (Simard et al, 2012d). In these experiments, the specific duration of MCAo and the dose of rtPA were chosen for their clinical relevance. Malignant edema was manifested as 8% and 15% increases in hemisphere volume measured at 10 and 24 hours, respectively, and was associated with 53% mortality at 48 hours. Administration of glibenclamide at 4.5 hours, at the time of recanalization and administration of rtPA, decrease hemispheric swelling to 4% and 8% at 10 and 24 hours, respectively, reduced mortality to 17%, and yielded significant improvements in neurologic function. Administration of glibenclamide at 10 hours after onset of ischemia (5.5 hours after recanalization plus administration of rtPA) decrease hemispheric swelling to 8% at 24 hours, reduced 48-hour mortality to 12%, and yielded significant improvements in neurologic function. In these experiments, lesion volumes were not changed by glibenclamide, consistent with 4.5-hour MCAo producing a maximum ischemic insult. However, examining the relationship between infarct volume and neurologic function was revealing. In the controls, a steep, highly significant negative correlation was found. By contrast, with glibenclamide, the association was much less steep and was not statistically significant, indicating that glibenclamide essentially dissociated functional outcome from infarct volume, presumably due to the significant reductions in edema and swelling.
The effect of glibenclamide was studied in a rat model of malignant cerebral edema involving 6-hour MCAo plus administration at the time of occluder withdrawal of a dose of rtPA (10 mg/kg intravenously) that is expected to be thrombolytic in rats (Simard et al, 2012b). Malignant edema was manifested as 25% increase in hemisphere volume and a high incidence of hemorrhagic transformation at 24 hours. Administration of glibenclamide at 6 hours, at the time of recanalization and administration of rtPA, decrease hemispheric swelling to 12% and significantly reduced scores measuring hemorrhagic transformation. Mortality and functional outcomes (neuroscores) were equally poor in rats without or with treatment with rtPA, with neuroscores of 7 to 8 in both groups (8 signifies death, 7 signifies coma). Glibenclamide significantly reduced mortality and improved neuroscores, both without or with treatment with rtPA, with neuroscores of 3 to 4. The best functional outcomes were observed in rats treated with a thrombolytic dose of rtPA plus glibenclamide. The beneficial effects of glibenclamide endured for the 2 weeks of observation.
Finally, protective effects of glibenclamide in the context of cerebral ischemia have been reported in two studies in which the effect was unlikely to be due to Sur1-NCCa-ATP channels, since there was no time for transcriptional upregulation of the channel. The first study involved a model of global ischemia/reperfusion (15 min/60 min), in which glibenclamide pretreatment was found to: (1) reduce neutrophil recruitment; (2) restore pro-oxidant/antioxidant balance; (3) reduce the inflammatory mediators, TNF
The second study involved an
Humans
The effect of sulfonylureas on stroke in humans has been examined retrospectively in diabetic patients presenting with ischemic stroke, as well as prospectively in nondiabetic patients with large ischemic strokes. In the retrospective studies on diabetics, patients whose diabetes was managed without sulfonylurea drugs (controls) were compared with patients whose diabetes was managed with sulfonylurea drugs and who were maintained on sulfonylurea drugs after hospitalization for stroke.
The medical records of patients with diabetes mellitus admitted to the hospital within 24 hours of onset of acute ischemic stroke presenting to the Neurology Clinic, Charité Hospital, Berlin, Germany, during 1994 to 2000 were analyzed (Kunte et al, 2007). After exclusions, the cohort comprised 33 patients taking a sulfonylurea (glibenclamide, glimepiride, or glibornuride) at admission through discharge (treatment group) and 28 patients not on a sulfonylurea (control group). The primary outcome was a decrease in NIHSS (National Institutes of Health Stroke Scale) of 4 points or more from admission to discharge or a discharge NIHSS score=0, either of which is considered as a ‘major neurologic improvement’. The secondary outcome was a discharge modified Rankin Scale score of 2 or less, which signifies functional independence. No significant differences, other than stroke subtype, were observed among baseline variables between control and treatment groups. The primary outcome was reached by 36% of patients in the treatment group and 7% in the control group (odds ratio (OR)=7.4; 95% confidence interval (CI)=1.5 to 37;
Another retrospective study of diabetic patients from the Registry of the Canadian Stroke Network was reported at the International Stroke Conference in 2009 (Silver et al, 2009; Frank Silver, personal communication). Of 15,814 patients screened, 2,448 with diabetes were admitted to hospital within 24 hours of symptom onset. For patients who had been on and continued on sulfonylurea drugs, compared with controls not on sulfonylureas, the likelihood of in-hospital mortality was less (OR=0.51; CI=0.37 to 0.71;
Recently, a multicenter, prospective, open label, Phase IIa trial of RP-1127 (glyburide for injection, Remedy Pharmaceuticals, Inc.) was completed (ClinicalTrials.gov Identifier: NCT01268683) (Kevin N Sheth and colleagues, personal communication). This clinical trial tested the effect of RP-1127 in 10 patients with a severe anterior circulation ischemic stroke (baseline infarct volume 82 mL; mean, 102±23 mL; baseline NIHSS score, 19±8) at high risk for malignant cerebral edema. Of the 10 patients, 1 died, even after decompressive craniectomy. The incidence of malignant edema was 20%, compared with 88% in a prospective observational study of patients with infarct volumes 82 mL (Thomalla et al, 2010). Moreover, 8/10 patients did not require osmotherapy, intubation, or decompressive craniectomy, and there were no clinically significant parenchymal hematomas (‘PH1/PH2’), in contrast to parenchymal hematoma rates of ∼30% in the DEFUSE and EPITHET trials in patients with a malignant profile (Mlynash et al, 2011). Most remarkable, the proportion of patients with 30-day modified Rankin Scale scores 4 was 90%, compared with 24% (at 12 months) in control patients from a pooled analysis of decompressive craniectomy trials (Vahedi et al, 2007), or 29% (at 90 days) for patients with infarct volumes >70 mL (Sanak et al, 2006). A larger clinical trial studying the effect or RP-1127 in patients with large ischemic strokes is anticipated.
Sulfonylurea Block of Sulfonylurea Receptor 1 in Perinatal Cerebral Ischemia
The effect of glibenclamide was examined in a rat pup model of neonatal hypoxia-ischemia, induced by unilateral carotid ligation plus 2- or 2.5-hour exposure (moderate and severe insults, respectively) to 8% O2 in 10-day old pups (modified Rice-Vannucci model) (Zhou et al, 2009). Unlike the forgoing experiments in rats that used constant infusion of low-dose glibenclamide, in these studies, glibenclamide (10
Sulfonylurea Block of Sulfonylurea Receptor 1 in Subarachnoid Hemorrhage
Subarachnoid hemorrhage initiates numerous injury cascades including oxidative stress, inflammation, and vasospasm (Macdonald et al, 2007; Simard et al, 2010a; Sehba et al, 2011).
The effect of glibenclamide was examined in a model of SAH induced by unilateral carotid filament puncture of the internal carotid artery (Simard et al, 2009a). Subarachnoid hemorrhage caused a large increase in barrier permeability (extravasation of immunoglobulin G) and disrupted the normal junctional localization of the tight junction protein, zona occludens 1. Glibenclamide significantly reduced both effects. In addition, SAH caused large increases in markers of inflammation, including TNF
Sulfonylureas and the Blood–Brain Barrier
As reviewed above, the treatment of acute CNS injury with glibenclamide may be highly beneficial. Normally, however, glibenclamide and other sulfonylureas do not accumulate in the brain in physiologically meaningful concentrations (Tomiyama et al, 1999). This accounts for the observation that, despite decades of clinical use in diabetic patients, there are no reports of side effects involving the CNS that are attributable directly to sulfonylureas (as distinct from CNS effects of hypoglycemia), even though the brain contains many regions with neurons that express KATP (Sur1-Kir6.2) channels (Liss and Roeper, 2001).
The net accumulation of sulfonylureas in the brain is governed by passive transport into, and active transport out of the brain. Transport across the blood–brain barrier (BBB) has been studied for the first-generation sulfonylurea, tolbutamide (Takanaga et al, 1998; Koyabu et al, 2004). The net accumulation of tolbutamide in the brain of rats is much lower than in other organs or tissues, and is almost nil compared with propranolol, a
Passive transport from the blood across an ‘intact’ BBB into the brain is determined by lipid solubility. As weak acids, glibenclamide (pKa, 5.3) and tolbutamide (pKa, 5.4) exist in ionized (hydrophilic, impermeant) and nonionized (lipophylic, permeant) forms. According to the Henderson-Hasselbach relationship (a.k.a., the pH-partition hypothesis), an equilibrium is established between the ionized and nonionized forms. For a weak acid, the ratio of ionized to nonionized forms is 10(pH−pK):1. For a weak acid like glibenclamide with a pKa of 5.3, the ratio of ionized to nonionized at pH 7.3 will be 100:1, i.e., the ionized, impermeant form will dominate at neutral pH. Thus, poor lipid solubility at neutral pH, combined with active transport out of endothelial cells and into the blood, results in a very low net accumulation of sulfonylureas in the normal brain.
The situation is quite different in ischemic brain tissues. Ischemia is associated with high glycolytic activity resulting in lactic acidosis that can drive extracellular pH to values ∼6.5 (Nedergaard et al, 1991). The phenomenon termed as ‘ion trapping’ occurs when there is a large permeability difference between ionized (impermeant) and nonionized (permeant) species of a drug across a membrane (Raghunand and Gillies, 2000). When extracellular pH is appreciably lower than intracellular pH, a weak acid will tend to concentrate in the more alkaline compartment, in our case, intracellularly in endothelial cells and neurons.
Apart from pH-dependent passive transport across an ‘intact’ BBB, glibenclamide also may be passively transported into the brain after ‘breakdown’ of the BBB. Breakdown of the BBB induced by ischemia leads to the passive transport of plasma across the compromised BBB, i.e., the formation of vasogenic edema. Glibenclamide, which is highly protein bound in the circulation (Skillman and Feldman, 1981), thus would be transported into the brain during the formation of vasogenic edema.
Empirical evidence indicates that glibenclamide indeed is transported from blood into the ischemic brain. Accumulation of the fluorescent analog, BODIPY-glibenclamide, in ischemic brain was reported after MCAo (Simard et al, 2006): in control tissues contralateral to the infarct, fluorescent drug was excluded, but in the ischemic tissues, the microvessels and neurons appeared bright with fluorescent drug (see Figure 6 of Simard et al (2006)). Independent confirmation of preferential uptake of glibenclamide into ischemic brain recently was published (Ortega et al, 2012): only 0.025% of injected [3H]glibenclamide entered the control brain, but after permanent MCAo (with the vessel still occluded), this value increased three-fold to 0.077% in the ischemic tissues.
The greater penetration of sulfonylureas into ischemic brain, due either to low pH or to breakdown of the BBB, has the effect that weak acids such as sulfonylurea drugs ‘selectively’ target injured tissues over normal tissues. As a result, relatively low doses of drug can be used to obtain a favorable therapeutic effect, while minimally affecting insulin secretion in the pancreas.
Conclusion
Recent advances have broadened our understanding of the role of Sur1-NCCa-ATP channels in acute CNS injury, especially its role in oncotic cell swelling and oncotic (necrotic) cell death, and its role in microvascular dysfunction marked by edema formation and delayed secondary hemorrhage. The channel is transcriptionally upregulated
Sur1 was discovered several decades ago and since has been the subject of intensive investigation due to its role in forming KATP channels, which are critically important in diabetes. As the present review suggests, the role of Sur1 in disease may be significantly larger than heretofore appreciated, due not to its association with Kir6.2, but to its involvement with a nonselective cation channel, possibly Trpm4. The lives of many patients already have been improved enormously by pharmacological manipulation of Sur1. If the preclinical and early phase clinical work reviewed here continues on its present trajectory, then it is likely that many more lives will be affected, perhaps even saved, by pharmacological manipulation of Sur1 in CNS injury.
Ethics statement
All of the experiments reviewed herein that were performed in the laboratory headed by the senior author adhered to the following criteria: (1) all experiments were performed in accordance with the relevant guidelines and regulations as stipulated in the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals; (2) in all cases, specific experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Maryland; and (3) all efforts were made to minimize the number of animals used and their suffering.
Footnotes
Acknowledgements
The authors acknowledge the enormous contributions made by the many scientists with whom we have been privileged to collaborate, and who coauthored the numerous publications describing this work during the last decade.
Disclosure/conflict of interest
JMS holds a US patent (#7,285,574), ‘A novel nonselective cation channel in neural cells and methods for treating brain swelling’, and is a member of the scientific advisory board and holds shares in Remedy Pharmaceuticals. No support was provided by Remedy Pharmaceuticals to JMS for this project.