
Abstract
Mitochondria play a central role in cell fate after stressors such as ischemic brain injury. The convergence of intracellular signaling pathways on mitochondria and their release of critical factors are now recognized as a default conduit to cell death or survival. Besides the individual processes that converge on or emanate from mitochondria, a mitochondrial
THE HYPOTHESIS: BOLSTERING MITOCHONDRIAL HEALTH BY TARGETED ORGANELLE TURNOVER CAN IMPROVE NEURAL ISCHEMIC OUTCOMES
Ischemic injury induces a complex array of environmental alterations, including calcium overload, reactive oxygen species generation, activation of intracellular signaling pathways, excitotoxicity, and inflammation. Virtually all of these conditions involve mitochondria at some level—whether in calcium buffering, disruption of energetic homeostasis, or translocation and release of potent signaling molecules. A large literature has focused on mitochondria as a major receiving center for cellular signaling, 1 but the question arises—does bolstering mitochondrial health and/or replacing dysfunctional and potentially toxic mitochondria with functional mitochondria confer neuroprotection? Although the concept is simple, the current state of knowledge related to mitochondria as a malleable organelle is nascent, with tools that are currently too limited in scope to satisfactorily address the issues. Furthermore, mitochondrial dynamics are likely to be influenced by both cell-type and specific stimuli, as emerging evidence has indicated that phosphorylation of critical proteins involved in dynamics are differentially phosphorylated under various cellular contexts and different gene isoforms are present in different tissue types.2,3 Although we will discuss what appear to be fairly general mechanisms, we must bear in mind that the particular response and control systems are by no means solidified across all contexts. Finally, evidence that mitochondrial dynamics play a role in various neural diseases, including cerebral ischemia, will be discussed with the concept that understanding the processes may lead to novel therapeutic approaches.
GENERAL OVERVIEW OF MITOCHONDRIAL DYNAMICS
The traditional model of mitochondria as static, bean-shaped organelles has vastly changed within the past few years. Mitochondria have now been better understood to go through an array of dynamic changes, including biogenesis and selective degradation, shape changes involving fission and fusion from a reticular-like mitochondrial cellular network, and rapid transport along the cell body to extremities. All of these processes are intertwined with each other, resulting in a dynamic balance to maintain the overall health of mitochondria within the cell. We will present in this section a general overview discussing the concepts of these dynamic changes as well as important mechanisms thus far identified.
Mitochondrial ‘Biogenesis’ or Replacement
The concept of and research regarding mitochondrial biogenesis has been ongoing for decades. Even the simple question of what actually defines ‘mitochondrial biogenesis’—whether it is intrinsic to mitochondria themselves (fully autonomous), wholly separate from preexisting mitochondria (nonautonomous), or somewhere in between (partially autonomous)—has been continuously addressed starting from biochemical lipid synthesis studies beginning 40 years ago. 4 In addition, the more recent findings of mitochondrial network fission and fusion events has turned the simplistic concept that damaged mitochondria can be replaced into an open debate on the precise definition of ‘replacement.’ Is there fission of existing mitochondria with selective replacement of damaged sections and constituents or complete replacement of proteins, lipids, and/or mitochondrial DNA (mtDNA)? The prevailing sentiment generally supports a partially autonomous model of biogenesis, 5 wherein newly synthesized proteins and other components (e.g., lipids) are incorporated into existing mitochondria, which is somehow linked to fission to restructure and/or divide. However, most of the studies thus far have not yet incorporated more than a few ‘markers’ of biogenesis, and thus are highly limited in interpretation. Despite these limitations, mitochondrial dynamics related to the term ‘biogenesis’ can be assessed by the combined use of several outcomes, many of which are now investigated in the context of brain pathology. The use of single end points by themselves cannot lead to conclusive evidence of mitochondrial biogenesis, as each end point has significant limitations and overlapping functions in cell biology. Thus, the combinatorial use of as many parameters as possible is the only viable method of exploring the biogenic process. 6
Several key points underlie biogenesis assessments (Figure 1). Mitochondrial proteins are encoded in both the nuclear and mitochondrial genomes, necessitating transcriptional activation in both organelles for fully functional mitochondria. This bigenomic origin of mitochondrial proteins is well documented and several critical components have been identified, but the exact mechanism and coordination of intra- and extra-mitochondrial processes are still poorly understood. The mitochondrial transcription factor A (Tfam) is encoded in the nucleus and is required for initiation of mtDNA replication, initiation of transcription of mitochondrial-encoded genes, as well as the organization, and possibly copy number, of the mtDNA genome.5,7 Although several other mitochondrial transcription factors have been identified, Tfam appears to be the major regulator of transcription. The expression of Tfam is primarily under the control of the nuclear transcription factor Nrf-1 (nuclear respiratory factor 1), which also induces the transcription of nuclear-encoded mitochondrial genes essential for the electron transport chain. The replication of mtDNA and the induction of both nuclear- and mitochondrial-encoded mitochondrial genes are necessary for mitochondrial biogenesis, and support the concept that induction of Tfam and possibly Nrf-1 are indispensable elements in biogenesis. To this end, upregulation of Tfam has been observed under several neuroprotective settings. 8
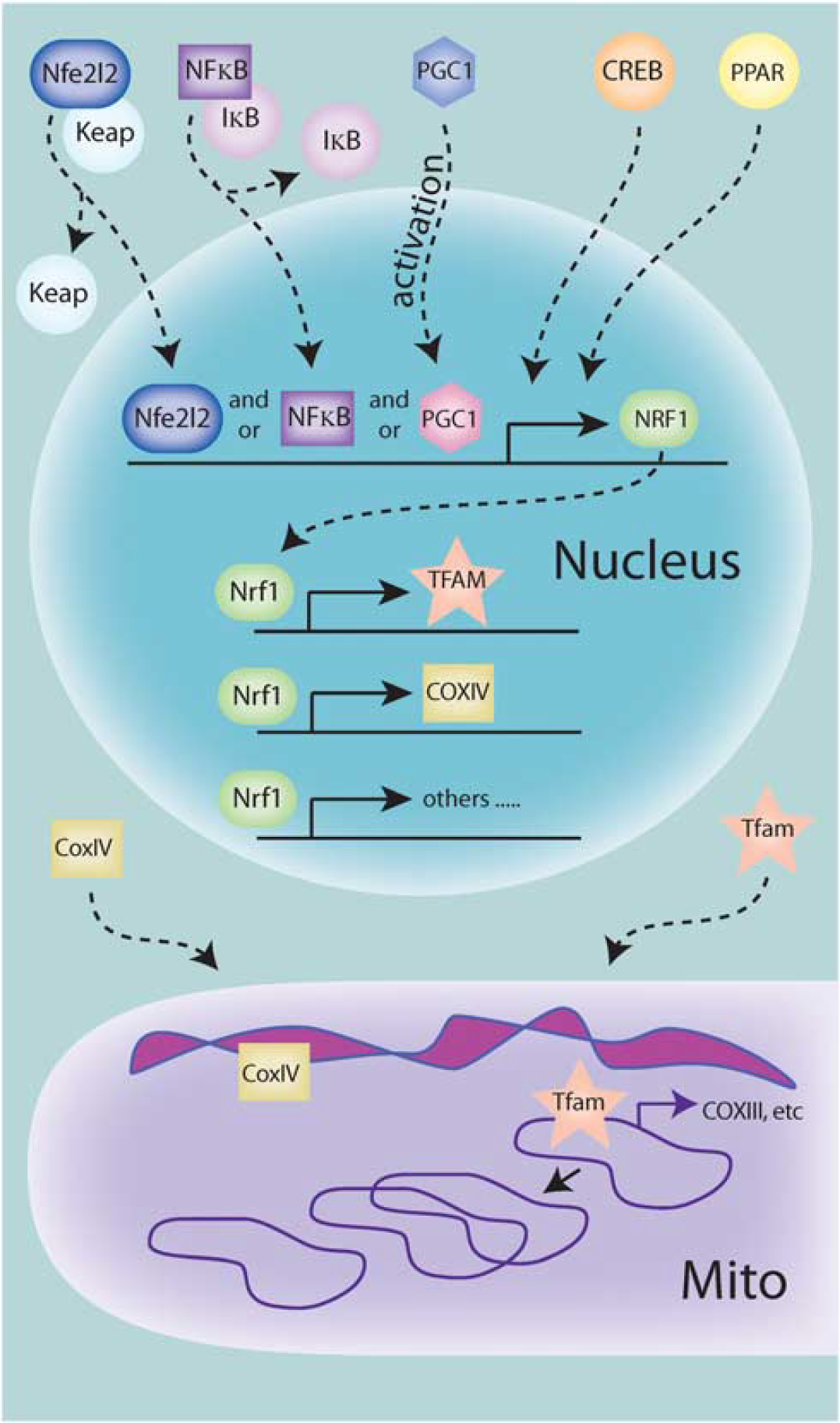
Signaling leading to transcriptional regulation of mitochondrial biogenesis. Various upstream signaling pathways can contribute to the activation of the transcriptional program necessary for biogenesis. So far, PGC1-α (peroxisome proliferator-activated receptor (PPAR)-γ coactivator 1-α), nuclear factor-κB (NF-κB), and Nfe2l2 can translocate to the nucleus either by dissociation of tethering proteins or posttranslational modification, and are then capable of binding to the promoter region and promoting transcription of nuclear respiratory factor 1 (NRF-1). Nrf-1 can then bind to and promote the transcription of mitochondrial transcription factor A (TFAM) and many other nuclear-encoded mitochondrial proteins, such as subunits of the electron transport machinery. These proteins are then translated and imported into the mitochondria. Tfam then can bind to and initiate transcription of the mitochondrial genomes, which includes mitochondrial-encoded subunits of the electron transport machinery. Tfam, in addition to other proteins, also acts in the control of mitochondrial DNA (mtDNA) copy number, including replication when needed.
The upstream induction of Nrf-1 has been mostly attributed to the activity of PGC1-α (peroxisome proliferator-activated receptor (PPAR)-γ coactivator 1-α), a nuclear transcription factor that is highly regulated by posttranslational modifications including, but not limited to, phosphorylation, acetylation, and sumoylation.
9
More recently, several other transactivators of Nrf-1 have been identified in nonneuronal cells, including NF-κB, Creb, and the NF-E2-related factor Nfe2l2 (Figure 1).10,11 Because of the complexity involved in the regulation (both positive and negative) of nuclear transcription factors and their transactivational activity, these transactivators may work in concert or separately under varying contexts.
9
Thus, assessment of mitochondrial biogenesis targeted solely at this level yields sorely limited interpretations. However, composite assessments of gene target synthesis
Sole reliance on induction of electron transport chain-related gene products may also be a limited proteomic approach to the determination of mitochondrial biogenesis. Several other proteins families, such as heat shock proteins, the mitochondrial transport system (TIM/TOM), and the sirtuins may also be indicators of mitochondrial biogenesis13–16 as well as the fission/fusion machinery discussed below. Although these proteins have proven to affect mitochondrial biogenesis under various conditions, it is still unclear which (if any) of these proteins may serve as a diagnostic benchmark of mitochondrial biogenesis induction.
In addition to the induction of gene products essential for mitochondrial biogenesis, it is likely that an (transient) expansion of mitochondrial volume and synthesis of mitochondrial lipids are also correlated with the process of biogenesis. Theoretically, increased newly synthesized mitochondrial lipids or volume may be observed when biogenesis precedes any significant clearance of damaged mitochondria. Cardiolipin is a unique phospholipid that is synthesized in and predominantly found within mitochondria. Its presence is essential in the maintenance of mitochondrial membrane composition as well as required for proper function of a number of mitochondrial proteins. 17 The biosynthesis of cardiolipin may thus be required either for mitochondrial biogenesis or for expansion of existing mitochondria. However, the measurement of cardiolipin content and synthesis—both of which would be necessary for accurate analyses—is not straightforward. Measurement of the synthase activity alone may mistakenly ignore a drop in available substrate. Direct quantitative measurement of cardiolipin content in brain is technically difficult, as it must be separated from more abundant phospholipids and typically involves a nonhistologic procedure. The most reliable technique remains mass spectroscopic analysis, including the more recently developed electrospray ionization-mass spectrometry, 17 which can differentiate between various cardiolipin subspecies. However, to date, no reliable histologic tool can assess cardiolipin content within a cell to compare with changes in mitochondrial morphology. The use of nonyl acridine orange (NAO) was previously published to label cardiolipin, but the NAO signal was subsequently found to be altered by changes in membrane potential.6,18 Thus, particularly in the case of ischemic injury or sublethal preconditioning stimuli, NAO is not a reliable or quantitative marker of cardiolipin content. More recently, several studies have found coordination between the mitochondrial import protein system and cardiolipin biosynthesis.19,20 Improved understanding of the regulation of cardiolipin biosynthesis could lead to newly identified proteins used in the assessment of mitochondrial expansion and/or biogenesis.
Imaging techniques to detect and quantify an expansion of mitochondrial volume have improved but still possess significant limitations. Previously, researchers relied on serial electron microscopic (EM) sections in which the number of mitochondria was manually counted. However, given the current understanding of mitochondria as a large dynamic network rather than static individual organelles, counting mitochondrial numbers in two-dimensional EM sections is not highly informative in semiquantification of mitochondrial mass. With better fluorescent microscopic techniques, measurement of relative mitochondrial volume has become possible.
6
More recently, confocal imaging analysis of
In sum, the conclusive evaluation of mitochondrial biogenesis remains somewhat elusive. Molecular approaches to identify newly synthesized or upregulated critical gene products are perhaps the most straightforward technique, but can be confounded in the case of rapid turnover, clearance, and other effects independent of the biogenetic process. Lipid analyses are technically limited in heterogeneous cell populations, and voxel volume measurements may not distinguish between expansion and swelling. Additionally, the development of sensitive measurements of mitochondrial respiratory function is being proposed for assessment of biogenesis, 22 but without additional biogenic endpoints, increases in mitochondrial respiration by itself would be difficult to interpret. Thus, the use of a combinatorial approach to assess as many known elements in the theorized process of mitochondrial biogenesis will improve our understanding of the biogenic response and its potential role in ischemic neuroprotection.
Mitophagy: The Flipside of Biogenesis
As with biogenesis, the clearance of defective mitochondria is still under investigation, particularly in the case of pathogenic stress (for recent in-depth reviews, see Ashrafi and Schwarz 23 and Novak 24 ). Several mitochondrial contexts can lead to defective mitochondria. Asymmetric splitting of mitochondria can lead to a loss of membrane potential in a proportion of so-called daughter mitochondria that then exist in a depolarized (hence, defective) state. 25 This has been observed during cell division under stress-free conditions. 26 These depolarized mitochondria are reabsorbed or cleared via autophagy (Figure 2). Interestingly, a second scenario of specific mitochondrial clearance occurs through a natural cellular differentiation process and appears to be independent of mitochondrial depolarization, such as erythrocytic maturation.27–29 Third, stress—both sublethal and pathologic—can impair existing mitochondria in such a way that targets depolarized mitochondria for degradation.30–32 The simultaneous assessment of these scenarios (cell cycle, maturation, and stress) that target mitochondria for clearance may reveal shared mechanistic pathways or important divergences.
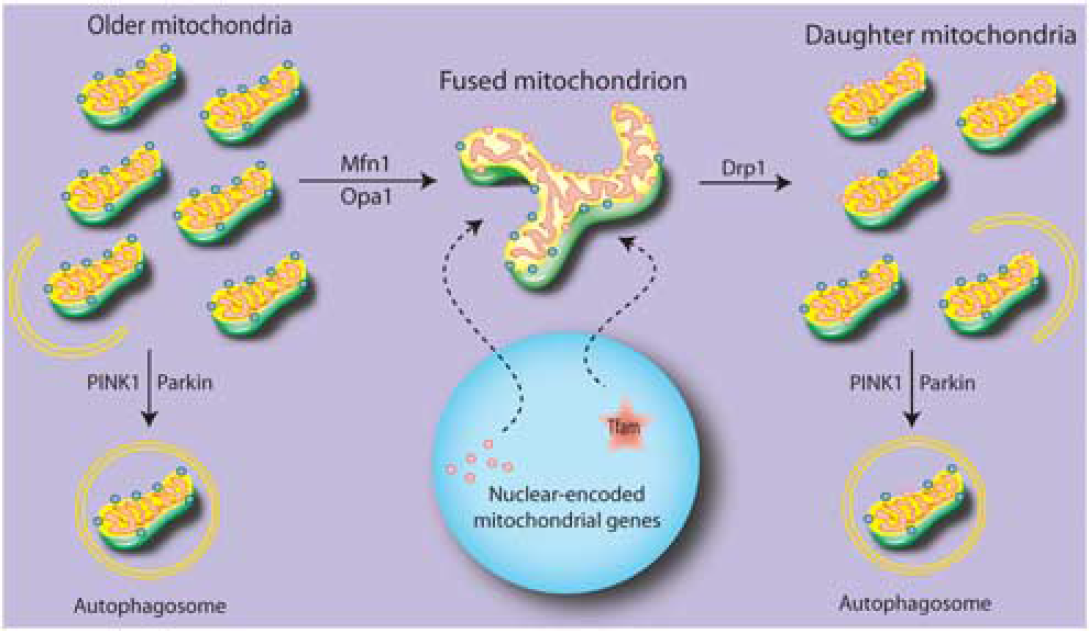
Proposed flux for mitochondrial replacement. Under normal circumstances, older elements of existing mitochondria may be sequestered and either targeted for degradation or fused to the mitochondrial network. Newly translated proteins are upregulated by activation of gene expression in the nucleus, and transported to the mitochondrial network to allow for the splitting of daughter mitochondria. These daughter mitochondria are heterogeneous, and those failing to maintain membrane potential are targeted for degradation.
Autophagy is a cellular process that clears intracellular components, including organelles, by formation of an internal isolation membrane that surrounds the cellular components, creating an ‘autophagosome’ that then fuses with lysosomal structures for degradation. Although mitochondria can be found within autophagosomes along with other intracellular targets, more recent studies have uncovered an autophagic process aimed solely toward mitochondria. The selective elimination of mitochondria through a targeted autophagic-like system was named ‘mitophagy’.
33
Specialized proteins, likely involving several overlapping players, appear to target mitochondria. These proteins in turn can modify existing mitochondrial proteins and/or bind to receptor molecules on developing autophagic membranes to recruit and tether the autophagosome to engulf the mitochondria. However, the particular proteins that carry out this process are likely to be highly context specific. Originally identified in yeast, a large majority of yeast autophagic proteins are involved in the mitophagic process, whereas six specific yeast proteins are involved selectively in mitophagy.
34
These specialized mitophagic proteins do not appear to participate in the general autophagic process. For example, Kissova
The molecular signals to initiate mitophagy in nucleated mammalian cells and target mitochondria to autophagosomal membranes are still being explored, and are likely to vary with context. Several proteins are involved in the mitophagic process in certain contexts, including Nix, Parkin, PINK1, and FUNDC1, as well as in the recruitment of the autophagic machinery, such as Atg11 and p62. Notably, the PINK/Parkin pathway has gained particular attention (Figure 3). In a general theoretical model that incorporates several independent studies (for an excellent review, see Jin and Youle 36 ), PINK1 translocates to the mitochondrial outer membrane via its mitochondrial localization signal. Healthy mitochondria internalize PINK through both the outer and inner translocases, at which point it is further processed and released into the cytosol and/or sent to the proteasome for degradation.37,38 However, depolarized mitochondria fail to fully import PINK1 through the inner translocase, leading to accumulation of PINK1 on the outer mitochondrial membrane. This accumulation leads to a recruitment of Parkin to the mitochondria, activation of both the kinase and E3 ubiquitinase activity of Parkin, and subsequent formation of ubiquitin chains on various substrates, such as VDAC, TOM, Mfn, and FIS1. These ubiquitin modifications then may lead to the recruitment of ubiquitin-LC3 adaptor molecules and the autophagic machinery. However, the extent or requirement of each of these molecules in the mitophagic process remains to be determined, as do potential parallel or redundant mechanisms. Nix is known to be required for mitophagy in maturing erythrocytes, and other studies have also suggested an involvement for Nix in mitophagy under hypoxic conditions. The influence of Nix on the Parkin/Pink model is unclear. Nix contains an LC3-interacting region (LIR) and is capable of binding ATG8/LC3/GABARAP, leading to the speculation that Nix may function as a mitophagy receptor. 39 Bnip3, a homolog of Nix that localizes to the mitochondria and contains the LIR motif, has also been proposed to be a mitophagy receptor. 40 Undoubtedly, other receptors or mechanisms must exist for the recruitment of the autophagic membrane, as Nix-independent mitophagy has been shown. Despite this, Nix still may serve to influence the process under both normal and pathologic conditions.
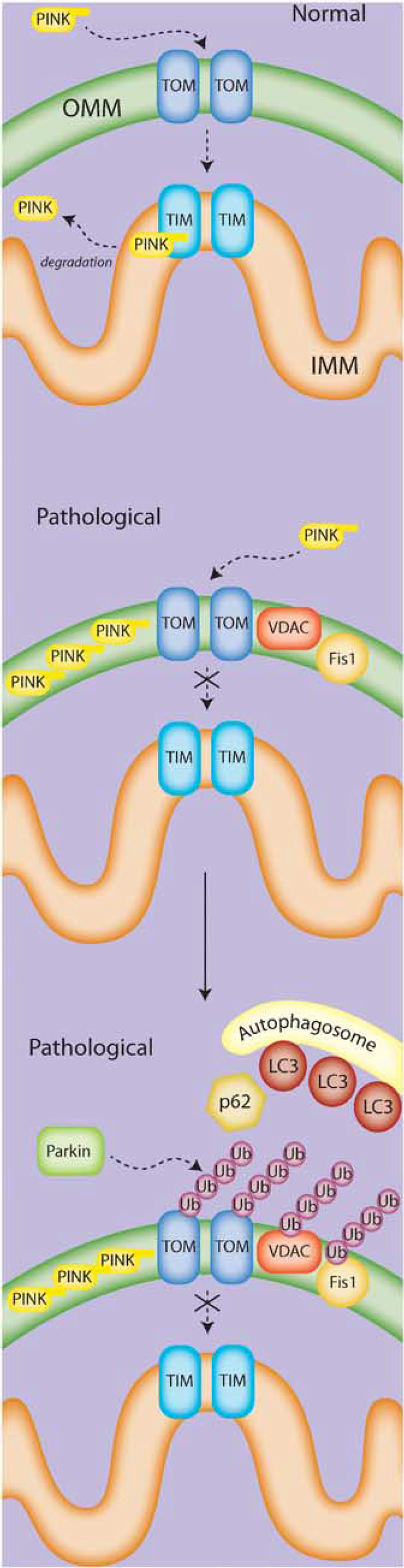
The PINK/Parkin mitophagy pathway. In the current model, normal mitochondria fully import PINK into the mitochondria via the TOM/TIM transport system, where it is cleaved and eventually degraded. Under pathologic conditions, PINK is only partially transported to the mitochondria, and accumulates on the outer membrane. The accumulation of PINK recruits Parkin to the mitochondria, where Parkin then acts via its E3 ubiquitinase activity to add ubiquitin chains to various mitochondrial proteins such as VCAC, Fis1, and Mfn. In an as-yet-undefined mechanism, this activity appears to lead to the recruitment of the autophagosome membrane and subsequent engulfment of the targeted mitochondria.
Perhaps even more so than biogenesis, the tools available to assess mitophagy are extremely limited in scope, and have only limited versatility. In neurons in particular, where mitochondrial alterations occur over a wide spatiotemporal plane, capturing and quantifying mitophagy remains challenging. A variety of techniques have been used to attempt to quantify mitophagy (reviewed in Zhu
Components of Mitochondrial Dynamics: Fission and Fusion
More recent mitochondrial studies have now underscored that any discussion of mitochondrial turnover would be incomplete without the inclusion of mitochondrial fission and fusion dynamics. In contrast with the archaic description of mitochondria as static, individual organelles, advanced three-dimensional microscopy has revealed a new model of mitochondrial structure as a highly organized and dynamic network, capable of rapidly shifting from an elongated tubular structure to fragmented independent organelles. Under nondeleterious conditions, neuronal mitochondria are fairly motile and appear to exist in a rather interconnected network. There is evidence of fission events and mitochondrial movement along the axon. However, the implications of these dynamic changes are still poorly understood. Current hypotheses include the concept of transport of ‘weaker’ mitochondria (e.g., mitochondria unable to maintain the membrane potential) toward the soma for degradation,45,46 delivery of mitochondria to distal regions of the cell as a result of synaptic activity changes,
47
and the possibility of mitochondrial movement as an organelle signaling mechanism. However, many of these hypotheses have yet to be resolved, with evidence for conflicting data
48
(reviewed inHollenb
Although the contexts of mitochondrial dynamics are still under intense scrutiny, several major underlying molecular mechanisms have been defined (Figure 4). Mitochondrial fusion occurs via a two-step process (reviewed in Palmer
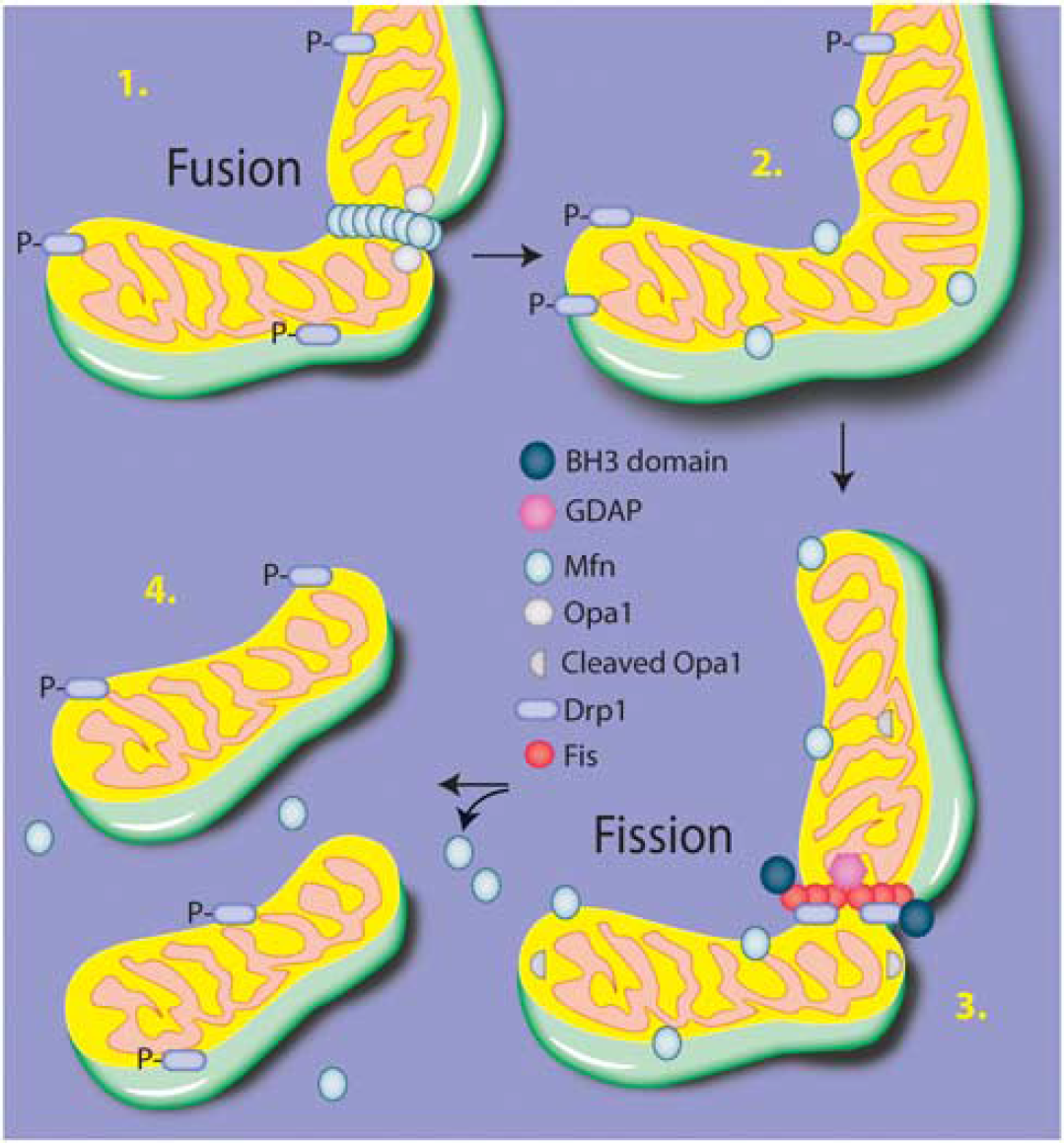
Fusion and fission. The overall execution of mitochondrial structural changes lies primarily with the dynamin-like GTPases Opa1 for fusion and Drp1 for fission. Briefly, Mfn dimerizes on the adjacent outer mitochondrial membranes and creates a tether between the fusing membranes. Cardiolipin is hydrolyzed, resulting in a curvature of the inner membrane, and the inner membrane protein Opa1 then acts via a GTPase motor activity to coordinate the fusion of the membranes. To progress to fission, Drp1 translocates from the cytosol to the outer membrane and associates with Fis1, forming an oligomeric ring-like structure. Using a GTPase motor activity, Drp1 constricts the membrane, leading to separation of the mitochondria.
Fission of mitochondria also requires motor-like processes, best characterized by dynamin-related protein 1 (Drp1). Drp1 is a member of the dynamin family of GTPases that translocates from the cytosol to the outer mitochondrial membrane. On associating with a receptor protein such as Fis1 at the mitochondrial membrane, Drp1 executes the scission of membranes by forming an oligomeric ring-shaped structure around the membrane region and pinching off the targeted mitochondrial region. This process is regulated by ganglioside-induced differentiation-associated protein 1 (GDAP), and may be affected by the translocation of members of the Bcl-2 family, particularly BH3-only members such as such as Bid, Bak, Bax, and BNIP1.53–56 In addition to biochemical markers of fission and fusion events, live cell imaging has been valuable at assessing both the rate of fission/fusion and morphologic features. 57 However, as the basic understanding of the regulation of these events is still largely unknown, multiple parameters should be assessed.
The correlate between fission/fusion and biogenesis/mitophagy has been postulated and explored under several conditions. For example, a recent study indicates that Mfn1/2 must dissociate from mitochondria for mitophagy to proceed. Depletion of Drp1 in Purkinje neurons led to hyperfused mitochondria and accumulation of autophagy markers adjacent to mitochondria, 58 indicating that completion of mitophagy may require not only dissociation of fusion proteins but also competent fission. In addition to mechanistic overlap, assessment of mitochondrial volume expansion may not be able to distinguish between incorporation of newly synthesized mitochondrial elements during biogenesis and fusion of existing elements, or between fission and mitophagy that may limit such volume expansion. Thus, for accurate interpretation of mitochondrial organellar dynamics, a more holistic picture must be developed at some point wherein fission/fusion and biogenesis/mitophagy are simultaneously considered.
Stimulation and Control of Mitochondrial Dynamics
The functional control of mitochondrial dynamics lies primarily in the coordination of events in the nucleus and mitochondria, although reports of involvement of other organelles such as the endoplasmic reticulum (ER) are emerging.59,60 As mentioned above, mitochondrial proteins are encoded in both the nuclear and mitochondrial genomes, and thus require a bigenomic coordination of events. However, virtually all of these proteins can be posttranslationally manipulated by a wide array of upstream signaling events, introducing a broad assortment of control points that sense local intracellular environments and quickly shift responses. Consistent with this, transcription factors often associated with cell survival have been implicated in affecting mitochondrial dynamics. For example, both Creb and NF-κB have been associated with the transactivation of NRF-1 and mitochondrial biogenesis in nonneural settings. 11
Conceivably, mitochondrial responses to ischemic injury can be initiated by two factors that stimulate changes in the organellar dynamic: a global cellular, nuclear-initiated (anterograde) response and a local, mitochondrial-initiated (retrograde) response. In reality, particularly in cases of cell stress, these two modes are likely to overlap significantly. In a more global scenario, signaling pathways sensitive to alterations in the cellular environment can elicit (e.g., Creb, NF-κB, Sirt1), converge on the nucleus, and activate pertinent mitochondrial transcription factors, such as PGC1-α, PPAR, and NRF-1, leading to transcription of mitochondrial genes and mtDNA replication.9,11 The nuclear-directed process has been shown by directly manipulating cytosolic or nuclear components and observing subsequent alterations in mitochondria. Elements of a global coordination of mitochondrial dynamics can also be observed in the course of the cell cycle, erythrocytic maturation, and possibly as part of inflammatory stimuli.27,28,61,62
In the second (and not necessarily distinct) scenario, local environmental or signaling changes could impinge directly on the mitochondria, leading to signaling back to the nucleus and other cellular compartments to initiate mitochondrial biogenesis and/or clearance. For example, environmental stimuli may lower membrane potential in specific mitochondrial regions. In some studies, the depolarized mitochondria appear to then undergo fission, 25 be targeted for retrograde transport, 45 and perhaps be cleared by an as-yet poorly understood mechanism, possibly by mitophagy or reabsorption into the mitochondrial network. 25 In addition, it is likely that direct signaling effects on mitochondria could cue signaling back onto the nucleus for stimulation of relevant transcription factors and mitochondrial gene expression, as has been described in the receiver/integrator model. 1 Interestingly, the sirtuin family resides in both the nuclear and mitochondrial compartments, and is highly sensitive to perturbations in intracellular environments from changes in, for example, NAD/NADH ratios or redox states.63,64 Additionally, SIRT1 can activate PGC1-α. 65 Thus, the sirtuin family may represent sensors in close proximity to the bigenomic arms essential for biogenesis.
The generation of mitochondrial hydrogen peroxide has also been proposed to instigate a series of signaling events leading to NRF-1 promoter transactivation and nuclear transcription of mitochondrial proteins. In this model, the NF-E2-related factor (Nfe2l2) is dissociated from its cytosolic tether, Keap1, via H2O2 oxidation and translocates to the nucleus. 66 Nfe2l2 is capable of binding to and transactivating the NRF-1 promoter in cardiomyocytes, which is associated with increased NRF-1 expression and subsequent transcription of Tfam. 10 As an example of how the antereograde and retrograde response leading to mitochondrial biogenesis may overlap, Nfe2l2 can also transactivate Hmox1, leading to the upregulation of heme oxygenase-1 (HO-1). HO-1 increases endogenous levels of carbon monoxide, which may trigger the production of mitochondrial H2O2, and thus cycle back to the oxidation of Keap1 and translocation of Nfe2l2. 67 Interestingly, the activation of Nfe2l2 may also be initiated by ER stress and lead to HO-1 activation, NRF-1 and TFAM expression, and mtDNA synthesis. 60 Although these signaling pathways have yet to be conclusively assessed in neural settings, a number of these molecules are heavily involved in neural ischemic injury. Thus, the scenarios present interesting avenues to explore.
In addition to signaling cascades, the mitochondrial dynamics machinery can also elicit an organellar response to environmental cues. Opa-1 can be processed under a loss of mitochondrial membrane potential, and is rendered unable to support mitochondrial fusion. 68 The processing of Opa-1 can also occur directly through mitochondrial proteases. 68 Similarly, Drp1 is sensitive to environmental alterations and undergoes posttranslational modifications, including phosphorylation, S-nitrosylation, ubiquitylation, and sumoylation, 69 all of which rely on upstream modulators sensitive to ionic conditions. The posttranslational changes can either increase or decrease the activity of Drp1, and, in turn, affect the fragmentation of mitochondria.
NEURAL DISEASE: A ROLE FOR MITOCHONDRIAL TURNOVER AND DYNAMICS
Despite the current nascent state of the assessments of mitochondrial biogenesis, mitophagy, and dynamics, a solid bank of evidence suggests that mitochondria, as an organelle, undergo flux both in the context of neural disease and injury and neuroprotective strategies, and that these fluxes may underlie the development of a neuroprotective state. Although originally identified in Charcot–Marie–Tooth (CMT) disease, many of the relevant molecules and processes involved in mitochondrial dynamics have been identified in Parkinson's disease (PD), Huntington's disease (HD), amyeloid lateral sclerosis (ALS), and cerebral ischemic models (Table 1). Furthermore, mitochondrial dynamics are closely correlated with several preconditioning stimuli, and may underlie at least part of the ischemic tolerant state.
Neural disease-specific alterations in mitochondrial dynamics
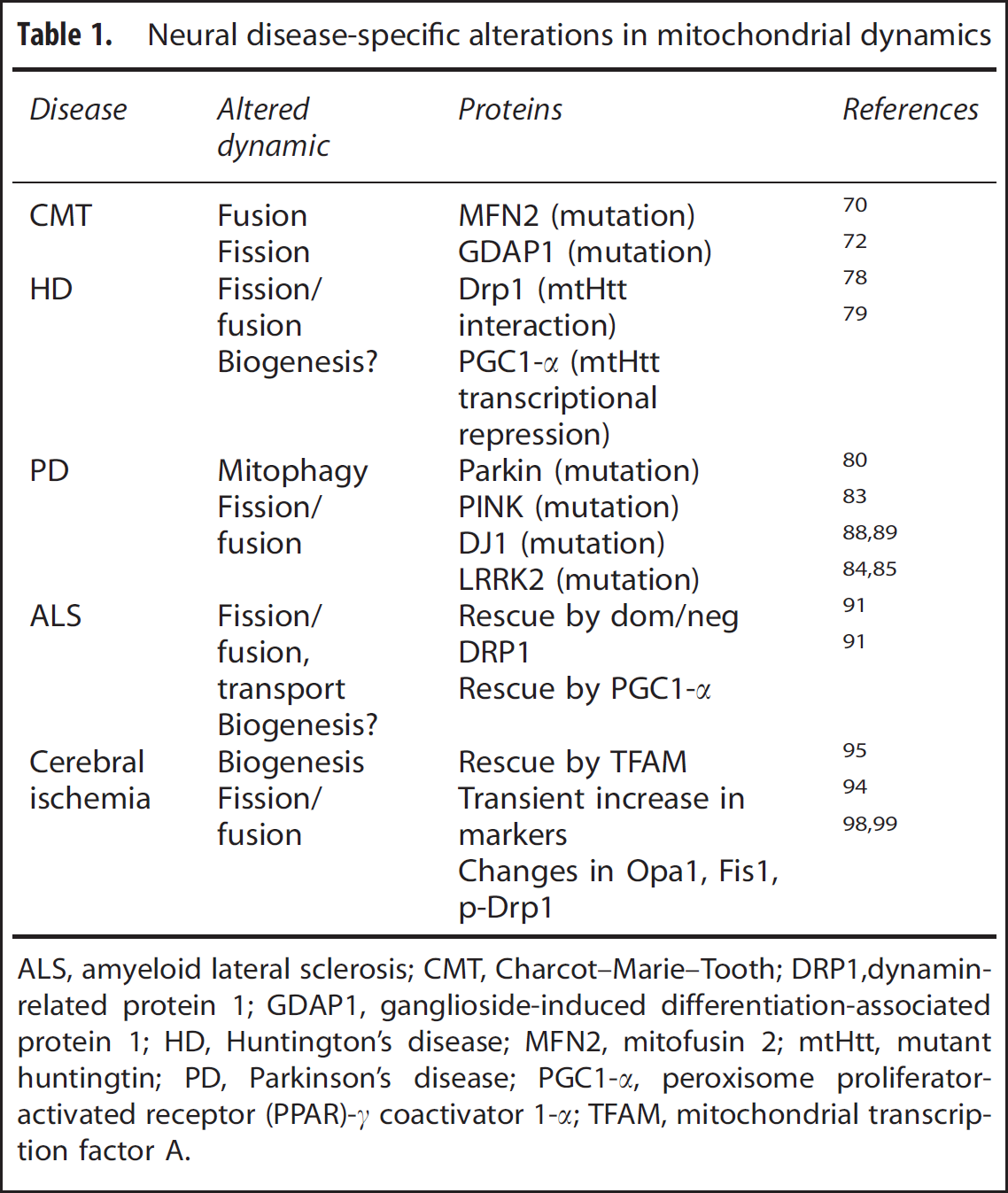
ALS, amyeloid lateral sclerosis; CMT, Charcot–Marie–Tooth; DRP1,dynamin-related protein 1; GDAP1, ganglioside-induced differentiation-associated protein 1; HD, Huntington's disease; MFN2, mitofusin 2; mtHtt, mutant huntingtin; PD, Parkinson's disease; PGC1-
Charcot–Marie–Tooth Disease
One of the earliest indicators of a role for aberrant mitochondrial dynamics in neurodegenerative disease was derived from the analysis of genetic mutations leading to CMT disease, a group of inheritable diseases affecting peripheral nerves. CMT diseases can be caused by a number of different mutations in various genes, are subdivided according to clinical phenotype, and now include more recent classifications based on genetic profiles and gene–family subgroupings. A striking number of the associated mutations occur in mitochondrial-related proteins, including proteins involved in the regulation of mitochondrial dynamics. A large subset of patients with CMT type 2A (CMT2A) were found to have a mutations in the
Huntington's Disease
In addition to CMT, disruption of mitochondrial dynamics appears to play some role in the pathology of HD. HD arises from the aberrant inclusion of an expanded glutamine repeat within the
Parkinson's Disease: Defects in Mitochondrial Clearance or Fission/Fusion?
Mitochondrial dysfunction has long been postulated to be integral to PD, but the exact pathology remains poorly understood. In a departure from earlier studies focused on the electron transport chain, several proteins critical to mitochondrial dynamics have now been implicated in both familial PD and in animal models of PD, including Parkin, DJ1, PINK, and the leucine-rich repeat kinase 2 (LRRK2). The involvement of a number of mitochondrial dynamics-related proteins in familial PD argues that, at least in a subset of PD cases, disruption of mitochondrial dynamics—particularly turnover—may underlie pathologic elements of the disease.
As mentioned above, Parkin has recently been identified as a protein bridging mitochondria to the autophagosome machinery, possibly through its E3 ligase activity or by an adaptor-like mechanism. Parkin was originally identified as a gene carrying genetic mutations associated with a subset of recessive PD. 80 Parkin contains both a kinase domain and an E3-ligase RING domain. 81 Although several studies have implicated Parkin in mitophagy, its precise role is still controversial. Many of the PD-associated mutations in Parkin lead to a loss of function, either by disrupted folding, mistranslocation, or reduction in enzymatic activity. 82 This loss of function is postulated to then lead to abnormal accumulation of dysfunctional mitochondria and may contribute to the observed mitochondrial pathology found in PD models. However, many of the mechanistic studies with Parkin have used overexpression of the wild-type or mutated forms, and thus fail to address whether alteration in mitochondrial clearance is the primary pathology or if overexpressed Parkin in other cellular compartments indirectly leads to abnormal mitochondrial clearance and/or pathology. Interestingly, a subset of familial PD was linked to mutations in PINK. 83 Given the close interaction between Parkin and PINK described above, it is likely that mutations in these two proteins may affect the same pathway in mitochondrial dynamic dysregulation.
Other PD-related proteins have also been implicated in altered mitochondrial morphology. For example, LRRK2 is a cytosolic kinase involved in a variety of cellular responses, and is also localized to the mitochondrial membrane. Mutations within LRRK2 are the most common cause of the familial autosomal-dominant form of PD and have been found in sporadic PD cases as well,84,85 with the most common mutation leading to an increase in kinase activity. 86 Although the targets of LRRK2 kinase activity are involved in a variety of cellular activities, several recent studies suggest that LRRK2 containing the PD-associated mutation increases mitochondrial fission via coordination with DLP1. 87 Further studies are necessary to understand the precise role of LRRK2 in mitochondria, including identification of potential substrates for its kinase activity in the mitochondrial framework.
Mutations in DJ-1 leading to a loss of its function have also been associated with autosomal recessive PD. 88 Although its precise role in mitochondrial dynamics is still unclear, PD-related DJ-1 mutations or DJ-1 knockout led to disrupted mitochondrial morphology,89,90 and was associated with aberrant mitochondrial clearance. 90 These studies and others now suggest that mitochondrial dynamics are an interesting and relevant aspect of PD pathology.
Amyeloid Lateral Sclerosis
Amyeloid lateral sclerosis is a progressive neural disease affecting the motor neurons of the spine, with an unclear etiology that largely appears to be sporadic, but a subset of cases have an identified genetic component. Mutations in Cu/Zn superoxide dismutase (SOD) have been found in a subset of familial-based ALS patients. Although SOD and its mutant form appear to be involved in a wide range of cellular processes and pathologies, mutant SOD (mtSOD) overexpression has also been associated with fragmented mitochondria and decreased axonal transport in cultured cells,91–93 including isolated spinal motor neurons. Interestingly, in cultured neurons transfected with mtSOD, fragmentation and defective trafficking could be reversed by cotransfection of a dominant-negative DRP1, which inhibits fission, 91 suggesting that the fragmented mitochondrial structures are because of impairment in the mitochondrial dynamic process, rather than a generalized cytoskeletal pathology. Not only did the dominant-negative DRP1 overexpression reverse the dysfunction in mitochondrial dynamics, it also led to increased cell survival. 91 However, it is as yet unclear as to how mutant SOD1 disrupts mitochondrial dynamics. Over-expression of either Sirt3 or PGC1-α also reduced mtSOD1-induced fragmentation and cell toxicity, 91 but whether this effect is via biogenesis or via other downstream targets of PGC-1α is unknown.
Cerebral Ischemia: Mitochondrial Dynamics Affect Outcomes
As has been outlined above and in many other reviews, control of mitochondrial organellar dynamics is orchestrated at multiple levels. The multiple stages of injury and repair that occur after cerebral ischemia would thus likely continually alter and be affected by mitochondrial dynamics. Therefore, understanding these dynamics and how to exploit them in the context of ischemic progression could have significant implications for therapeutic strategies. A rapid upregulation of critical components of mitochondrial biogenesis, including Tfam, mtDNA, and components of the electron transport machinery, occurs in the acute period after neonatal hypoxia ischemia, 94 significantly before the onset of cell death. Transgenic overexpression of Tfam decreased the number of TUNEL (TdT-mediated dNTP nick end labeling)-positive cells after transient forebrain ischemia. 95 Selective downregulation of Parkin was also observed after focal ischemia before evidence of neuronal death. 96 Global cerebral ischemia induces an elongation of hippocampal mitochondria distinct from swelling, 97 and alterations in Opa1, Fis1, and phosphorylated Drp1 occur over an extended reperfusion period after focal ischemia.98,99 Additionally, knockdown of the fission protein Drp1 blocked toxicity in a glutamate-induced oxidative stress model in HT22 cells, 100 and inhibitors of Drp1 reduced infarct in a transient focal ischemia model. 100 Mitochondrial biogenesis/fusion and altered fission after ischemia may reflect compensatory adaptations to mitigate the damage associated with the multistage reperfusion period after the ischemic episode. Thus, the timing of the biogenesis-related machinery and regulation of fission before death may be effectors of cell fate.
Preconditioning: Mitochondrial Dynamics as Neuroprotection
Perhaps more central to the concept of mitochondrial dynamics as preventative targets, many of the classic preconditioning stimuli used to establish an ischemic tolerant state also affect mitochondrial turnover and dynamics. The specific mechanisms of preconditioning, wherein a sublethal stimulus confers protection against subsequent ischemia, have remained elusive and likely involve a complex subcellular, cellular, and possibly systemic response in which the mitochondrial organelle may play a central role. Repeated observations now reveal that, in addition to upregulation of antioxidant systems and a heat shock response, preconditioning alters the mitochondria organelle beyond its traditional role in cell death/survival signaling. In support of the targeting of the mitochondria as an organelle for neuroprotection, resveratrol, which activates SIRT1 and leads to the deacetylation and thus activation of PGC1-α, has neuroprotective effects when administered before cerebral ischemic insults,101–103 and also induces markers of mitochondrial biogenesis in other systems. 104 Exercise is correlated with both ischemic neuroprotection and induction of genes related to mitochondrial biogenesis.105–107 In addition, dietary supplementation with omega-3 polyunsaturated fatty acids is effective at promoting ischemic neuroprotection,108–110 and has also been shown to induce expression of genes required for mitochondrial biogenesis in white fat. 111 Sublethal inflammatory stimuli—including lipopolysaccharide (LPS)—are also capable of preconditioning against subsequent cerebral ischemia112,113 and induce activation of nuclear transcription factors, such as Nrf-1, as well as upregulation of Tfam and mtDNA via CREB activation in hepatocytes. 11 Our lab has shown that LPS preconditioning in cortical neurons leads to transiently increased mitochondrial biogenesis in a time frame and concentration that is conducive to ischemic protection. 114 Additionally, transient knockdown of TFAM, a critical component of mitochondrial biogenesis, blocked the ischemic protection afforded by LPS preconditioning. 114 Thus, it is quite possible that targeting the mitochondrial organelle may help establish an ischemic tolerant state.
Additionally, recent evidence in cardiac ischemic preconditioning indicates that Parkin, the mitophagy-related protein described above and most commonly associated with mutations leading to PD, may be critical to ischemic preconditioning in cultured cardiomyocytes. 42 Overexpression of Parkin led to decreased mitochondrial loss (measured by Tom70 immunofluorescence), and Parkin knockout resulted in a loss of preconditioning-afforded ischemic cardioprotection.
ISSUES IN MITOCHONDRIAL DYNAMICS AND NEURAL DISEASES
There still remain unexplored avenues and significant caveats to implicating mitochondrial organelle dysfunction in neural disease. A major limitation to the above findings—and to the general research on mitochondrial dynamics—relates to the roles that many of the critical proteins involved in dynamics (e.g., Drp1, MFN2) or turnover (e.g., PGC1-α, Parkin) play in other cellular processes, such as peroxisome dynamics.115–117 Until mitochondria-specific, technically simple assays are developed, our understanding of mitochondrial dysfunction in brain disease will lag behind. However, the development of mitochondrial-targeted photoactivatable fluorophores is leading to the improvement of experimental designs monitoring mitochondrial dynamics,118–120 including the cell-specific expression of these fluorophores in transgenic mice. 121 The unique structure of neurons, with their fairly extensive projections and localized, subcellular environmental differences, appear to lead to heterogeneous mitochondrial phenotypes even within a single cell 122 that could confound interpretations based solely on cell lysate experimental designs. Furthermore, a critical unexplored issue is the potential difference in mitochondrial responses in the various cell types of the brain, particularly during injury. Studies using EM in several CMT2A cases indicated mitochondrial abnormalities74,123 that appear unique to the cell type (nerve axon vs. myelin sheath). 74 The recent development of transgenic mice expressing a mitochondrial-targeted fluorescent protein (dsRed2) under the control of the neuron-specific promoter Thy1 may help to address some of the technical limitations of studying mitochondrial responses in brain. 121 This is particularly relevant to ischemic conditions, as ischemic pathology encompasses a wide range of cell types, each demanding differing energetic loads, antioxidant capacity, and calcium buffering. This issue is also relevant to a variety of other conditions that may alter the dynamic response and control of cell survival by mitochondria. The interplay between neural fate and mitochondrial dynamics was recently described wherein alterations in mitochondrial fission/fusion (e.g., Opa1 deficiency) altered the expression of glutamate receptors, SOD, and Bcl-2 family members. 124 Thus, mitochondrial dynamics may be an effector of injury. In short, the examination of mitochondrial dynamics in affecting and/or effecting neural disease remains an underexplored field.
SUMMARY
The response of mitochondrial dynamics to environmental changes under sublethal or lethal conditions is still poorly understood, particularly in the context of the brain, and is often incompletely assessed. Assessment strategies based solely on signaling are confounded by an incomplete knowledge of effectors and suppressors of target molecules, promiscuous signaling pathways, and multiple levels of feedback in signaling. Although activity of these molecules may be correlated with biogenesis, mitophagy, or changes in dynamics, conclusive arguments require further exploration and knockdown or knockout strategies. Strategies based on content over time (e.g., mitochondrial volume, mtDNA, respiratory chain components) are useful to describe the flux between biogenesis and mitophagy/clearance, but are limited in the case of rapid turnover. Additionally, content data can be difficult interpret. For example, volume measurements based on microscopy must be rigorously controlled, and molecular constituents are not always kept at a constant ratio between individual mitochondria. Furthermore, molecular or lipid content assessments alone are inadequate in uncovering dynamic events such as fission or fusion, which may occur independent of biogenesis or clearance. Therefore, a highly combinatorial approach is necessary to more fully describe the mitochondrial morphology and changes over time. This approach would incorporate molecular analyses measured over time, live-image confocal microscopy for movement and fission/fusion over time, and EM (the gold standard for mitophagy). These techniques are both labor intensive and highly specialized, often beyond the resources of a given lab, and can be confounded in the mixed cell setting, such as brain. The recent development of transgenic mice expressing a mitochondrial targeted label (dsRed2) under the control of the neuron-specific promoter Thy1 may help to address some of the technical limitations of studying mitochondrial response in brain. 121 However, given the constraints in combinatorial studies, it has become necessary to pool from the literature to assemble a more holistic sense of the mitochondrial response to and impact on cellular changes.
Despite the limitations in assessment strategies, an aggressive pursuit of the mitochondrial organelle will be of great significance to the field of acute neural injury, such as stroke. The multiple layers at which ischemic injury affects critical mitochondrial processes, and the plasticity of the mitochondrial organelle in the face of environmental challenges, render the mitochondrion a multimodal target to help protect against or recover from ischemic neural death. Indeed, the complex nature of the different phases of neural injury—sublethal preconditioning, excitotoxicity, inflammation, apoptosis, and so on—repeatedly implicates mitochondria function/dysfunction. Mitochondrial dynamics under these conditions may differ significantly from those in the normal cellular environment, which is unlikely to experience such extreme fluctuations in milieu. Thus, limiting experimentation to nonpathologic environments to lay the groundwork on mitochondrial dynamics is likely to miss the full range of organelle dynamics and the specific role that the mitochondrion plays in maintaining homeostasis when the environment is disrupted. Furthermore, the multicellular nature of the nervous system will demand cell-specific targeting of mitochondrial dynamics, a field that remains virtually uncharted.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
ACKNOWLEDGEMENTS
The authors thank Pat Strickler for secretarial support.