
Abstract
A major challenge in developing stroke therapeutics that augment adaptive pathways to stress has been to identify targets that can activate compensatory programs without inducing or adding to the stress of injury. In this regard, hypoxia-inducible factor prolyl hydroxylases (HIF PHDs) are central gatekeepers of posttranscriptional and transcriptional adaptation to hypoxia, oxidative stress, and excitotoxicity. Indeed, some of the known salutary effects of putative ‘antioxidant’ iron chelators in ischemic and hemorrhagic stroke may derive from their abilities to inhibit this family of iron, 2-oxoglutarate, and oxygen-dependent enzymes. Evidence from a number of laboratories supports the notion that HIF PHD inhibition can improve histological and functional outcomes in ischemic and hemorrhagic stroke models. In this review, we discuss this evidence and highlight important gaps in our understanding that render HIF PHD inhibition a promising but not yet preclinically validated target for protection and repair after stroke.
Keywords
Introduction
Stroke is a leading cause of death and disability worldwide, yet few proven options exist for treatment. Because of recurrent lack of success at the human bedside, the clinical stroke community has become understandably deliberate, almost tentative, in considering evolving therapeutic approaches. One general approach of increasing interest to clinical neuroscientists, aided by the molecular and genomic revolutions, is the use of drugs that activate endogenous adaptive programs. Indeed, activators of the Nrf2-mediated antioxidant response (Calkins et al, 2009; Smirnova et al, 2011), peroxisome proliferator-activated receptor γ-mediated metabolic adaptation and antiinflammatory programs (Zhao et al, 2006), and histone deacetylases inhibitor-induced neuroprotective and repair cassettes (Sleiman et al, 2009; Langley et al, 2009) are examples of how this general approach has been applied. Excitingly, with each of these targets, there is a clear trajectory toward the human bedside. Here, we critically review the hypoxic adaptive response as a therapeutic target in stroke and discuss the complexities of inducing compensation to reach a new level of homeostasis after stroke. Specifically, the review focuses on a fascinating group of iron, 2-oxoglutarate, and oxygen-dependent dioxygenases called the hypoxia-inducible factor prolyl-4-dioxygenases (HIF PHDs). Hypoxia-inducible factor prolyl hydroxylases (PHDs) are the enzymatic gatekeepers of the adaptive response to hypoxia and thus qualify as oxygen sensors (Kaelin, 2011). They are able to transduce momentary changes in oxygen availability into stabilization of a host of proteins, including the hypoxia-inducible transcription factors. Stabilization of these proteins leads to activation of a host of adaptive responses that are transcriptional and posttranscriptional. These adaptive responses are poised to act at local, cellular, and systemic levels to address the discrepancy in oxygen demand and supply (Ratan et al, 2007). Inhibitors of the HIF PHDs, which permit the activation of hypoxic adaptation under conditions of normoxia, have been shown to be protective in ischemic and hemorrhagic stroke models, but there is still significant work to do to make this a viable therapeutic approach for stroke.
Hypoxia-inducible factor family transcription factors
The HIF transcription complex, which is activated by low oxygen tension, controls a diverse range of cellular processes including angiogenesis, erythropoiesis, bronchodilation, and cellular metabolism targeted at increasing oxygen delivery to tissues. Hypoxia-inducible factor was purified in the early 1990s in a heroic effort by Semenza and Wang (1992) using a DNA affinity column composed of the hypoxia response element (5′-[A/G]CGTG-3′). The HIF response element exists in many of the genes, including erythropoietin (EPO) known to be upregulated in response to hypoxia. A heterodimeric protein binds to this response element and consists of a hypoxia-regulated α subunit and a constitutively expressed β subunit (also known as the aryl hydrocarbon receptor nuclear translocator). The regulated subunit, HIFα exists as three isoforms HIF-1α, HIF-2α, and HIF-3α. Hypoxia-inducible factor-1α is ubiquitously expressed, while HIF-2α and HIF-3α have more restricted expression patterns (Maxwell et al, 1999; Maynard et al, 2003). Hypoxia-inducible factor-2α has been shown to be expressed in central nervous system (CNS) cells in vitro and in vivo (Trollmann and Gassmann, 2009). Little is known about HIF-3α, but recent studies indicate that this isoform has complex roles in hypoxia (Li et al, 2006). Hypoxia-inducible factor is a member of the bHLH (basic helix-loop-helix)—PER (period), ARNT (aryl hydrocarbon nuclear translocator), SIM (single minded) (PAS) family of proteins. The bHLH domain mediates the dimerization and DNA binding, and PAS domain facilitates additional dimerization between the family members. The central region of HIF-1α is identified as oxygen-dependent degradation (ODD) domain, which is critical for oxygen-regulated protein degradation. The C-terminal of HIF-1α contains two transactivation domains that mediate interaction with coactivators like CBP (CREB-binding protein) and p300 (Kallio et al, 1998).
Hypoxia-inducible factor prolyl hydroxylases as gatekeepers of the adaptive response to hypoxia
Under normoxic conditions, HIF-1α is continuously transcribed and translated to HIF-1α protein. However, HIF-1α is rapidly hydroxylated and rapidly degraded by the ubiqutin-proteosome pathway. Hypoxia-inducible factor-1α stability is regulated via the activity of a class of oxygen, 2-oxoglutarate, and iron-dependent enzymes known as the HIF prolyl-4-hydroxylases (Bruick and McKnight, 2001). Hypoxia-inducible factor PHDs hydroxylate the two specific proline residues pro402 or pro564 (amino-acid positions designated in humans) within the oxygen degradation domain (ODD) of HIF-1α (Bruick and McKnight, 2001; Kaelin and Ratcliffe, 2008). Hydroxylation of HIF-1α allows it to bind to the VHL (Von Hippel Lindau) tumor suppressor protein that acts as a recognition component of E3 ubiquitin ligase complex. Hydroxylated HIF-1α is polyubiquitinated at three lysines in the central ODD domain and directed to the 26S proteosome for degradation (Paltoglou and Roberts, 2007). The half-life of HIF-1α is <5 minutes in normoxia (Huang et al, 1996). As intracellular oxygen levels drop below 200 μmol/L or iron levels become limiting, HIF PHDs fail to hydroxylate the proline residues within the α subunits of the HIF-1 protein. Hydroxylation is required for the polyubiquitylation and degradation, thus the nonhydroxylated HIF-1α becomes stabilized and translocates to the nucleus. Hypoxia-inducible factor-1α can partner with its constitutively expressed partner HIF-1β to form HIF-1α/HIF-1β heterodimers. This heterodimer can bind to hypoxia response elements like a key in a lock and recruit distinct transcriptional coactivator complexes, including p300/CBP and SRC (steroid receptor coactivator) to effect compensatory changes in gene expression (Figure 1). Hypoxia-inducible factor-1α and HIF-2α proteins are closely related and are regulated in a similar manner by oxygen tension, bind to similar sites on the DNA, but can differ in the coactivators they recruit. For example, NEMO (nuclear factor-κb essential modulator) is specific for HIF-2α, which enhances HIF-2α-mediated transcription. Hypoxia-inducible factor-1α and HIF-2α thus have overlapping but clearly separate functions. Hypoxia-inducible factor-1 is better known as a regulator of hypoxic adaptation, while HIF-2 was first defined as a regulator of oxidative stress.
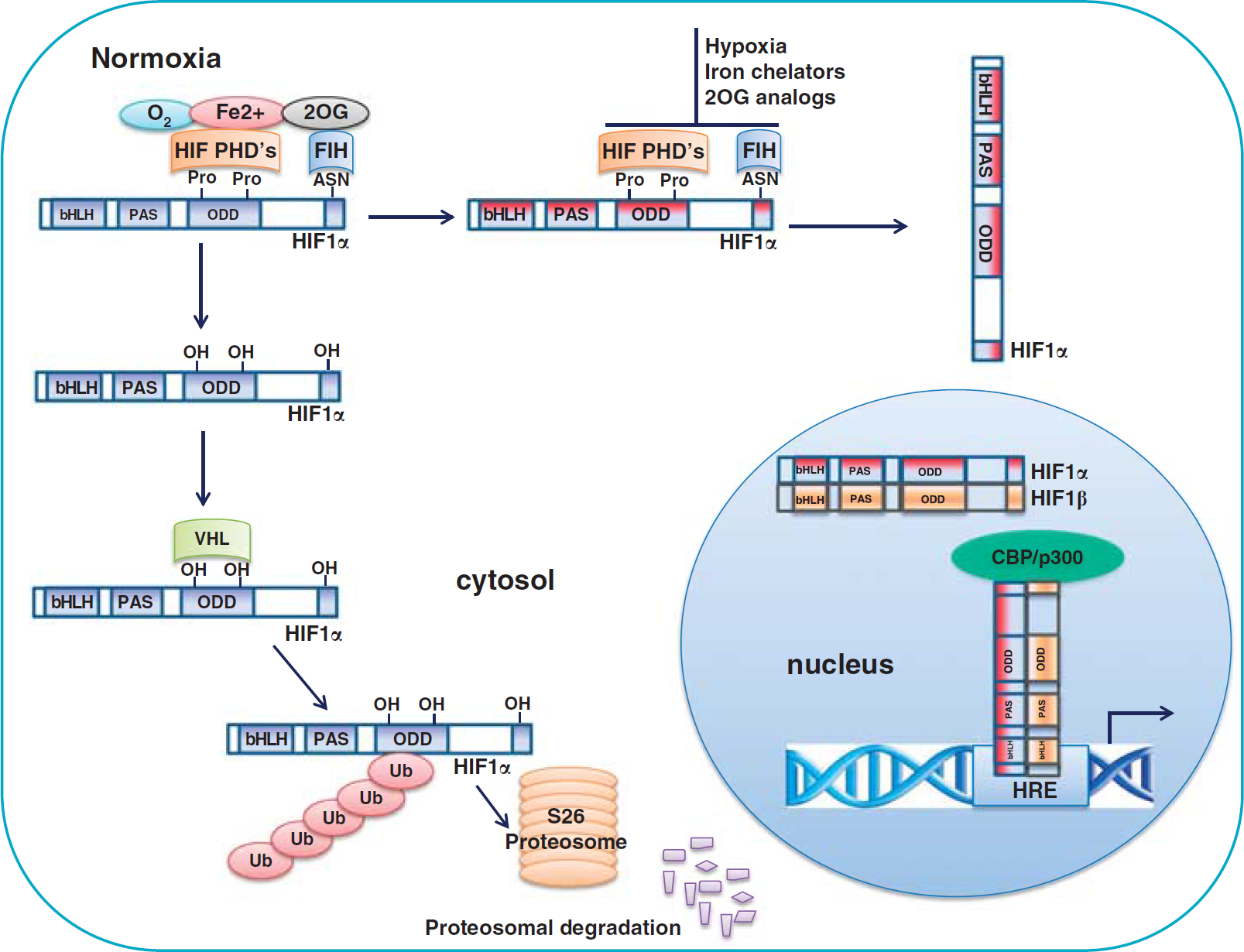
Hypoxia-inducible factor (HIF) prolyl hydroxylation is a major mechanism for modulating HIF stability and activity. Under normoxia, HIF prolyl hydroxylases (PHDs) in the presence of iron (Fe2+) and 2-oxoglutrate (2OG), hydroxylate the proline residues in the oxygen degradation domain (ODD) of HIF-1α (blue color), which recruits the tumor suppressor protein, the von Hippel Lindau protein (pVHL) to bind and initiate the proteolysis of HIF-1α by acting as a recognition component of ubiquitin ligase complex leading to proteosomal degradation. Factor inhibiting HIF (FIH) hydroxylates asparagine residues in the C-terminal transactivation domain (C-TAD) and prevents the interaction of transcriptional coactivators p300/CBP (CREB-binding protein). Under hypoxia, iron insufficiency, or 2OG depletion, HIF PHD's are inhibited, HIF-1α (red color) stabilized, translocates to nucleus and heterodimerizes with constitutively expressed HIF-1β, binds to the hypoxia responsive elements and induces the expression of target genes.
Hypoxia-inducible factor prolyl hydroxylase domain enzymes regulate many substrates in the adaptive hypoxic response
Prolyl hydroxylation is the most well-studied regulator for HIF levels and is catalyzed by nonheme iron dioxygenases known as the HIF PHDs (Bruick and McKnight, 2001). In addition to iron, HIF PHD's also require 2-oxoglutarate. 2-Oxoglutarate is produced in mitochondria via the tricarboxylic acid cycle and can be transported out of the mitochondria via the malate/aspartate shuttle. These shuttles serve to maintain redox homeostasis in the inner mitochondrial membrane. The dependence of HIF PHDs on iron and 2-oxoglutrate (2OG) in addition to oxygen suggests that these enzymes may have evolved not only as oxygen sensors but also as iron or 2-oxoglutarate sensors. One speculation is that since iron is required for the synthesis of Fe/S cluster proteins in the mitochondria and 2-oxoglutarate is a key substrate in the tricarboxylic acid cycle to produce electron equivalents for mitochondrial respiration, that defects in any of the these factors leads to mitochondrial insufficiency and activation of cellular programs that rely less on respiration (Lu et al, 2011).
In humans, there are estimated to be over 60 2OG-dependent dioxygenases (Loenarz and Schofield, 2011). The HIF PHDs are among the most well studied of the identified 2OG-dependent dioxygenases, and we previously used peptide inhibitors and interfering RNAs to implicate this subfamily in neuronal cell fate after injury. All of these studies have been in vitro, thus, as we discuss in detail later, the jury is still out on which if any of the 2OG-dependent dioxygenases is relevant in stroke.
Hypoxia-inducible factor PHDs began as a single isoform in flies and worms and diverged to three isoforms in mice and humans (HIF PHD1–HIF PHD3). These enzymes require oxygen and 2OG as substrates and iron as cofactors for prolyl hydroxylation. A reducing agent like ascorbate is also needed to keep the iron in the active site in the reduced state. In the HIF prolyl-4-hydroxylases, 2OG undergoes decarboxylation to form succinate and CO2 concomitant with the hydroxylation of the substrate, which is from proline to trans-4-hydroxyproline residues of the ODD domain of the HIF-1α (Mazumdar et al, 2010).
The structure of the three HIF PHD isoforms share a common motif, a double-stranded β-helix core fold also known as jelly-roll fold consists of eight β-strands (Clifton et al, 2006; Ozer and Bruick, 2007). They require Fe (II) for catalytic activity, which binds to a His1-X-Asp/Glu-Xn-His2 motif, these residues being His313, Asp315, and His374 for human HIF PHD's (McDonough et al, 2006). All three PHD isozymes can hydroxylate the motif LXXLAP in HIF. This motif is being identified in a growing number of substrates outside of HIFs, including Iκ B, RNA polymerase and the β-adrenergic receptor. The ability to modify distinct substrates is likely the result of divergence of the N-terminal sequences of the HIF PHDs, as the three isoforms share homology in the C-terminal catalytic domain (Huang et al, 2002). Hypoxia-inducible factor PHD's also differ in their expression patterns, tissue distribution, subcellular localization, and their ability to hydroxylate HIF-1. With all three isoforms, HIF PHD activity is reduced if the oxygen concentrations fall below 120 μmol/L and hydroxylation of HIF-1α is proportionately reduced. Even though the well-studied collagen prolyl-4-hydroxylase has affinity for O2, the affinity is too low to make these enzymes responsive to pathological or physiological changes in oxygen tension. Moreover, collagen PHDs do not hydroxylate proline residues in the ODD of HIF-1α, because they recognize the recurrent PPG motif in collagen rather than an LXXLAP motif in HIF and other substrates (Jaakkola et al, 2001).
Isoforms of hypoxia-inducible factor prolyl-4-hydroxylase enzymes: nomenclature
Prolyl Hydroxylase-1
Confusion has developed surrounding PHD isoforms because there is not single accepted nomenclature. Prolyl hydroxylase-1, also known as egg-laying defective nine homolog-2 (EGLN-2) or HIF PHD-3 (HPH-3) is primarily localized to the nucleus (Metzen et al, 2003). Under nonstress conditions, it is highly expressed in the testis, moderately in the liver, and low levels in the heart, brain, and kidney (Lieb et al, 2002). In situ hybridization studies reveal a diffuse expression in the cortex of mice. Hypoxia-inducible factor PHD1 contains an NLS (nuclear localization signal) (Steinhoff et al, 2009; Yasumoto et al, 2009), and an actin-regulated importin α/β-dependent extended NLS directs the nuclear import (Steinhoff et al, 2009). Prolyl hydroxylase-1 is a constitutively expressed protein that has no response to hypoxia or CoCl2 at an mRNA level but is induced by estrogen and stimulates cell proliferation (Cioffi et al, 2003).
Hypoxia-inducible factor-1α contains two oxygen-dependent domains (ODD), a C-terminal ODD (CODD) (around proline 402), and an N-terminal ODD (NODD) (around proline 564). Prolyl hydroxylase-1 can hydroxylate both N- and C-terminal ODD in HIF-1α, although it is unclear whether it does so in neurons under any condition. Two isoforms of PHD1 with molecular weights of 43 and 40 kDa, respectively, are generated by alternative translational initiation (Tian et al, 2006). Both PHD1 isoforms have similar HIF-1α prolyl-4-hydroxylase activity. While constitutive HIF activation can promote apoptosis or survival in a injury-dependent manner, HIF PHD inhibition in cells expressing the same oxygen stabilized form of HIF biases cells toward survival without affecting HIF levels. In cultured cortical neurons, PHD1 inhibition but not inhibition of other isoforms regulates normoxic oxidative stress-induced death in a HIF-independent and CREB-independent manner; we thus proposed a model in which the nondegradative oxygen-dependent prolyl hydroxylation of Rpb1 may selectively inhibit transcription of prodeath protein while augmenting prosurvival proteins (Siddiq et al, 2009).
Prolyl Hydroxylase-2
Prolyl hydroxylase-2 is also known as EGLN-1 or HIF PHD-2 (HPH-2). Prolyl hydroxylase-2 is localized primarily in cytoplasm (Metzen et al, 2003; Soilleux et al, 2005) and is highly expressed in the heart, testis and moderately in the brain, kidney, and liver (Lieb et al, 2002). Hypoxia-inducible factor PHD2 is believed to be the primary regulator of the HIFα transcription factors, but all these studies have been performed outside the CNS (Appelhoff et al, 2004; Berra et al, 2003; Epstein et al, 2001; Lieb et al, 2002). Like HIF PHD1, HIF PHD2 hydroxylates both N- and C-terminal ODD in HIF-1α (Appelhoff et al, 2004). Studies have shown that all the three HIF PHD isoforms can hydroxylate specific prolines in the HIF subunits in a strongly conserved LXXLAP sequence motif to degrade the protein. Inhibition of HIF PHD2 but not PHD1 or PHD3 by siRNA (small interfering RNA) is sufficient to upregulate HIF-1α in normoxia in various cancer and primary cell types. As mentioned, no one has established that similar isoform preference for HIF exists in neurons. Hypoxia-inducible factor PHD2 deletion leads to accumulation of HIF-1α but not HIF-2α in mouse liver and kidney tissue, while PHD1/PHD3 double knockouts lead to preferential HIF-2α stabilization but not HIF-1α in the liver (Takeda et al, 2007, 2008).
Prolyl Hydroxylase-3
Prolyl hydroxylase-3 also known as EGLN-3 or HIF prolyl hydrolase-1 (HPH-1) is localized in both cytoplasmic and nuclear compartments (Metzen et al, 2003) and highly expressed in the heart and liver and moderately in the brain and kidney (Lieb et al, 2002). These data reflect total RNA averaged over the whole organ, and may not reveal local pockets of elevated expression, particularly in the brain. Prolyl hydroxylase-3 expression can be upregulated by hypoxia, hypoxia mimetics, or in sympathetic neurons by growth factor deprivation as a necessary part of the apoptotic program (Lomb et al, 2007; Appelhoff et al, 2004). Prolyl hydroxylase-3 is less active in mediating HIF-1α hydroxylation compared with PHD2 (Berra et al, 2003). Prolyl hydroxylase-3 has been shown to promote degradation of the transcription factor ATF4, suggesting that ATF4 may be hypoxia responsive. ATF4 is a leucine zipper transcription factor that has been shown to have prodeath and prosurvival roles in response to oxidative stress or endoplasmic reticulum stress in neurons (Lange et al, 2008). Prolyl hydroxylase-3 appears not to be necessary for oxidative death of cortical neurons, and given the sympathoadrenal dysfunction associated with germline deletion of PHD3, conditional knockouts will need to be used to assess the role of this PHD in ischemic or hemorrhagic stroke (Koditz et al, 2007; Xie et al, 2009) (Figure 2).
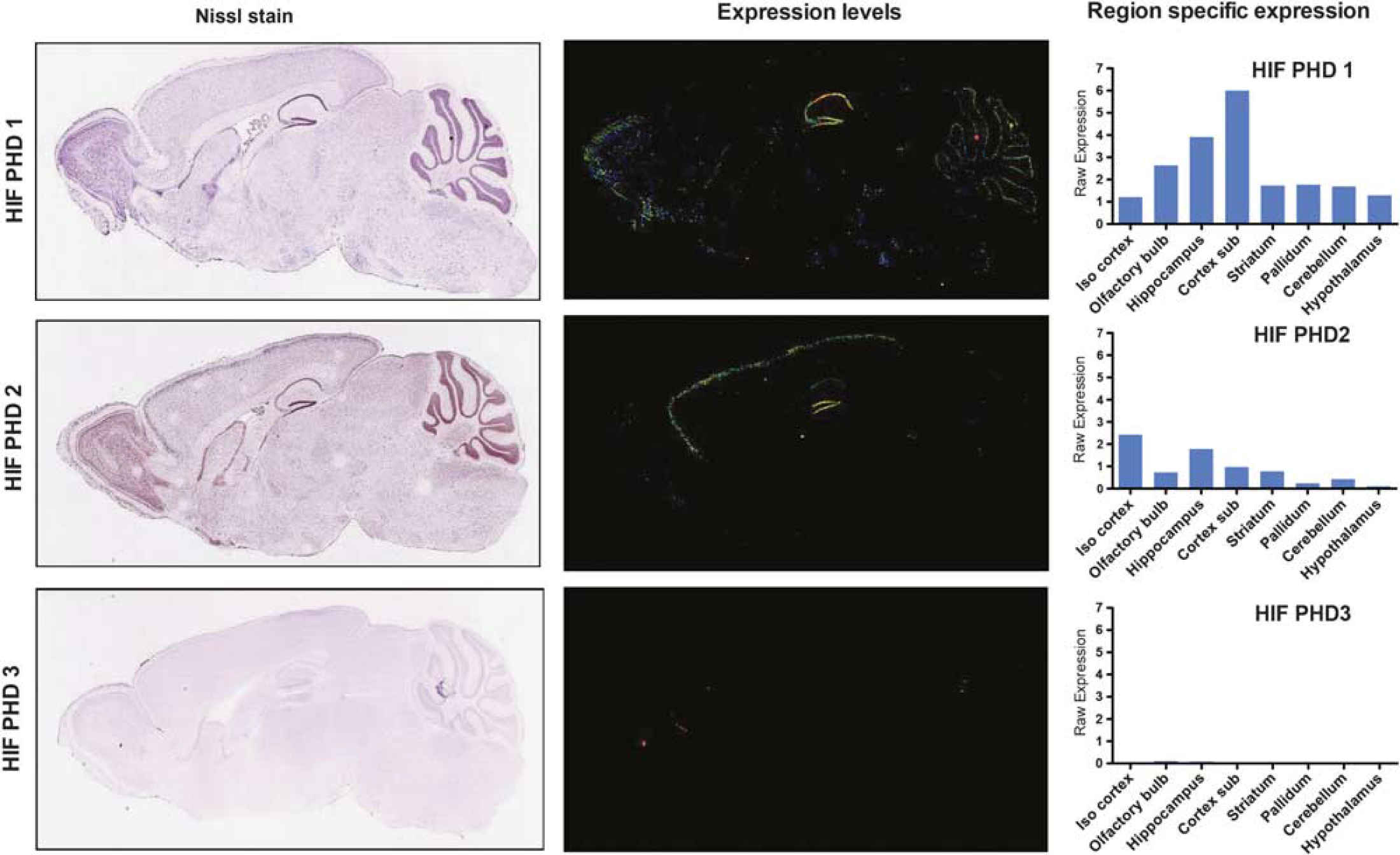
Hypoxia-inducible factor prolyl hydroxylases (HIF PHDs) expression in the mouse brain. Left panel shows representative Nissl staining images in the mouse brain. Middle panel shows the HIF PHDs 1, 2, and 3 gene expression levels in the mouse brain. Right panel show the quantification of region specific expression of HIF PHD1, 2, and 3 in the mouse brain. (Reproduced with permission from Allen Mouse Brain Atlas. Seattle (WA): Allen Institute for Brain Science © 2009. Available from: http://mouse.brain-map.org).
Factor inhibiting hypoxia-inducible factor
Factor inhibiting HIF (FIH) is not a PHD but rather an asparaginyl hydroxylase, although both enzymes interact with HIF. It is localized in cytoplasm and nucleus and highly expressed in the breast, testis, ovary, and kidney. A recent study in rats suggests that FIH is widely distributed in the CNS (Fukuba et al, 2008) and its conditional deletion in the brain influences metabolism and insulin sensitivity throughout the body. Similar to HIF PHDs, the structure of FIH consists of a jelly-roll-like β-barrel formed from eight β-strands, and the iron-binding site of FIH contains the conserved two-histidine, one-carboxylate triad (His199, Asp201, and His279) (Elkins et al, 2003; Lee et al, 2003). Also, like HIF PHD's this nonheme, iron, 2OG, and oxygen-dependent dioxygenase catalyzes the hydroxylation of the asparagine residue in the C-TAD (C-terminal transactivation domain) of HIF. This restricts binding of a host of coactivators to HIF-1 or HIF-2 (Lando et al, 2002; Hewitson et al, 2002). Recent studies suggest that PHD inhibition and FIH inhibition occur concomitantly in response to significant depression of oxygen tension and lead to increases in gene expression. Interestingly, PHD inhibition alone appears to be sufficient for stabilizing HIF and augmenting gene expression although the constellation is distinct from those induced by collective PHD and FIH inhibition. Future studies will definitively establish whether canonical PHD inhibitors including iron chelators such as desferoxamine also inhibit FIH. Our recent data based on comparison of the crystal structures of FIH and PHDs suggest that some potent inhibitors of HIF PHDs have no effect on FIH. While this review mainly focuses on HIF PHDs, these issues need to be considered as one develops small molecule inhibitors of HIF PHDs as drugs (Figure 3).
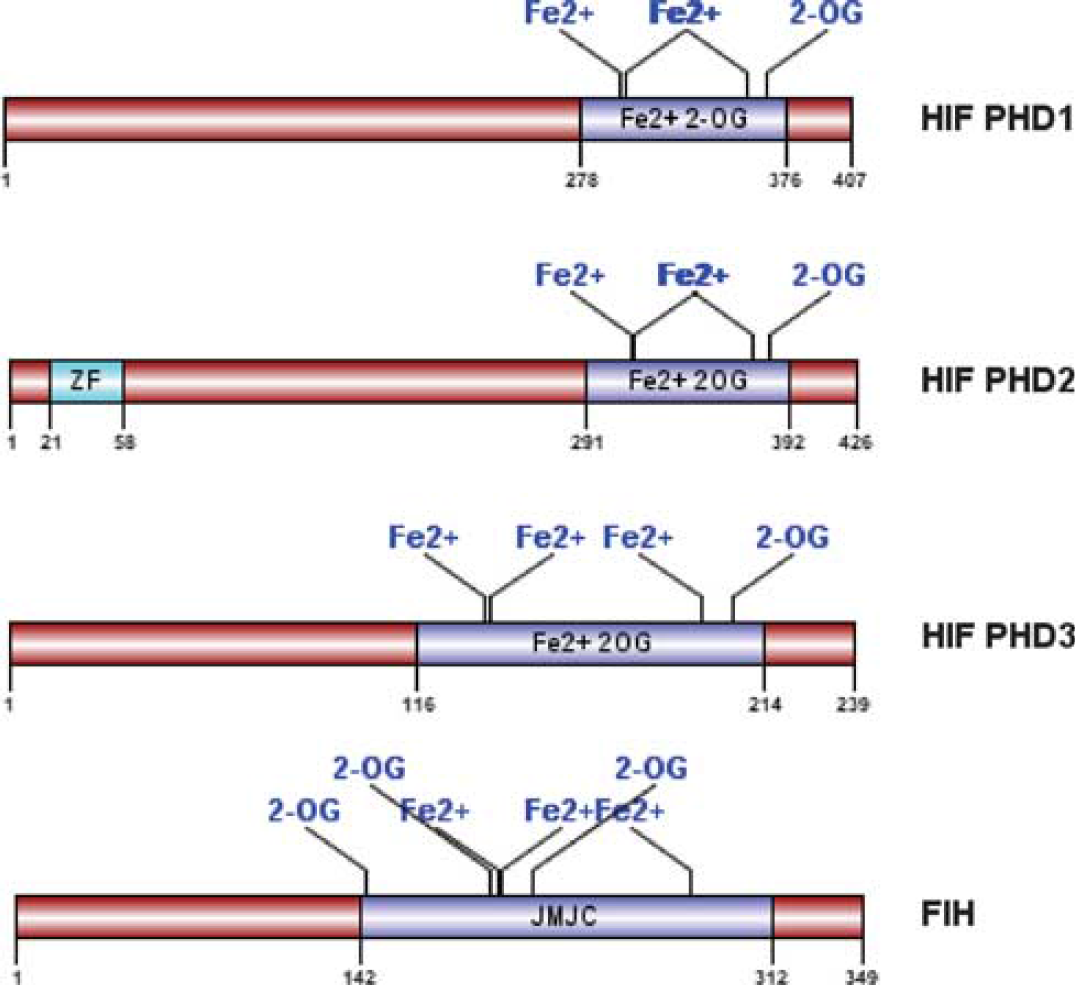
Domain structures for hypoxia-inducible factor prolyl hydroxylases (HIF PHDs) and factor inhibiting HIF (FIH), an asparagine hydroxylase. Schematic diagram of protein domain structures of HIF PHD1, 2, 3, and FIH with functional motifs/sites.
Pharmacological inhibition of hypoxia-inducible factor prolyl hydroxylases as a strategy for stroke
Drugs that activate hypoxic adaptation entered the realm of stroke therapeutics long before HIF-1 was purified or the HIF PHDs were identified as dominant modulators of HIF stability. Indeed, deferoxamine (DFO), an iron chelator, is a well-known inhibitor of the HIF PHDs and is known to stabilize HIF in almost every cell type. However, DFO and other structurally diverse chelators were used for treatment of acute and chronic neurologic diseases in animals and humans since the mid-1980s for reasons completely independent of their hypoxia modulating properties. Indeed, almost two decades ago, a promising study describing the beneficial effects of intramuscular DFO in a 2-year study of Alzheimer's disease progression in humans was published (Crapper McLachlan et al, 1991). Hurn et al (1995) demonstrated that DFO could ameliorate metabolic failure in dogs following stroke. The impressive effects of DFO on biological end points in stroke models and its tantalizing effects in rodent and human studies of other diseases catalyzed our interest in examining whether iron chelators abrogate oxidative death in neurons via their ability to inhibit the production of Fenton-driven hydroxyl radical formation. At the outset of our studies in 1993, our goal was to identify the target(s) for iron chelators for two reasons: (1) To identify a way to manipulate the target(s) independent of iron chelation to prevent the theoretical toxicities associated with binding iron as there are numerous iron-dependent enzymes within the cell. Critical iron-dependent enzymes include the iron sulfur cluster proteins in the mitochondria, as well as cyclooxygenase in the cytoplasm. (2) Identifying a target, if a single or family of proteins, could aid in the appropriate dosing of molecules like DFO in the critical sites of action, including the brain.
Iron chelators: a window into the hypoxic adaptive response
While a number of groups recognized the potential import of HIF-1 to stroke, our interest in hypoxic signaling as a target for protection by iron chelators was serendipitous, but richly formative. While using release of LDH (lactate dehydrogenase) as an indirect measure of cell death in a well-established oxidative stress model, we noted that iron chelators prevent LDH release but also increased cell-associated LDH activity by 400%. As LDH is a glycolytic enzyme that converts pyruvate into lactate to regenerate nicotinamide adenine dinucleotide +, we looked at whether DFO induces other glycolytic enzymes. We ultimately demonstrated that iron chelators were transcriptionally upregulating all glycolytic enzymes examined via HIF-dependent pathways (Zaman et al, 1999). Message levels for other HIF target genes including p21 waf1/cip1 and Epo were also upregulated by iron chelators. These findings gave rise to a model in which iron chelators induce neuroprotection in normoxic conditions by mimicking hypoxia, stabilizing HIF-1, and activating the genetic program of hypoxic adaptation. Key evidence supporting the model was that the lowest concentration of DFO required to protect all the cells was the lowest concentration that would activate hypoxic adaptation. Moreover, DFO could be added well after the onset of the death stimulus and still protect all the neurons (Zaman et al, 1999). Finally, cobalt chloride had been previously shown to act as a hypoxia mimic and it, like DFO, stabilized HIF and protected neurons from oxidative death. Moreover, in vitro and in vivo studies using DFO or cobalt chloride in several models showed dramatic neuroprotection or reductions in infarct volume by pretreatment with DFO or cobalt chloride 24 hours before stroke onset (Sharp et al, 2004; Gidday et al, 1994; Bergeron et al, 2000; Semenza, 2000; Prass et al, 2002).
A major prediction of the original model was that forced expression of an oxygen stabilized HIF-1 with a constitutively active transactivation domain (VP16 acidic activation domain) should be protective against oxidative death, while molecular deletion of HIF in vitro or in vivo should increase cell death and infarct size. In fact, we found that HIF potentiated death in response to oxidative death, an apparent confirmation of findings by Federoff and colleagues in an in vitro ischemia model. We demonstrated that HIF's prodeath effects in neurons could be attributed to induction of the established prodeath proteins BNIP3 and PUMA. Accordingly, siRNA deletion of HIF-1α prevented oxidative death. However, the prodeath effects of HIF are context dependent, and in response to other death stimuli including DNA damage and endoplasmic reticulum stress, HIF was protective (Aminova et al, 2005, 2008). The role of HIF in stroke appears to be similarly context dependent, as in both ischemic and hemorrhagic stroke some laboratories have reported prosurvival roles and others prodeath roles (Halterman et al, 1999; Helton et al, 2005; Baranova et al, 2007). Collectively, the findings argue against a linear relationship between iron chelation and HIF activation, and prompted us to revise the original model in favor of a greater focus on HIF PHD inhibition (Figure 4).
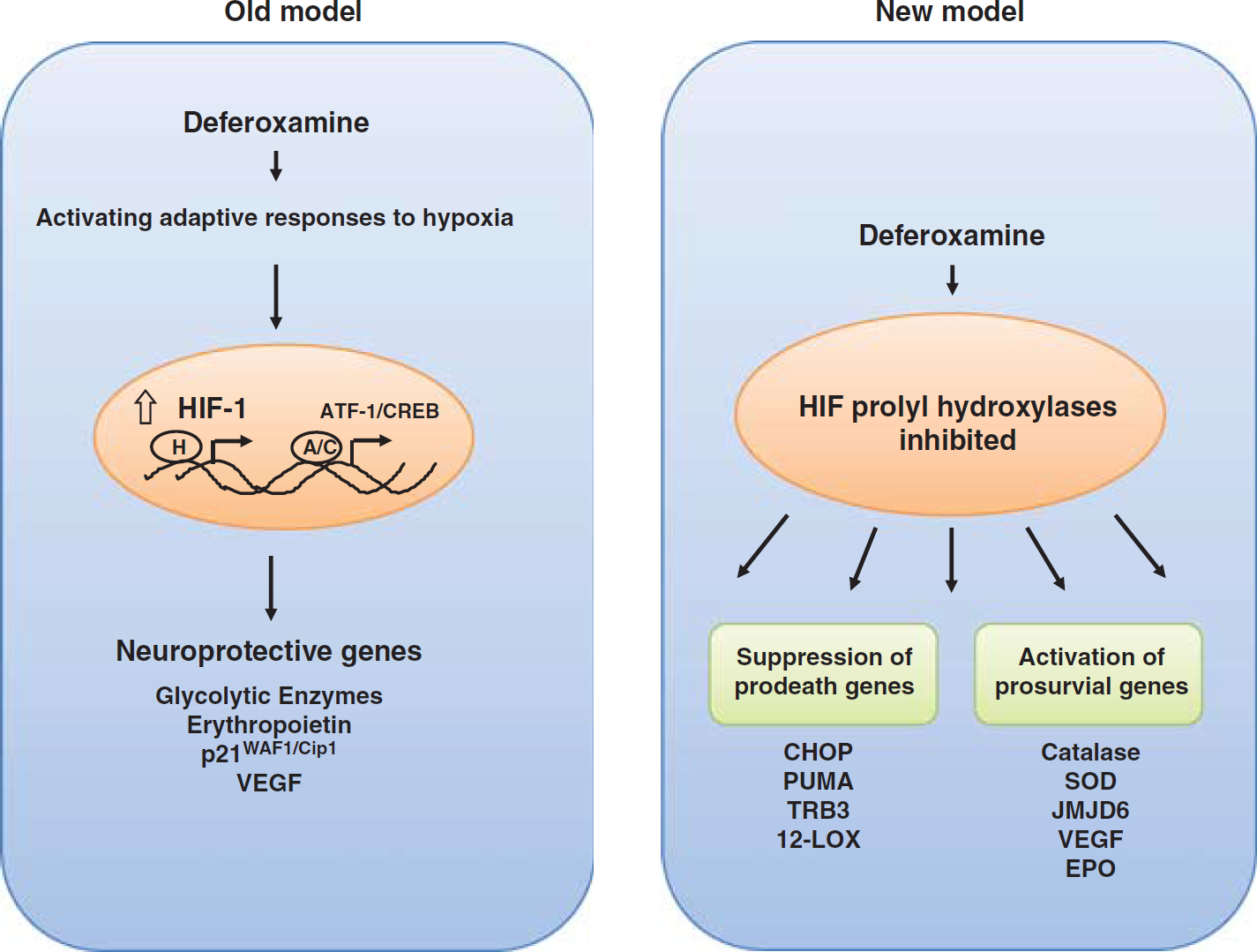
Mechanisms of stroke neuroprotection by iron chelators: ‘old’ and ‘new’ model. Schematic representation reflects the evolution in our thinking regarding the mechanisms of neuroprotection by iron chelators. In the old model, deferoxamine (DFO) activates adaptive responses to hypoxia via the induction of hypoxia-inducible factor (HIF)-1 and ATF1/CREB, leading to induction of neuroprotective/neurorestorative genes such as enolase, erythropoietin, vascular endothelial growth factor (VEGF), and p21. In the old model, HIF is the focal point of therapeutic efforts. In the new model, DFO inhibits the HIF prolyl hydroxylases (PHDs), and suppresses prodeath genes like Puma, Chop, and Trib3 via modification of RNA polymerase II and activates prosurvival genes such as Catalase, SOD, and JMJD6 via activation of HIF-2α. HIF-1α may have a small if not antagonistic role in stroke protection and repair.
Hypoxia-inducible factor prolyl hydroxylase inhibition as an on-target effect of iron chelator-mediated neuroprotection
As mentioned, our interest in hypoxic adaptation derived from observations that iron chelators could activate the HIF pathway in neurons. A subsequent seminal discovery by three groups paved the way for clearer understanding of the role of iron in oxygen sensing. Kaelin and Ratcliffe (2008) independently used proteomic approaches to identify prolyl hydroxylation as the essential, oxygen-dependent posttranslational modification of HIF-1α that mediated its interaction with the VHL complex, and its subsequent degradation. Using genetic approaches, the HIF PHDs were soon identified by Bruick, McKnight, and Ratcliffe as dioxygenases that are dependent on iron and oxygen, thus providing a molecular link between iron sensing and oxygen sensing (Bruick and McKnight, 2001; Schofield and Ratcliffe, 2005). The findings afforded us and other members of the clinical neuroscience community several new tools to address the notion that iron chelators work in stroke via PHD inhibition.
A first prediction of the new model was that small molecules that inhibit the HIF PHDs independently of the iron-binding site should also be protective in vitro or in vivo. Indeed, competitive inhibitors of 2-oxoglutarate such as 3,4,dihydroxybenzoic acid (DHB) were as expected, good inhibitors of the HIF PHDs that stabilized HIF. We found that DHB (10 μmol/L) protected oxidative death in vitro even when added hours after the onset of the injury stimulus and that it did so independent of any effects of on cellular iron (Siddiq et al, 2005). Intraperitoneal injection of DHB at concentrations that stabilize HIF and activate HIF-dependent gene expression (180 mg/kg) in the CNS reduced infarct volumes by 46% in a model of permanent focal ischemia, a paradigm where PHD inhibitors that bind iron were also protective. Two subsequent studies showed that the 2-oxoglutarate analog, dimethyl oxaloylglycine at 40 mg/kg or DHB (200 mg/kg) could be delivered at the time of reperfusion or 6 hours after the onset of ischemia and still reduce infarct volumes and improve behavior (and increase cerebral blood flow in the affected hemisphere while preserving tissue and improving behavior (Baranova et al, 2007; Nagel et al, 2011). The initial study demonstrated that the protective effects of HIF PHD inhibitors were only partially abrogated by specific deletion of HIF-1α in the brain. While these studies suggest that HIF PHD inhibition is a promising target for stroke therapy, all the inhibitors used to date likely inhibit many 2-oxoglutarate-dependent dioxygenases outside of HIF PHDs. Indeed, there are currently 60 known 2-oxoglutarate-dependent dioxygenases including the jumanji family of histone demethylases and the TET family of DNA-modifying proteins. Current studies are underway to determine the contribution of these proteins to the salubrious effects of global PHD inhibitors (Mole et al, 2003; Mecinovic et al, 2009; Rose et al, 2010, 2011; McDonough et al, 2005).
The most compelling data to date that HIF PHDs can directly participate in cell death induced by injury stimuli found in stroke has come from in vitro studies. Our laboratory used a cell permeant peptide inhibitor of all three HIF PHD isoforms but not the collagen PHDs to prevent oxidative death of cortical neurons (Siddiq et al, 2005). RNA interference showed that deletion of HIF PHD1 but not deletion of HIF PHD2 or HIF PHD3 can provide protection against oxidative death of neurons (Siddiq et al, 2009). Conditional deletion of all HIF PHDs has been shown to affect cell viability in response to developmental stimuli in sympathetic neurons (HIF PHD3; Bishop et al, 2008), ischemia in muscle (HIF PHD1; Aragones et al, 2008), and ischemia in the heart (HIF PHD2; Holscher et al, 2011), but these studies while promising, seem to have limited import for posttreatment protection. Future studies using a combination of conditional knockouts and specific pharmacological inhibitors will clarify whether isoform selective HIF PHD inhibition is a viable treatment strategy after stroke.
We should hasten to add that compensatory upregulation of other isoforms in the context of isoform-specific deletion raises that possibility that global (pan-isoform) HIF PHD inhibition may be superior in its posttreatment effects to isoform selective inhibitors. Indeed, compensatory EPO upregulation in the setting of chronic kidney failure in mice only happens when all HIF PHD isoforms are selectively deleted in the liver, an alternate site of Epo production (Minamishima and Kaelin, 2010). Because of the highly conserved C-terminal PHD domain between isoforms, small molecule, isoform selective inhibitors will likely affect interactions between the more divergent N-terminal domain and the growing number of substrates outside of HIF known to interact with individual PHD proteins. One putative HIF PHD2 inhibitor has been developed, but further validation of this compound and development of others will require a combination of traditional test tube assays involving recombinant HIF PHD isoforms, novel cell based assays, and in silico modeling using the crystal structures of each of the HIF PHDs (Tegley et al, 2008; Smirnova et al, 2010). Currently, a crystal structure is only publically available for HIF PHD2, and thus the optimal development and validation of inhibitors of other isoforms requires more information on the cell and structural biology of HIF PHD1 and HIF PHD3 in CNS cell types (Table 1).
Small molecule inhibitors of 2-oxoglutarate-dependent dioxygenases including HIF PHDs and their beneficial roles in experimental stroke models
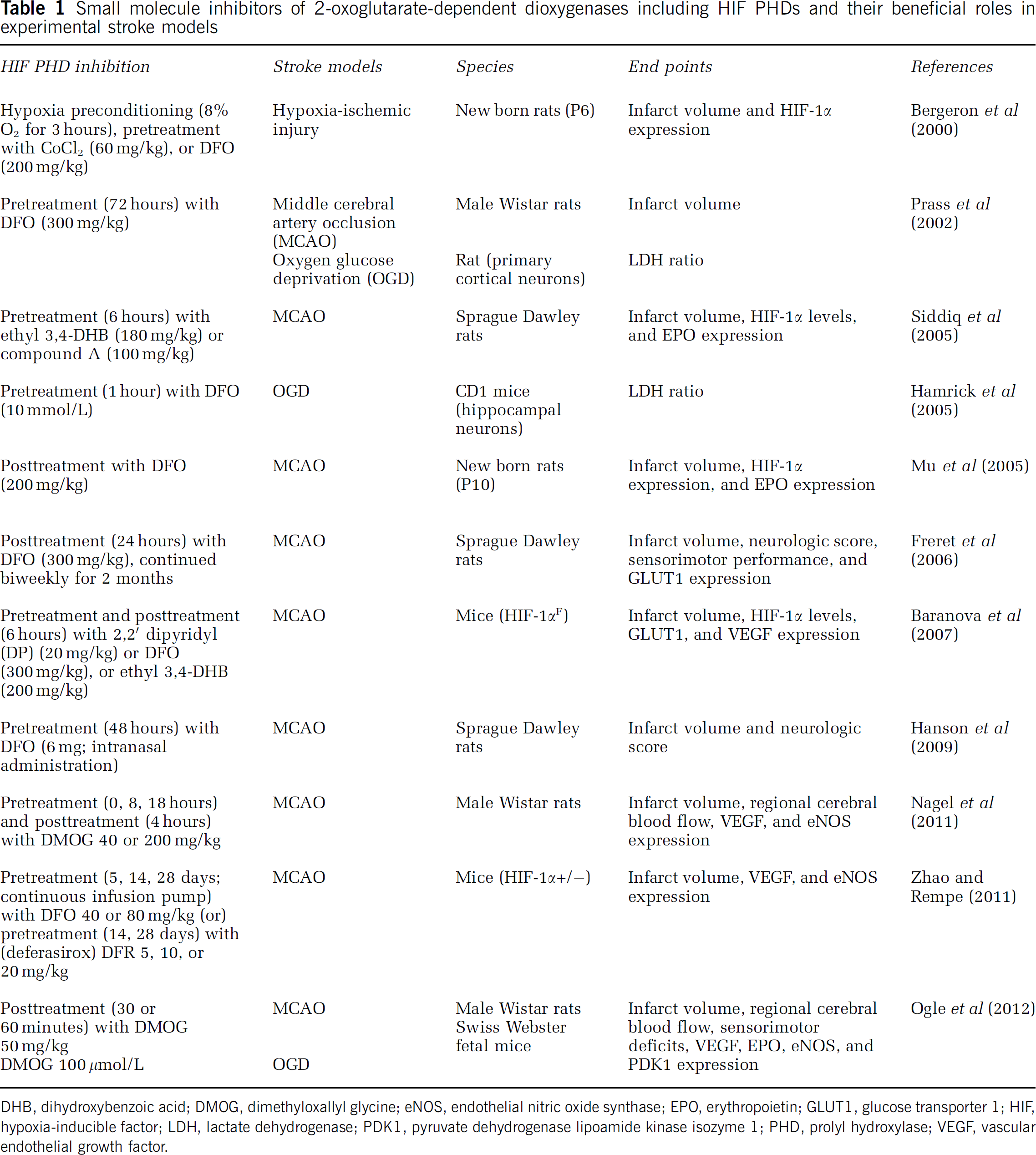
DHB, dihydroxybenzoic acid; DMOG, dimethyloxallyl glycine; eNOS, endothelial nitric oxide synthase; EPO, erythropoietin; GLUT1, glucose transporter 1; HIF, hypoxia-inducible factor; LDH, lactate dehydrogenase; PDK1, pyruvate dehydrogenase lipoamide kinase isozyme 1; PHD, prolyl hydroxylase; VEGF, vascular endothelial growth factor.
Do hypoxia-inducible factor prolyl hydroxylase inhibitors act in the central nervous system or in the periphery?
Prior studies using quantitative polymerase chain reaction and immunoblotting demonstrate that HIF PHD isoforms are expressed in neurons and glia in vitro. Initial analysis of HIF PHD expression showed that they are expressed in the brain (Lieb et al, 2002). In situ hybridization of individual HIF PHD isoforms (taken from the Allen mouse brain atlas) illustrate the heterogeneous distribution of expression of the isoforms (Lein et al, 2007). Hypoxia-inducible factor PHD1 is most highly expressed in the hippocampus, olfactory bulb, and striatum; moderately in the cerebellum and cortex; and lower levels in the hypothalamus and midbrain. By contrast, HIF PHD2 is highly expressed in the cortex, hippocampus, striatum, and olfactory bulb. Interestingly, HIF PHD3 expression is low all the brain regions. It is important to note that these studies reflect expression under steady-state conditions. As HIF PHD2 and HIF PHD3 are hypoxia regulated, the distribution of expression likely changes after stress. However, this has not been formally evaluated. These studies suggest that systemic administration of HIF PHD inhibitors could modulate HIF PHD activity in the brain. Previous studies from our group and others have demonstrated increases in HIF stability and HIF-dependent gene expression in the whole brain tissue following systemic administration of putative HIF PHD inhibitors. By contrast, other studies using low concentrations of HIF PHD inhibitors have failed to detect HIF stabilization and augmentation of HIF target genes, despite showing evidence of protection from CNS ischemia. These findings are consistent with several possible models, all of which have yet to be explored in detail. First, it is possible that HIF PHD inhibition is protective independent of HIF activation, and thus using HIF target gene expression is not a sensitive or specific proxy for HIF PHD inhibition. Second, it is possible that HIF PHD inhibition in the periphery induces HIF-dependent expression of proteins such as Epo that then penetrate the CNS to protect against ischemia and enhance repair. These models are not mutually exclusive, but they highlight the need for better tools to monitor the pharmacokinetics of HIF PHD inhibition in the brain. Hypoxia-inducible factor protein levels are a notoriously insensitive inverse measure of HIF PHD activity (less HIF PHD activity equals more HIF protein). Monitoring HIF target genes using quantitative polymerase chain reaction is sensitive only if the changes occur in a large number of cells synchronously in the brain, which may not be the case. One potential alternative is to use in vivo bioluminescence imaging of a fusion protein containing the ODD domain of HIF-1 and luciferase. If HIF PHDs are inhibited, the ODD-luciferase construct is stabilized and can be visualized dynamically in living mice by intraperitoneal injection of the luciferin substrate. More precise estimates of HIF PHD activity in the distinct brain regions can be obtained by dissecting fresh tissue and measuring luciferase activity in a luminometer. Preliminary studies indicate that classical HIF PHD inhibitors (DFO, ciclopriox, DHB) do not inhibit HIF PHD activity in the brain in uninjured animals using in vivo bioluminescence imaging. Although it is possible that the expected breakdown of the blood–brain barrier (BBB) would allow HIF PHD inhibitors to cross following injury, this needs to be examined experimentally. Collectively, these observations argue that the role of HIF PHD inhibition in neural protection and repair still is yet to be formally evaluated, and the question of whether this is a good target for ischemic stroke remains unclear.
Is hypoxia-inducible factor prolyl hydroxylase inhibition a potential target for hemorrhagic stroke?
Hemorrhagic stroke constitutes about 10% to 15% of all strokes; however, it is estimated that between 32% and 52% of patients succumb to the disease soon after its onset (Zahuranec et al, 2010). One class of agents that has garnered significant attention as therapeutics for ‘bleeding in the brain’ are iron chelators. The simple rationale behind the use of drugs that bind iron is as follows: Bleeding into the parenchyma of the brain not only compresses surrounding tissue to limit blood flow, but also results in the breakdown of red blood cells leading to release of hemoglobin and its oxidation by product, hemin (Regan et al, 2004; Dang et al, 2011). Hemin is taken up into neurons and glia where it is degraded by heme oxygenase liberating biliverdin, iron, and carbon monoxide. Classically, iron is believed to participate in the generation of ‘hydroxyl radicals.’ Hydroxyl radicals interact with all biomolecules at diffusion-limited rates and thus are capable of oxidizing cell constituents (Robinson et al, 2009). Iron chelators theoretically neutralize the formation of hydroxyl radicals by sequestering iron and preventing its interaction with peroxide. Based, on this model, a number of groups have demonstrated that iron chelators are effective at stemming neuronal death and promoting functional recovery in models of intracerebral hemorrhage. The effectiveness of iron chelators raise the question of whether these agents are acting to stem injury and enhance repair via HIF PHD inhibition. Preliminary observations suggest that noniron chelating HIF PHD inhibitors can abrogate hemin toxicity in vitro consistent with this possibility.
In an in vivo experimental subarachnoid hemorrhage model, DFO reduced the brain malondialdehyde content and induces the recovery of Na–K ATPase activity in guinea pigs (Bilgihan et al, 1994). Further, Richard Keep, Guohau Xi, and coworkers have extensively studied the effects of DFO in autologous blood infusion model of intracerebral hemorrhage and subarachnoid hemorrhage rat models and reported that DFO posttreatment reduces the brain nonheme iron concentration, iron-handling protein expression, oxidative stress, brain edema, neuronal death, and behavioral deficits in rat and piglet models (Nakamura et al, 2004; Gu et al, 2009; Okauchi et al, 2010; Lee et al, 2010).
Are hypoxia-inducible factor prolyl hydroxylase inhibitors useful for stimulating stroke recovery in addition to their putative effects on neuroprotection?
At the cellular and molecular level, neural plasticity in response to injury may be enhanced by a number of established biological manipulations that overcome intrinsic and environmental barriers to repair (Ratan and Noble, 2009). A particularly promising approach is the recognized ability of newly born neurons in the subventricular zone to deviate from their normal migration pathway, toward regions of damage (Jin et al, 2003; Kojima et al, 2010). Necrotic cell damage appears to be antagonistic for these events, whereas targeted apoptosis is clearly permissive (Sohur et al, 2006). Accordingly, damage in the stroke ‘core’ may be more difficult to reconstitute as compared with the ‘penumbra,’ where apoptotic death likely dominates. The proliferation, survival, differentiation, and integration of replacement neurons have been extensively evaluated in the CNS, and numerous lines of evidence support the viability of this approach for brain repair (Greenberg, 2007; Kernie and Parent, 2010; Kojima et al, 2010; Madri, 2009; Ohab et al, 2006; Zhang et al, 2008). Indeed, EPO, VEGF (vascular endothelial growth factor), SDF-1 (stromal-derived factor-1), and fibroblast growth factor are able to induce a systematic bias toward repair and functional recovery (Cui et al, 2009; Maric et al, 2007; Wang et al, 2004). An unanswered question is whether adaptive plasticity can be stimulated in the damaged brain by activating (via stabilization of single transcription factor family-HIFs) a coordinate cassette of genes that control endogenous cell replacement. Hypoxia-inducible factor is a heterodimeric transcription factor that is stabilized by hypoxia or growth factors following stroke (Chavez and LaManna, 2002; Ratan et al, 2004). Stabilized HIF translocates to the nucleus to bind to hypoxia response elements in a cassette of genes that mediate adaptive responses to ischemia, including EPO, VEGF, Trk B, and SDF-1. Over many years, many laboratories, including our own have focused on the role of HIF PHD inhibition and HIF-dependent and HIF-independent pathways to neuroprotection. However, their ability to stimulate recovery in chronic stroke has not been evaluated.
As mentioned above, drugs that selectively target HIF PHDs can be delivered systemically or intracerebroventricularly to augment transcriptional changes associated with hypoxic adaptation (Siddiq et al, 2005), including upregulation of EPO and VEGF. Since EPO does not pass readily into the CNS if the BBB is intact, manipulating HIF PHD activity in the CNS provides a mechanism to generate these proteins locally and theoretically circumvent BBB penetration problems of peptide growth factors (Hermann, 2009). We postulate that the global inhibition of HIF PHDs or conditional deletion of HIF PHD2, the isoform most associated with HIF regulation, will facilitate recovery in the postacute stroke phase via stabilization of HIF and upregulation of genes involved in the proliferation of NSC (neural stem cell), enhancing the proliferation and integration of newborn neurons. Conditional knockouts will also provide a tool to validate the specificity of our drug treatment. Indeed, recent studies show that HIF can regulate lymphoid enhancer factor-1 and T-cell factor-1; downstream effectors of Wnt/β-catenin activation; conditional deletion of HIF-1 impairs Wnt-dependent processes including NSC proliferation, differentiation, and neuronal maturation (Mazumdar et al, 2010). In addition, high doses of EPO are able to enhance angiogenesis, neurogenesis, and functional recovery following stroke (Ding et al, 2010; Iwai et al, 2010; Osredkar et al, 2010), even though <1% of EPO appears to cross the BBB (Zhang et al, 2010). Moreover, hypoxia increases the migration of progenitors to sites of injury via HIF-mediated induction of SDF-1 (Li et al, 2009). These and other genes, including VEGF are induced by HIF's activity as a sensor of hypoxia and ischemia. Pharmacological inhibition of HIF PHDs, using a small molecule that can penetrate the BBB will enable us to activate HIF to synthesize cassettes, rather than single proteins, that are hypothesized to be critical for brain repair. We also postulate that activation of a broad program of genes (70 to 100) in distinct cell types (neurons, glia, endothelial cells) will be better at stimulating biological effect compared with the separate application of growth factors or other repair proteins (Ratan et al, 2007). In addition, the breadth of this program is not likely to increase toxicity, since we are augmenting an endogenous program of adaptation.
Conclusion
In this review, we have tried to highlight the promise and potential of HIF PHD inhibitors for both protecting and repairing the brain following stroke. Current studies with selective but not specific inhibitors of the HIF PHDs are congruent with a role for these interesting enzymes. More specific pharmacological inhibitors of HIF PHDs are on the way that penetrate the BBB. These tools along with appropriate understanding of pharmacokinetics will permit evaluation of the role of these enzymes following stroke that will establish whether they have a clinically relevant therapeutic window. Conditional knockouts will be necessary to establish specificity of compounds once they are identified, but will have limited value for validating posttreatment protection and repair. It is important to emphasize that HIF PHD inhibition does not equal HIF activation; HIF is only one of a number of growing substrates known to modulate via HIF PHD activity (Mikhaylova et al, 2008). Chemical screens involving novel assays and an assortment of chemical libraries provide optimism that HIF PHD inhibition as a preclinical target should be validated or refuted in short order (Smirnova et al, 2010). The ability of HIF PHD inhibition to target many proteins and genes by flipping a single switch suggests that this is a therapeutic intervention that can challenge the heterogeneity in stroke pathophysiology present in humans. Because it is a pathway that leverages endogenous adaptive programs, we would argue that the breadth of the response will not be doomed by an increased likelihood of toxicity.
Footnotes
Acknowledgements
The authors acknowledge the support of the Adelson Foundation, National Institute of Health (P01 NIA AG014930, project 1 to R.R.R.) and the Burke Foundation and helpful discussion with Ambreena Siddiq, Leila Aminova, Juan Chavez, Philipp Lange, Manuela Basso, Sama Sleiman, Thong Ma, Hossein Aleyasin, Timothy Schallert, Irina Gazaryan, Natalia Smirnova, Guhua Xi, Richard Keep, and Lewis Morgenstern.
Disclosure/conflict of interest
The authors declare no conflict of interest.