
Abstract
Although relatively few in number compared to astrocytes and neurons, microglia demonstrate multiple, varied neuroimmunological functions in the central nervous system during normal and pathological states. After injury to the brain or spinal cord, microglia express beneficial pro- and anti-inflammatory phenotypes at various stages of recovery. However, prolonged microglial activation following injury has been linked to impaired parenchymal healing and functional restoration. The nature and magnitude of microglial response to injury relates in part to peripheral immune cell invasion, extent of tissue damage, and the local microenvironment.
Glial cells were distinguished from neurons during the nineteenth century. Their varied morphology and distinctive functions were subsequently observed by pioneers such as Ramon y Cajal who classified astrocytes. Cajal’s protégé, Pio del Rio-Hortega further described oligodendrocytes and microglia. He labeled the latter as a ‘third element’ of the central nervous system (CNS), noting microglial heterogeneity and phagocytic functions. 1 During the twentieth century, properties of microglia versus other glial and peripheral immune cells were further defined. Astrocytes and oligodendrocytes were understood to emerge from distinct progenitors within the neural tube ectoderm, while microglia share a lineage with macrophages from the yolk sac. 2 Both phagocytose pathogens and release growth factors, cytokines, and chemokines; however, microglia express distinct cell surface biomarkers and exhibit calcium wave responses not observed in macrophages. 3 Microglia compose approximately 10% of CNS cells, but this varies from 5% to 15% depending upon location and local environmental factors. In addition to their neuroprotective role against native and invasive pathogens, microglia detect alterations in the local microenvironment, contribute to myelin homeostasis, and participate in neuronal synaptic density/remodeling. 4 Normally microglia are observed with a ramified morphology (multiple thin, extending processes) when inactivated. Microglia have also been described as moderately activated with larger cell bodies and shorter processes in conjunction with inflammatory factor release, while some activated microglia in inflammatory immune responses appear spherical with significant pro-inflammatory cytokine/chemokine production. 5 Thus, while never in a truly quiescent state, microglia adapt their morphology and function to the local microenvironment to participate in CNS physiology, development, senescence and injury (Figure 1).
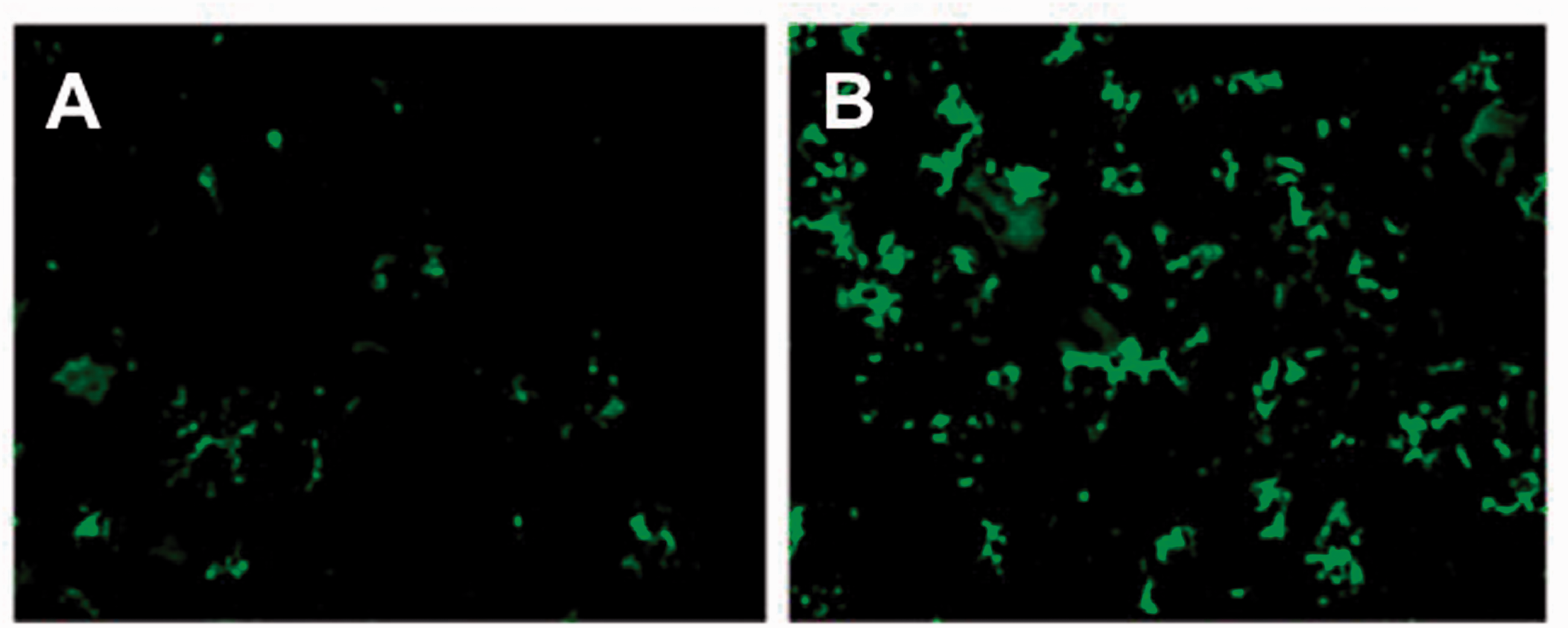
Immunofluorescent staining from 5 µm frozen sections (OX-42 primary antibody) demonstrating microglia in uninjured rodent spinal cord (a) in comparison with activated microglia (b) in the ventral horn just caudal to the site of SCI (200×). (Modified from Samantaray et al., Neurochem Res 2011; 36:1809–1816). 97
Microglia in inflammatory responses
Immune responses within the CNS are somewhat distinct from peripheral responses to injurious stimuli. For instance, antibodies present throughout most mammalian organ systems are largely absent in the CNS due to their size relative to the blood-brain barrier (BBB). Thus, microglia must rapidly distinguish local pathogens and function as antigen presenting cells for T lymphocytes as part of the innate cell-mediated immune response. 6 In addition to upregulated CD86, MHC II, and complement factor expression, activated microglia release both pro- and anti-inflammatory factors including TNF-α, IL-1α, MIP-1α, IL-1β, and IL-10. Specific microglial cytokines/chemokines may contribute to disruption of the BBB – allowing for infiltration of peripheral immune cells (monocytes, neutrophils, etc.) and further expansion of the inflammatory cascade. 7
Activated microglia also appear to have polarization states that are somewhat similar to the distinct functional phenotypes of peripheral macrophages. In response to microenvironmental stimuli, activated macrophages in the M1 polarization state participate in phagocytosis and produce pro-inflammatory cytokines and microbiocidal molecules. 8 The alternatively activated M2 phenotype is associated with repair of damaged cells and resolution of the inflammatory cascade. 9 Although not identical to macrophage polarization, microglial activation phenotypes have been described as ‘M1-like’ and ‘M2-like’ since they have similar pro- and anti-inflammatory profiles (Figure 2). 10 In both microglia and macrophages, various epigenetic mechanisms, signaling pathways, extracellular cytokine profiles, and transcription factors can influence polarization states. Pro-inflammatory activated microglia respond with phagocytosis and production of reactive oxygen species (ROS) and related by-products. 10 In specific scenarios, however, these pathways contribute to surrounding neuronal cell death. For example, microglial NADPH oxidase upregulation results in superoxide production, while nitric oxide (NO), glutamate and cathepsin B production contribute to disruption of plasma membranes. 11 Moreover, microglia that have been primed to previous inflammatory ligands can continue to present MHC II and create a chronic hyperinflammatory response to subsequent antigen presentation. 12 Xing et al. have recently shown that endothelial cells and astrocytes also provide differing signals to microglia that may determine their phenotype switch and beneficial versus deleterious effects. 13 Likewise, microglia demonstrate different molecular patterns and morphologies depending on disease type and brain region. 14 Thus, a microglial-mediated chronic pro-inflammatory state can contribute to local neuronal degeneration.
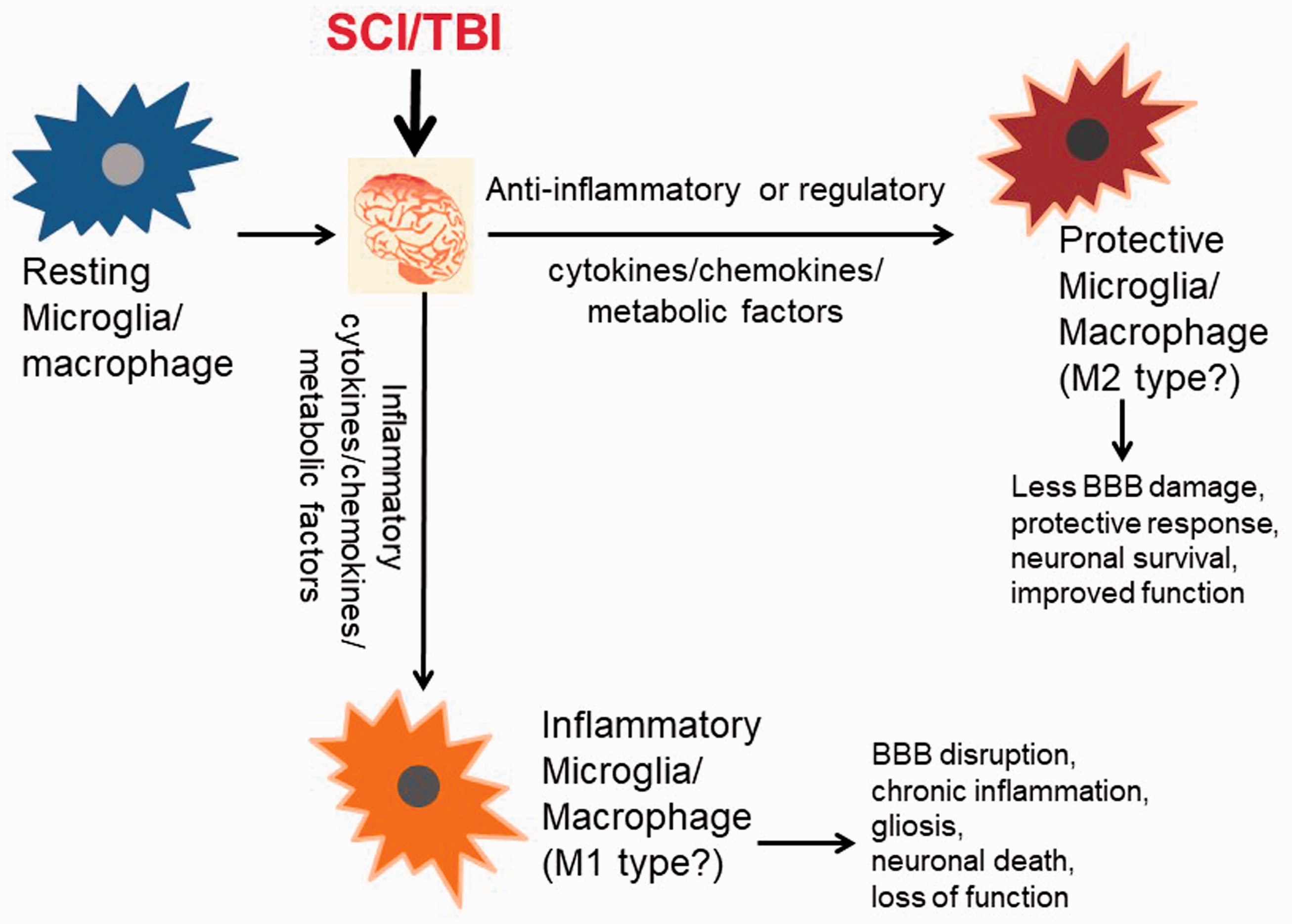
Depiction of divergent microglial inflammatory phenotypes following CNS injury.
Role of microglia in spinal cord injury
Disruption of the spinal cord parenchymal architecture at the site of impact (primary injury) triggers local cell death via disruption of cell membranes, ionic homeostasis dysregulation, ischemia, neurotransmitter accumulation, oxidative stress, and multiple injurious cascading pathways. Ramified microglia adjacent to the site are thereby activated, and more distant microglia can migrate to the area of injury. This migration is coupled with extensive microglial proliferation at the lesion site; both processes are most notable during the first 14 days after injury. During the acute phase, neutrophils have also been observed to migrate to the injury site in large numbers by day 1 following injury. In addition to release of ROS and pro-inflammatory factors, neutrophils phagocytose the damaged cellular debris. 15 By post-injury day 3, dendritic cells, natural killer cells, T lymphocytes, and monocytes are observed in larger numbers. 16 , 17 During this period, cavitation at the injury epicenter may develop due to necrosis or apoptosis of endogenous neuronal/glial cells. This cell death progression is spatially contained to some extent by glial scar encapsulation. Phagocytosis during the first three days after injury is largely due to microglia, but recent evidence suggests infiltrating macrophages account for most phagocytic activity from days 7 to 42. 18 Microglia appear to more efficiently clear phagocytosed cellular debris, while macrophages are themselves more likely to undergo cell death. After approximately one week, the anti-inflammatory component of the immune response begins to promote repair and tissue regeneration; often after several weeks, redundant infiltrating immune cells are cleared from the injury site. 19
Microglial response to injury is in part modulated by interactions with nearby glial and nonglial cells. Under normal conditions, microglia communicate with neurons via the CD200 receptor to maintain a non-activated state. 20 Structural damage to the spinal cord leads to a local accumulation of normally intracellular damage-associated molecular patterns (DAMP) such as nucleic acids, hemoglobin, oxidative phosphorylation metabolites, etc. 21 Microglia are activated by DAMP binding to toll-like receptors (and other pattern recognition receptors on microglial cell surfaces); these ligands also activate astrocytes and attract peripheral immune cells to the injury site. 22 Secreted factors such as cytokines from these immune cells further contribute to microglial activation whereby the latter upregulate signaling pathways for production of immunomodulatory receptors, transcription factors, and cytokines/chemokines that further promote the immune response. 23
Since the BBB is often disrupted, peripheral immune cells also contribute to the inflammatory cycle and interact with activated microglia and reactive astrocytes to form a glial scar 4–15 days after injury. 24 , 25 This secondary injury component includes deposition of chondroitin-sulfate proteoglycan (extracellular matrix protein) by reactive astrocytes that inhibits axonal regeneration. 26 Microglial and astrocytic processes contribute to a mesh-like (fibrous/glial) structure that includes infiltrating immune cells such as fibroblasts which secrete laminin, collagen, and fibronectin. 27 Although glial scar formation impedes axonal regeneration, transgenic animal models without reactive gliosis have demonstrated impaired wound healing. 28 These models also demonstrate an organized accumulation of microglia around the lesion epicenter. Microglia form an intermediate layer between neutrophils/monocytes inside the lesion cavity and the migrating astrocytes just peripheral to the microglia. 29 This architecture may help isolate myeloid cells in the lesion core to prevent a more expansive penumbral inflammatory response. Depletion of microglia in these models resulted in more extensive neuron/oligodendrocyte death and reduced motor function recovery, while increased microglial proliferation at the lesion due to macrophage colony-stimulating factor (M-CSF) stimulation prompted improved locomotor recovery. 29 Thus, glial scar formation appears to be beneficial, but poorly regulated gliosis and M1 macrophage phenotype activity for extended time periods after injury prevent neuronal repair and related functional recovery 30 In humans, the extent of this functional recovery inversely correlates with the magnitude of pro-inflammatory cytokines found in cerebrospinal fluid during the acute phase. 31 In contradistinction to immune responses with macrophages in other organ systems, microglia can continue a pro-inflammatory response for extended periods. Spinal cord injury (SCI) animal models have demonstrated both proinflammatory M1 and anti-inflammatory M2 phenotypes in microglia one week after injury, but at four weeks the cells may remain activated with a pro-inflammatory marker profile at the lesion site. 32 Failure of the CNS to regenerate following SCI may be associated with improper macrophage/microglia transition from M1 to M2 phenotypes; thus, increased regeneration may result from an enhancement of M2 polarization of microglia.
Unlike macrophages, microglia do not typically express arginase-1 (anti-inflammatory enzyme) following injury. 33 Furthermore, anti-inflammatory macrophages introduced to an in vivo SCI site become pro-inflammatory, consistent with the surrounding microglia polarization phenotype. Similar findings in human SCI studies detect persistent pro-inflammatory immune responses through the chronic phase – preventing tissue repair and remodeling that would normally occur in peripheral tissues several weeks after crush injury. 34
Multiple lines of evidence suggest microglia and macrophages are largely responsible for modulating this pro-inflammatory response. For instance, activation of microglia in normal spinal cord using zymosan results in a localized inflammatory lesion. 35 Activated microglia also promote axon dieback in dystrophic neurons that physically contact them. 36 Simultaneously, microglia support axonal growth from a distance through activation of toll-like receptor 2 and upregulation of matrix metalloproteinase 2 (MMP-2). SCI models lacking MMP-2 demonstrate more extensive glial scarring/lesion expansion and functional deficits. 37 Indeed, animal models with induced microglial anti-inflammatory phenotypes after injury show less neurodegeneration and commensurate functional recovery. 30 Similarly, when the NOX enzyme (NADPH oxidase) is inhibited, the resulting decrease in ROS reduces oxidative stress, M1 microglial polarization, and resulting functional deficits. 38 Microglial posttranscriptional mechanisms also contribute to pro-inflammatory polarization. MicroRNA (miRNA)-based therapeutics include miRNA mimics and inhibitors which can modulate the expression of target genes in various disease models. 39 For example, miR-155 can bind to specific mRNAs, causing a pro-inflammatory response. 40 MiR-155 knockout mice demonstrate more robust axonal regeneration and functional recovery following SCI. 41 Conversely, miR-98 has been shown to reduce the prevalence of M1 microglia within the impacted area in stroke models. 42 miR-98 has also been found to attenuate BBB permeability and improve locomotor function following CNS ischemia. 42
In addition to microglial interaction with neurons following injury, microglia also participate in an interdependent relationship with astrocytes. Astrocyte cultures with less than 5% microglial cell composition can have dramatically altered inflammatory responses. 43 Transgenic models with microglial depletion demonstrate less dense glial scar formation with a more disorganized astrocyte architecture. In addition to the deficient limiting microglial border between myeloid cells and astrocytes, these animals lack microglial production of typical insulin-like growth factor 1 (IGF-1) which promotes astrocyte proliferation and migration to the lesion. 29 Likewise, various cytokines released by microglia at the lesion induce astrocytes toward either destructive or neuroprotective roles. TNF-α, IL-1α and C1q from microglia impede astrocyte support for neuronal function and survival. 44 TNF-α expression from activated microglia contribute to astrocytic glutamate production and resulting neuronal excitotoxicity. 45 However, when anti-inflammatory phenotype microglia release IL-10, binding of this ligand with IL-10 receptors on astrocytes triggers astrocytic release of transforming growth factor-β which attenuates further microglial activation. 46 Astrocytes also contribute to decreased microglial adhesion and inflammatory gene expression. 47 , 48 Likewise, astrocytes upregulate microglial pro-inflammatory responses to bacterial ligands like lipopolysaccharide. 49 , 50 Moreover, axonal regrowth and neuronal viability is enhanced after injury upon C3 removal.
Microglia also interact with oligodendrocytes in both normal and post injury environments. Products released from activated microglia promote oligodendrocyte death which peaks eight days post injury; these include glutamate, NO, IL-1β, and TNF-α. 51 , 52 Microglia-mediated oligodendrocyte apoptosis contributes to demyelination of spared axons in the adjacent penumbra; myelin interaction with complement factors present in the inflammatory response promotes this phagocytic response.53–55 While pro-inflammatory microglial responses promote oligodendrocyte death, they also support oligodendrocyte precursor cell proliferation and migration to the injury site for remyelination. 56 Microglia with anti-inflammatory phenotypes also appear to promote remyelination through the effects of activin-A. 57 Thus, this complex glial-glial interaction contributes to the extent of demyelination following SCI.
Role of microglia in traumatic brain injury
In the United States, most SCI leading to chronic functional deficits is related to motor vehicle accidents (individuals <30 years of age) and falls (people >65 years old). However, the pathophysiology of traumatic brain injury (TBI) neuroinflammation is more diverse due to motion dynamics of the brain and volume of parenchymal tissue affected in many TBIs. Penetrating brain injuries are likewise less common, but TBI related to acceleration forces is an important component in concussions, cerebral contusions, and blast injuries. In some patients, these primary injuries are also compounded by acute epidural/subdural hematomas and intracerebral hemorrhages which can compress nearby brain parenchyma – promoting further structural compromise. 58 As in the spinal cord, mechanical forces related to the primary brain injury then prompt secondary injury mechanisms including biochemical cascades over minutes to months. 59
As suggested above, due to the multiple anatomical and pathophysiological factors associated with TBI, a clearly defined progression from primary to secondary injury stages may be obscured. Most TBIs are classified as ‘mild’, but in a significant percentage of these patients the BBB can be disrupted, allowing for greater infiltration of peripheral immune cells. 60 Meningeal vascular leakage has been observed in approximately half of otherwise apparently normal post-concussion patients. 61 Moreover, BBB disruption allows for leakage of brain parenchymal debris into the peripheral vascular circulation, leading to systemic inflammatory response syndrome. 62 The resulting changes in circulating coagulants, cytokines, and leukocytes can trigger systemic immune dysfunction. 63 Thus, in TBI without BBB disruption, microglia likely play a relatively greater role in neuroimmune modulation during the secondary injury phase in the absence of significant numbers of peripheral immune cells.
During the secondary injury phase, microglial response to injury also depends on structural brain damage patterns which can be more diffuse than typically focal SCI lesions. Extensive networks of activated microglia have been observed near the injury site. 61 These appear to help clear debris and restore disrupted astrocytic glial limitans and BBB integrity. 64 When this response is blocked in experimental injury models, parenchymal destruction is more profound. 61 Similar responses are seen in focal compression and laser-induced brain injury models where microglia migrate to the site of astrocytic injury to provide a barrier between damaged tissue and intact penumbra. 65 Microglial responses (process extension toward the injury site) have been observed within minutes of injury, and local microgliosis is found in the area of injury years later. 65 , 66 Activated microglia can likewise be present in patients following blast injuries and mild concussions with persistent inflammatory cytokine/chemokine production for extended periods.67–69
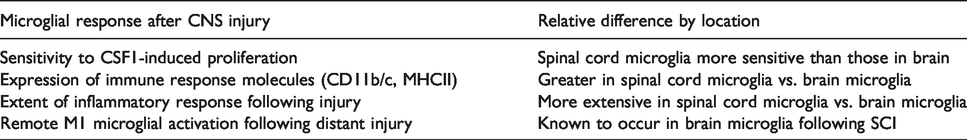
Regional differences between microglia in the brain and spinal cord also appear to contribute to different responses to CNS injury (Table 1). For instance, microglia in the spinal cord appear to be more sensitive to colony-stimulating factor 1 (CSF1) for proliferation than those in the cerebral cortex. 70 Microglia in the spinal cord also express higher levels of certain immune molecules (CD11b/c, MHCII) than those in the brain. 71 This complements data demonstrating a greater microglial inflammatory response to injury in both gray and white matter of the spinal cord compared to that in the brain. 72 Interestingly, microglia in multiple brain sites (e.g., hippocampus, cerebral cortex) become activated with an M1 phenotype following SCI; this is associated with neuronal loss in these brain regions. 73 Remote brain microglial activation and neurodegeneration are reduced with administration of cyclin-dependent kinase inhibitor CR8 following SCI. Whether cell cycle modulators are released by activated microglia near the site of SCI is currently speculative, but cysteine-cysteine chemokine ligand 21 (synthesized by damaged neurons at SCI lesion) has been shown to trigger remote microglial activation in the brain. 74 Thus, microglial location in the CNS appears to influence proliferation, receptor expression, and specific inflammatory responses to injury – even over significant anatomical distances.
Regional differences in microglial response to CNS injury.
Other individual factors such as age and sex of the subject may influence microglial response following TBI. Compared to younger subjects, microglia in older animals (less ramified morphology) demonstrate different M1/M2 phenotypical characteristics with poor responsiveness to injury. 75 Microglia from older mice show downregulation of more than 80% of genes which regulate detection of endogenous ligands such as purinergic molecules released upon neuronal death. 76 With respect to microglial responses in relation to varying gonadal steroid hormonal levels, TBI models in males have demonstrated peak microglial activation between days 1 and 7 post injury. In females, microglial activation is most notable between 3 and 7 days, with a subsequent anti-inflammatory response at 30 days after injury. 77 , 78
As observed in SCI, the interaction of microglia with infiltrating peripheral immune cells depends in part on the extent of injury and disruption of the BBB. Neutrophils have been largely localized in the meningeal and perivascular spaces within 24 hours after TBI, with parenchymal invasion more pronounced between days 3 and 5 in concert with monocytes and T cells. 61 , 79 Brain invading T cells may also be generated following SCI and TBI as in ischemic stroke models, where microglia and astrocytes interact with T cells to alter neuronal repair processes. 80 Reactive astrocytes also secrete cytokines/chemokines that regulate neuroinflammation. 81
Removal of neutrophils from TBI models results in less cerebral edema and parenchymal loss. 82 Microglia also interact with monocytes to promote the post-injury immune response; they may recruit monocytes into brain parenchyma within two days after injury. 83 , 84 Studies of brain injury responses in humans over subsequent years demonstrate persistent activated microgliosis in subjects with repetitive closed head injuries, suggesting this sustained neuroinflammatory response may also contribute to chronic traumatic encephalopathy. 85 This progressive neurodegeneration is also associated with adverse effects on naive astrocytes, microglia, and neurons.86–89 Thus, the neuroinflammatory responses of microglia in TBI contribute to both beneficial and potentially deleterious effects on parenchymal restoration, depending in part on microenvironmental factors, peripheral immune cell invasion, and extent of tissue damage.
Future applications
Recent innovations related to stem cell transplantation may provide for therapeutic applications in human SCI/TBI treatment. Human pluripotent stem cell-derived macrophage progenitors have been xenografted into neonatal mouse brains. The resulting microglia in the chimeric mouse brains appear to maintain their human microglial identity and characteristic gene expression patterns. 90 Microglial transplants into human CNS injury sites, however, require microglia which express specific cytokine profiles at specified timepoints following injury. Indeed, M2 phenotype microglia (induced with IL-4 exposure) transplanted into mouse SCI models contribute to significantly more motor function recovery and axonal transport than observed in controls or SCI animals receiving transplanted M1 phenotypic microglia. 91 However, translational application of these techniques from rodent to human CNS injury is limited to some degree by characteristic differences between microglia from rodents compared to those from humans. For instance, transplanted human microglia age differently and retain specific expression patterns of immune response and neurological disease-risk genes not present in their murine counterparts. 90 Moreover, cytokine expression profiles are unique in mouse, rat and human microglia following CNS injury. Following glucose and oxygen deprivation, microglia in rats exhibit upregulation of multiple cytokines/chemokines, while many of these are downregulated in murine microglia. 92 Human microglia demonstrate a more varied cytokine response following oxygen/glucose deprivation. This microglial heterogeneity after injury has been recently better characterized by single cell microglia RNA sequencing studies; microglial nuclei from frozen CNS tissue can also be analyzed. 93 Nuclear microglia transcriptomes are now compiled in multiple databases (brainsat.eu, microgliasinglecell.com, etc.) which can provide valuable information for future studies of microglial gene expression in CNS injury models. 94 Genome-wide transcriptomic analysis results have also recently challenged the singular M1/M2 microglial phenotype classifications. Single cell RNA sequencing has identified a disease associated microglia (DAM) subtype which is characterized by elevated lipid metabolism and phagocytic activity. 95 Other subtypes such as necroptotic microglia (very proinflammatory/immunogenic) and dark microglia (associated with oxidative stress and aging) have recently been described. 96 These recent developments therefore suggest effective future therapeutic applications for SCI/TBI will require more comprehensive understanding of microglial heterogeneity in the setting of CNS injury.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Support for this work was provided by U.S. Veterans Administration (1I01BX002349-01/2I01BX001262-05/1I01BX004269-01) and S.C. State Spinal Cord Research Fund (SCIRF-2015P-01/SCIRF-2015P-04/SCIRF-2015-I-01/SCIRF-2016 I-03/SCIRF2018 I-01).
Declaration of conflicting interests
The author(s) declared no conflicts of interest with respect to the authorship and/or publication of this article.