
Abstract
Uncoupling protein 2 (UCP2) is upregulated in the brain after sublethal ischemia, and overexpression of UCP2 is neuroprotective in several models of neurodegenerative disease. We investigated if increased levels of UCP2 diminished neuronal damage after global brain ischemia by subjecting mice overexpressing UCP2 (UCP2/3tg) and wild-type littermates (wt) to a 12-min global ischemia. The histopathological outcome in the cortex, hippocampus, striatum, and thalamus was evaluated at 4 days of recovery, allowing maturation of the selective neuronal death. Global ischemia led to extensive cell death in the striatum, thalamus, and in the CA1 and CA2, and less-pronounced cell death in the CA3 and dentate gyrus (DG) hippocampal subfields. Histologic damage was significantly lower in the ventral posterolateral VPL and medial VPM thalamic nuclei in UCP2/3tg animals compared with wt. These thalamic regions showed a larger increase in UCP2 expression in UCP2/3tg compared with wt animals relative to the nonprotected DG. In the other regions studied, the histologic damage was lower or equal in UCP2/3tg animals compared with wt. Consequently, neuroprotection in the thalamus correlated with a high expression of UCP2, which is neuroprotective in a number of models of neurodegenerative diseases.
Introduction
Accumulating evidence has shown that uncoupling protein 2 (UCP2) is neuroprotective (Bechmann et al, 2002; Clavel et al, 2003; Diano et al, 2003; Mattiasson et al, 2003; Sullivan et al, 2003; Vogler et al, 2005), in models of focal cerebral ischemia (Mattiasson et al, 2003), traumatic brain injury (Mattiasson et al, 2003), Parkinson's disease (Andrews
Uncoupling proteins (UCPs; thermogenins) are encoded by nuclear DNA and are located in the inner membrane of the mitochondria. Uncoupling protein 1 is located in brown adipose tissue, where it is clearly involved in mitochondrial uncoupling and thermogenesis. The role of the other UCPs (UCP2–4, UCP5/BMCP1 (brain mitochondrial carrier protein 1)) is less clear, but one of their functions is to translocate protons from the intermembrane space to the matrix of the mitochondria (Mattiasson and Sullivan, 2006), although the extent of mitochondrial depolarization mediated by these proteins is presently unknown. The UCPs, UCP2, UCP4, and BMCP1, are all expressed in the central nervous system. UCP4 and BMCP1 have around 30% similarity with UCP1, whereas UCP2 shares about 56% amino-acid identity with UCP1, and has a high homology between rat, mouse, and humans (Hidaka et al, 1998). Under normal circumstances, UCP2 is expressed predominantly in neurons in several brain regions in both rodents and primates (Horvath et al, 1999; Arsenijevic et al, 2007), although microglia, residing monocytes of the brain (Bechmann et al, 2002; Clavel et al, 2003), invading monocytes and neutrophils (Arsenijevic et al, 2007), cells of the choroids plexus (Richard et al, 1999) as well as endothelial cells (Fink et al, 2005) also express this mitochondrial uncoupling protein. In mouse, it is expressed in several parts of the fore-, mid- and hindbrain, including the hypothalamus (Horvath et al, 1999; Richard et al, 1998, 2001), thalamus (Richard et al, 1998; Horvath et al, 1999), and cerebellum (Richard et al, 1998). UCP2 is expressed at low levels in the mouse hippocampus (Richard et al, 1998), whereas hippocampal expression is higher in rat (Richard et al, 2001).
Excitotoxic cell death is believed to be one of the central pathogenic mechanisms behind human neurodegenerative disorders, including cerebral ischemia. The cell death mechanisms are complex and not fully understood, but include glutamate toxicity, which leads to deranged ion homeostasis with K+ efflux and Ca2+ influx. The influx of Ca2+ is a key event in acute brain injuries, which affects signaling cascades within the cell, such as second messenger systems and protein kinases. It also has an effect on mitochondrial function, integrity and production of reactive oxygen species (ROS), activation of caspase cascades, and changes in gene expression (Wieloch, 2002). Increased levels of intracellular calcium as well as oxidative stress may induce mitochondrial permeability transition (mPT), which leads to mitochondrial swelling and release of apoptogenic factors that activate cell death cascades (mitochondria-mediated cell death). Inhibition of mPT by Cyclosporin A and some of its analogues is neuroprotective after brain trauma, as well as global and focal ischemia (Wieloch et al, 2007).
The molecular mechanisms behind the neuroprotective effect of UCP2 are not fully known. It has been suggested that UCP2 leads to a slight depolarization of the inner mitochondrial membrane, with a reduction in the rate of calcium uptake and production of ROS. This will subsequently reduce the probability of mPT and activation of cell death cascades (Richard et al, 2001; Mattiasson et al, 2003). This hypothesis is supported by the fact that a slight mitochondrial depolarization with 2,4-dinitrophenol is neuroprotective both
In this study, we investigated the neuroprotective effect of UCP2 after global ischemia in mice overexpressing UCP2 (Fuller et al, 2000). Using a model of global cerebral ischemia in mouse (Olsson et al, 2003), UCP2/3tg animals and wild-type (wt) littermate controls were subjected to 12 mins of global ischemia. The histopathological outcome in the cortex, hippocampus, striatum, and thalamus was evaluated at 4 days after injury, a time point where the ischemic lesion matured in this model.
Materials and methods
Animals
All experiments were approved by the Malmoe/Lund Ethical Committee on Animal Experiments. The animals were housed under diurnal light conditions and provided free access to food and water before surgery. UCP2/3-overexpressing mice (UCP2/3tg) were developed using an 80 kb Bacterial Artificial Chromosome containing the human UCP2/3 genes, including native promoters and
The Global Ischemia Model
The global ischemia procedure has previously been described in detail (Olsson et al, 2003) and applied in several studies (Olsson et al, 2004a, b, c). Briefly, animals were anaesthetized with halothane (O2/N2O, 30:70), intubated, administered muscle relaxant (norcuron) to avoid spontaneous breathing movements during ischemia, and connected to a small animal respirator. The pCO2 in respiratory end-tidal volume was measured with a capnometer to enable adjustment of the respirator to ensure physiologic blood gases. The common carotid arteries were exposed via a small ventral neck incision and were encircled loosely with a silk thread so as to later enable occlusion with a nontraumatic aneurysm clip. To estimate the density of ischemia in each hemisphere, the regional cerebral blood flow (rCBF) was recorded in both hemispheres with a two-channeled laser-Doppler flowmeter. Regional CBF was measured before, during, and for 5 mins after the ischemia with flexible optical filaments attached to the skull bone, 1 mm caudal to bregma and 4 mm lateral to the midline. Only hemispheres in which rCBF decreased below 10% of baseline within 1 min after occlusion were considered as ischemic and were included in the study. Individual hemispheres were not dependent on the contralateral blood flow (Olsson et al, 2003) and could be analyzed independently. Rectal temperature was kept at 37.2°C±0.2°C before, during, and shortly after ischemia by a homeothermic blanket. The brain was kept normothermic (37.1°C±0.1°C) during the whole period of ischemia with the help of a continuous flow of humidified warm air over the head of the mouse. The duration of ischemia by bilateral common carotid artery occlusion was 12 mins. Anesthesia was discontinued 2 mins before the end of ischemia. Laser-Doppler flow confirmed restoration of rCBF after clip removal. Five minutes after ischemia the laser-Doppler probes were removed, and the skin sutured. When the animals were able to breathe normally, after 10 to 30 mins, they were disconnected from the ventilator and placed in an incubator at 33.5°C to maintain normothermia for 24 h. The mice were extubated about 90 mins after the end of ischemia. Rectal temperature was monitored within the first 2 h after ischemia, and then at least two more times during the first day after surgery.
Measurement of End-Tidal CO2, Regional Cerebral Blood Flow, and Postischemic Temperature
The end-tidal pCO2 was monitored continuously with a capnometer from 15 mins before ischemia until 5 mins after the end of ischemia. The respirator was adjusted to allow a pCO2 of 35 to 40 mm Hg in arterial blood, corresponding to a value of 1.5% CO2 measured by the capnometer in a separate group of animals. Every animal was stabilized at 1.5% CO2 before the start of ischemia. The rCBF was monitored continuously from at least 5 mins before ischemia until 5 mins after the end of ischemia by laser Doppler. Changes in rCBF during ischemia (1 and 2 mins after occlusion) and 1 and 5 mins after the start of reperfusion were compared between UCP2/3tg and wt mice. Decreased rates of rCBF during ischemia were calculated as follows: (ischemia rCBF/preischemia rCBF) × 100. Postischemic temperature was rectally measured 0 to 2, 2 to 4, and 4 to 8 h after ischemia, when the animals were housed in an incubator.
Perfusion Fixation and Histologic Preparation
After 4 days of recovery, the animals were anesthetized in 4% halothane and transcardially perfused with 4% buffered formaldehyde at a flow of 3 to 5 mL/min after a short rinse with saline. The brains were removed and stored for at least 24 h in 4% formaldehyde at 4°C before dehydration and embedding in paraffin. Ten-micrometer coronal sections were cut and stained with celestine blue/acid fuchsin.
cRNA Probe Synthesis and In Situ Hybridization Histochemistry
To analyze the expression of mouse UCP2 (mUCP2) mRNA, a riboprobe was used (Richard et al, 1999). The UCP2 cRNA probe was generated from the 945-bp
A second UCP2 riboprobe, not cross-reacting with mUCP2 mRNA, was used for the detection of human UCP2 (hUCP2) transgene in the mouse brain. Amplification of the RNA sequence for hUCP2 was performed as previously described (Diano et al, 2003) (5′-CATCTCCTGGGACGTAGC-3′ and 5′-AGAGAAGGGAAGGAGGGAAG-3′). The resulting complementary DNA (1.1 kb), purified from agarose gel using the QIAquick gel extraction kit (Qiagen Inc., Valenica, CA, USA), was digested with
Quantification of UCP2 mRNA Expression
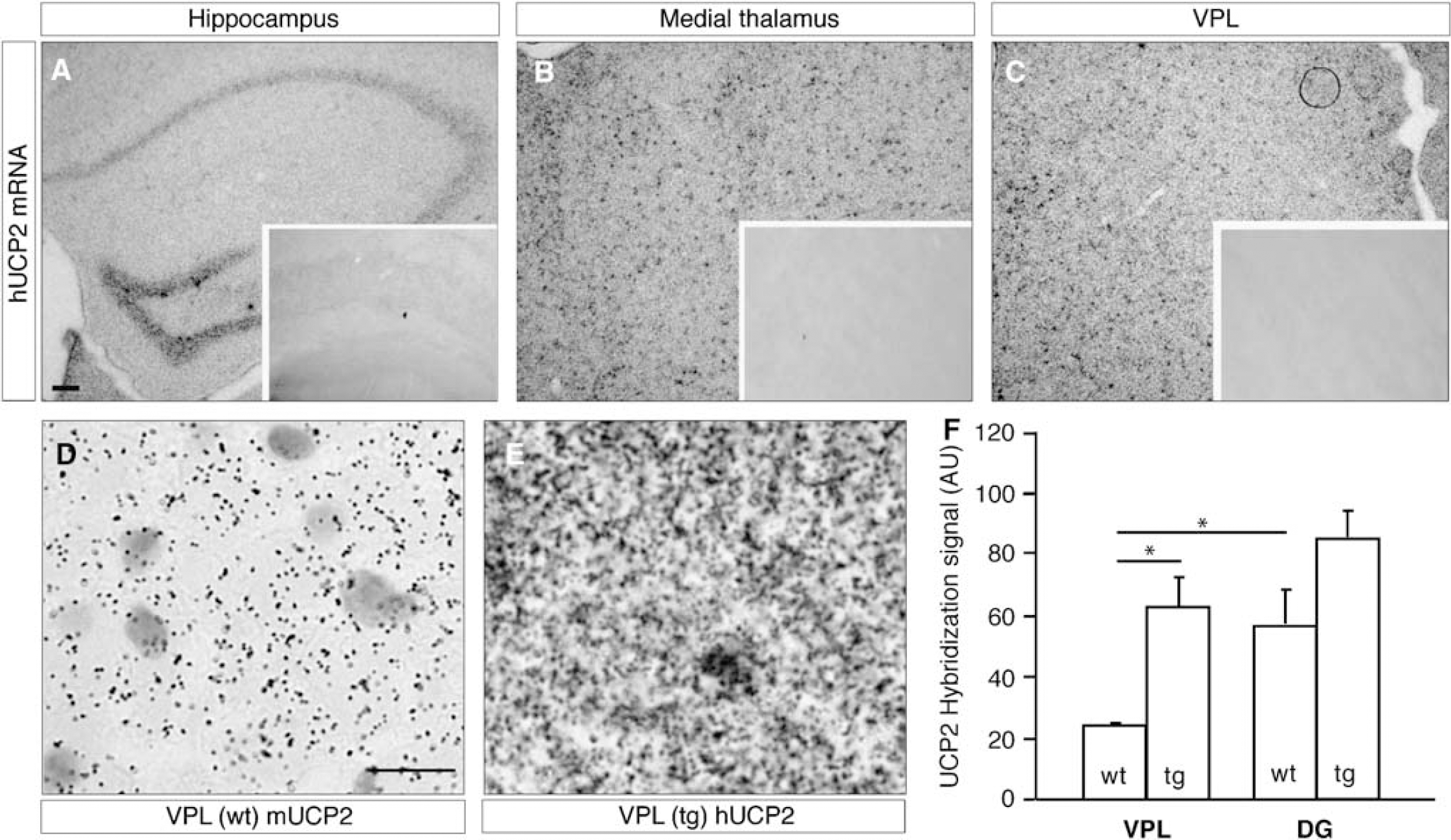
The density of the hybridization product for mUCP2 and hUCP2 mRNA was assessed in wt and hUCP2/3 transgenic mice. To digitally analyze, quantitate, and compare the amount of UCP2 mRNA, an Image-1/AT image processor (Universal Imaging Corp., West Chester, PA, USA) using an Olympus IMT-2 inverted microscope with dark-field optics (Olympus Corp., Lake Success, NY, USA) and a Hamamatsu CCD camera (Hamamatsu Photonics, Hamamatsu, Japan) were employed. Six sections per animal were selected from the same area to assess the intensity of the hybridization product (Arbitrary Optical Density). The total surface covered by the hybridization product was assessed within a test region measuring 2 × 105 mm2. The threshold for measurement was assessed for each slide by determining the background labeling over the third ventricle.
Assessment of Ischemic Damage
Light microscopy was used for the evaluation of the ischemic neuronal injury. The damage to the hippocampus, cortex, and thalamus was evaluated at coronal sections 1.7 mm caudal to bregma. In the sections, hippocampal neurons in the entire CA1 (∼500 cells), CA2 (∼50 cells), CA3 (∼280 cells), and dentate gyrus (DG; ∼400 cells) were counted, as were neurons in the barrel field of the neocortex (∼120 cells). Thalamic damage was evaluated in a 500 × 500 μm2 section (∼160 cells) placed 1.7 mm lateral to bregma and 3.2 mm ventral of the dura mater. This area corresponds to the ventral posterolateral (VPL) and medial (VPM) thalamic nuclei. In the hippocampus, cortex, and thalamus, the total number of neurons showing morphologic features of ischemic cell death (shrunken cell bodies, eosinophilic cytoplasm, and triangulated nuclei (see Figure 2) as well as normal-appearing neurons was counted. The damage was calculated as percentage of the total neuronal cell numbers in the respective structures. Striatal damage was assessed by visual assessment, confirmed by area measurement (by ImageJ) of the percentage of lesioned area 0.9 mm rostral to bregma using a 0 to 5 graded scale (0=no damage, 1=0% to 20%, 2=20% to 40%, 3=40% to 60%, 4=60% to 80%, and 5=80% to 100% of the total striatal area damaged).
Data Analysis
Individual hemispheres showed no dependency of the contralateral blood flow (Olsson et al, 2003). Therefore, all hemispheres were counted separately and in all graphs each circle represents one hemisphere (
Results
Physiologic Parameters
There was no difference in weight between wt and UCP2/3tg animals before surgery or 4 days after ischemia when mice were killed (Table 1). Regional CBF measured by laser Doppler in the outer layer of the barrel cortex in individual hemispheres before, during, and after ischemia did not show any difference between the experimental groups at any of the time points (Table 1). The body temperature during the first 8 h of reperfusion was similar between the groups (Table 1).
Physiologic parameters before and after global cerebral ischemia in UCP2/3tg and wt mice
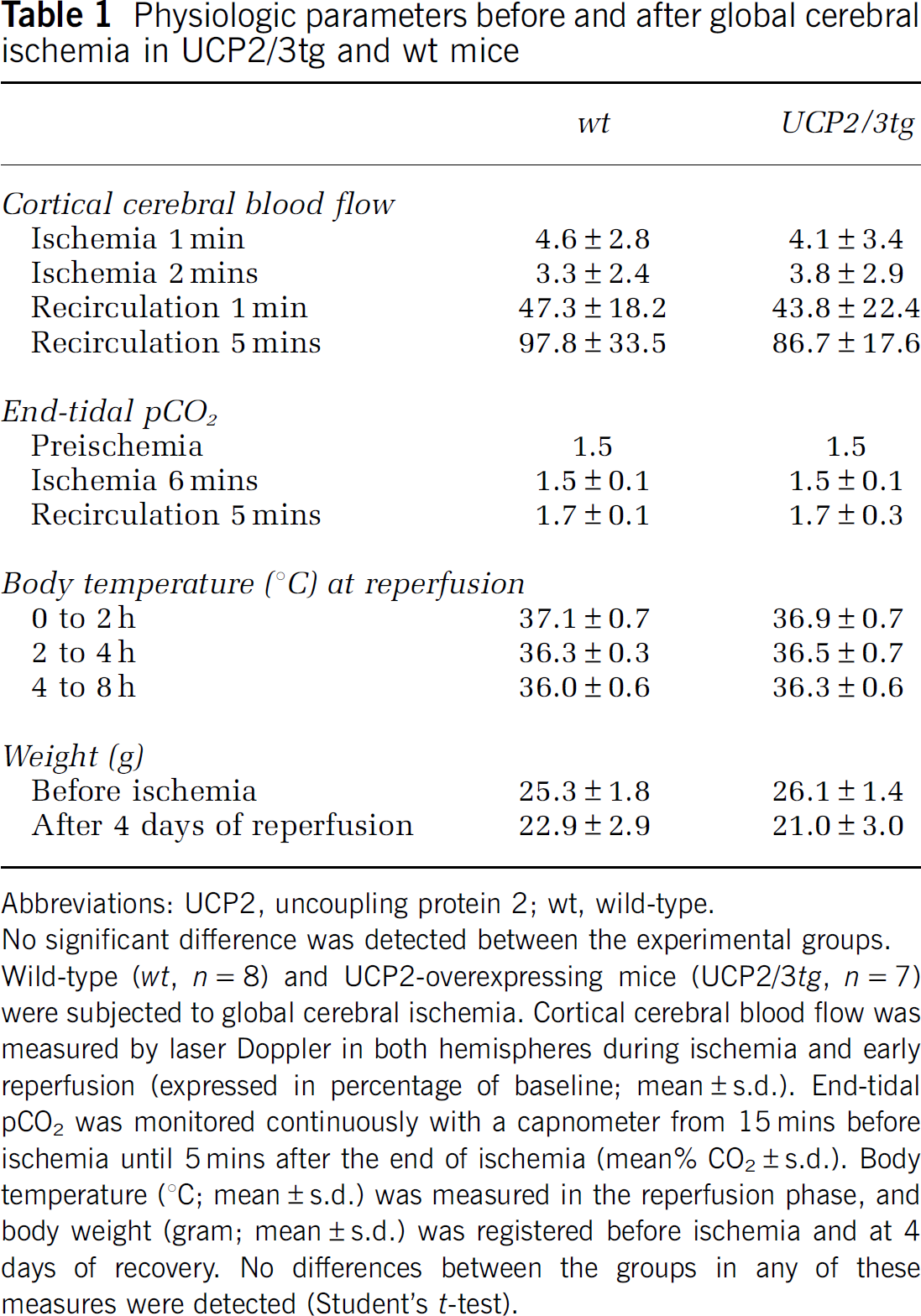
Abbreviations: UCP2, uncoupling protein 2; wt, wild-type.
No significant difference was detected between the experimental groups.
Wild-type (
Ameliorated Ischemic Cell Death in the Thalamic Nuclei of UCP2/3tg Mice
Representative photomicrographs of the hippocampal subregion CA1, striatum, and thalamus in UCP2/3tg mice and in wt littermates are displayed in Figure 1. The extent of ischemic cell death in the specific brain regions is shown in Figure 2. In the thalamic VPL and VPM, cell death was significantly decreased in UCP2/3tg mice compared with wt controls (19.4%±15.5% versus 47.1%±32.1%;
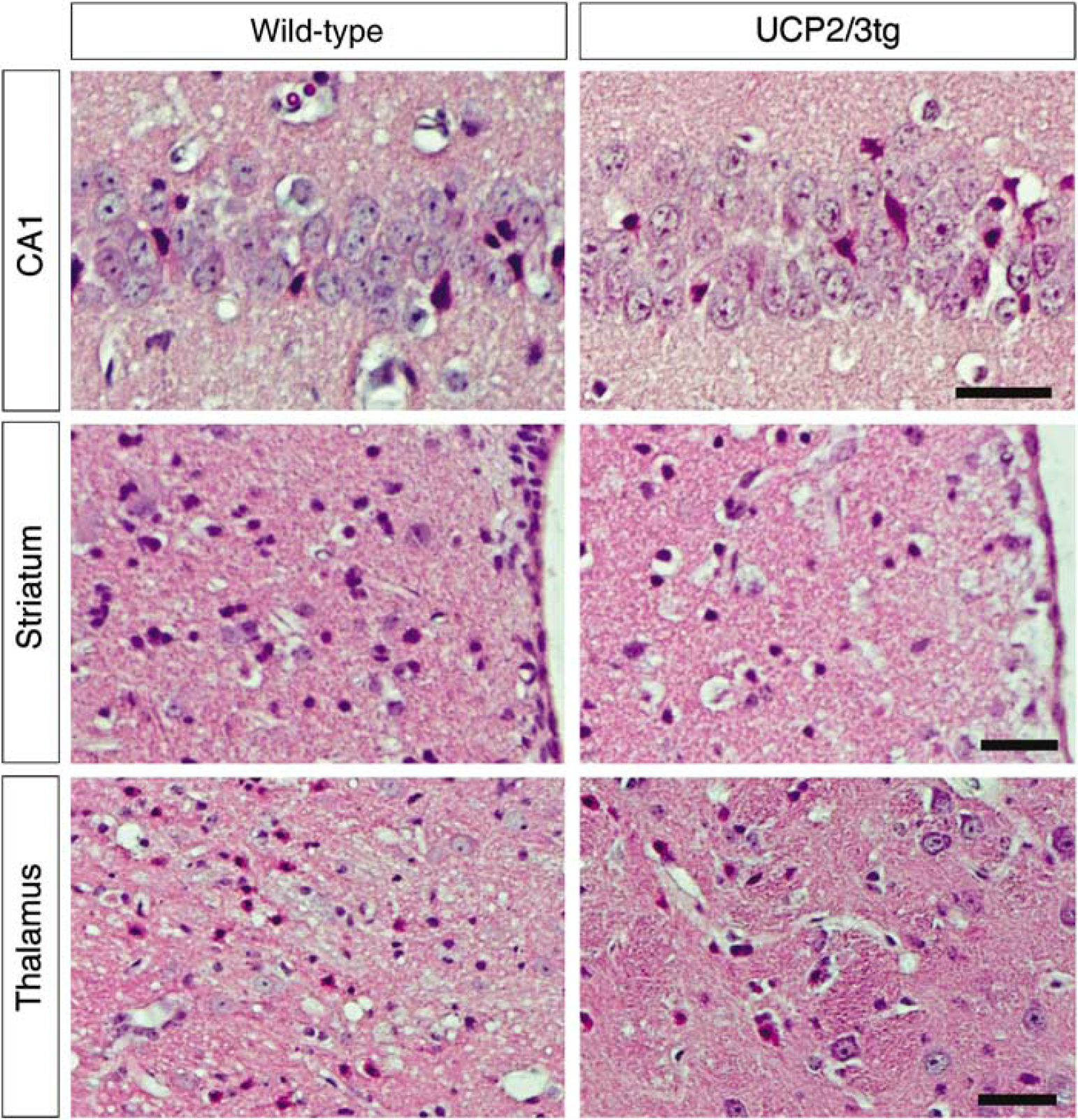
Representative pictures showing the hippocampal subregion CA1, the medial part of striatum next to the ventricle, and the ventral posterolateral and medial thalamic nuclei in wild-type and UCP2-overexpressing animals (UCP2/3tg). Normal-appearing neurons and dead neurons showing typical morphologic features of ischemic cell death with shrunken eosinophilic cytoplasms and pyknotic nuclei are seen. (Scale bar=35 μm).
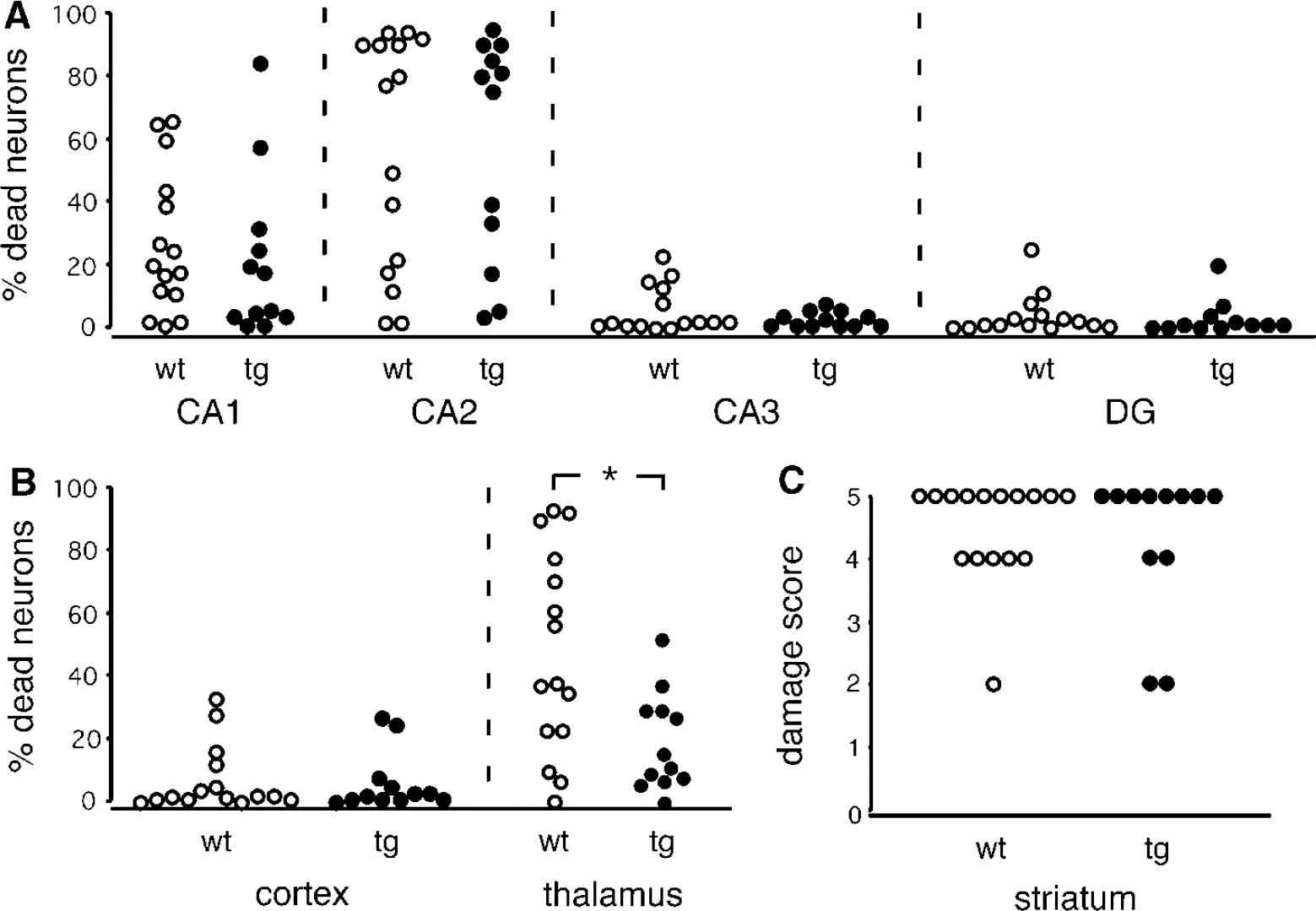
Ischemic cell damage in transgenic animals overexpressing UCP2 (UCP2/3tg;
Specifically High UCP2 mRNA Expression Levels in the Thalamic Nuclei of UCP2/3tg Mice
Similar to previous observations (Andrews et al, 2005b), no difference was observed regarding mUCP2 mRNA expression between wt and transgenic animals. Expression of mUCP2 was detected in the cortex, the CA1–3 and DG hippocampal fields, and the thalamus both in wt and UCP2/3tg mice (
Discussion
Ischemic tolerance or IPC is time- and protein-synthesis-dependent (Barone et al, 1998), represents the mobilization of endogenous neuroprotective pathways, and includes changes in the expression of a large number of genes (Kawahara et al, 2004). After IPC, UCP2 (but not UCP1, 3, 4, or BMCP1/UCP5) is upregulated
The molecular mechanisms behind UCP2-mediated neuroprotection are not fully understood. Previous experimental data indicate that the neuroprotective effect of UCP2 after an excitotoxic insult may be related to multiple effects of UCP2 on mitochondrial, and consequently cellular, metabolism. For example, UCP2 may induce a slight depolarization of the inner mitochondrial membrane, with a decreased electrophoretic uptake of calcium and reduced induction of mitochondrial mPT (Bechmann et al, 2002; Mattiasson et al, 2003; Sullivan et al, 2003). However, the magnitude of mitochondrial depolarization mediated by UCP2 is still unclear, and evidence for its uncoupling activity is obtained through measurements of mitochondrial respiration.
In the context of excitotoxic/ischemic brain injury, a mitochondrial depolarization could lead to a decrease in the electrophoretic movement of calcium ions into the mitochondria, preventing mitochondrial calcium overload and cytotoxicity (Castilho et al, 1998). Further, a reduction in the mitochondrial membrane potential, by increasing proton transport, may decrease the generation of ROS, presumably by decreasing the time of interaction between electrons and molecular oxygen, decreasing the formation of ROS (Skulachev, 1996). Even a slight depolarization (∼10 mV) of mitochondrial potential is sufficient to significantly decrease the production of ROS (Liu, 1997), suggesting that even small mitochondrial depolarizations, mediated by UCP2, may have important physiologic effects. Under intact conditions, UCP2/3tg mice were shown to have decreased levels of lipid peroxidation (Diano et al, 2003) and decreased amounts of free radicals (Andrews et al, 2005b) in neurons. This, together with a positive correlation between UCP2 levels and neuronal mitochondria number (Diano et al, 2003; Andrews et al, 2005b; Coppola et al, 2007) and brain ATP and ADP levels and ratios (Diano et al, 2003), suggests that UCP2/tg animals may have a higher ability to maintain mitochondrial function. A decrease in intramitochondrial calcium and ROS release as a consequence of UCP2 activation would decrease the probability of mPT, collapse of respiration, the release of apoptogenic factors such as cytochrome
The neuroprotective effects of UCP2 outlined above assume that there is some mitochondrial membrane potential left in the area-at-risk. This would be the case, for example, after focal ischemia or trauma, where the area surrounding the ischemic core or traumatic lesion (the penumbra) is still sufficiently perfused to maintain metabolism, although at a reduced rate. In the present model of global ischemia, blood flow in the brain is completely stopped for a period of 12 mins, leading to the consumption of all available oxygen and oxidizable substrates. ATP in the brain drops to <10% of normal levels within 2 mins of cardiac arrest (Folbergrova et al, 1990), and ATP levels are further reduced as ATP synthase is reversed in an effort to maintain mitochondrial membrane potential. In a matter of minutes, all available ATP and ADP are consumed, and the mitochondria depolarizes, suggesting that neuroprotection through a slight depolarization, mediated by UCP2, is no longer feasible. Possibly, increased levels of UCP2 may delay mitochondrial mPT by preventing mitochondrial calcium overload. Thereby, the collapse of oxidative phosphorylation is delayed, postponing the final depolarization of mitochondria and cellular energy crisis. UCP2 is neuroprotective in a number of models and species (Mattiasson and Sullivan, 2006), and it is therefore possible that UCP2 has other neuroprotective effects besides those outlined above, probably involving several cell types in the brain besides neurons. UCP2 has been suggested to affect neuronal plasticity (Yamada et al, 2006), which could be neuroprotective after cerebral ischemia by decreasing detrimental excitotoxic synaptic signaling or by stimulating functional recovery. UCP2 has also been implicated in substrate transport across the mitochondrial membrane (Criscuolo et al, 2006), which may be important for cell energy homeostasis under conditions of limited substrate and oxygen availability, that is, ischemia. UCP2 is expressed in endothelial cells (Cui et al, 2006), and may influence adhesion and migration of inflammatory cells across the endothelium. Our results show that there were no significant differences in CBF between the genotypes before, during, or after ischemia, but it is possible that UCP2 influences microvascular function or architecture in ways that were not detected. Further, UCP2 may reduce detrimental inflammatory response after ischemia by limiting acute microglial response (Bai et al, 2005), or stimulating the expression of neuroprotective genes (Mattiasson et al, 2003).
In rat, IPC leads to an increased expression of UCP-2, and the brain is protected against a subsequent longer global ischemia. Induction of UCP-2 by rosiglitazone also protected the rat brain against global ischemia (Chen et al, 2006). In the present study, the lack of significant protection in the cortex, hippocampus, and striatum after global ischemia in mice overexpressing UCP2 could be explained by the fact that UCP2 is maximally induced in some of these regions (cortex and hippocampus) in the wt animals. Neuroprotection in the thalamus correlated with a higher relative increase in UCP2 expression in UCP2/3tg compared with wt animals. Alternatively, the thalamus may suffer a less ischemic insult, sufficient to maintain some mitochondrial electron transport, thereby promoting a neuroprotective UCP-2 function as discussed above. In this case the less ischemic insult could be comparable to the hypoperfused area of the peri-infarct zone during focal occlusion of the middle cerebral artery, where overexpression of UCP-2 has been shown to be effectively protective (Mattiasson et al, 2003). Another possible explanation along this line is that the insult in the regions that showed no protection was too severe, with a rapid onset of energy depletion and mitochondrial membrane depolarization.
Based on the data from the present study, we conclude that after global ischemia in mouse, overexpression of UCP2 leads to significant neuroprotection in the thalamus. In the other brain regions studied, there was a trend toward neuroprotection, but the effect was not significant. The molecular basis for this protection is presently unclear. UCP2 can be induced by PPARγ-agonists, which are currently used as antidiabetic drugs. Treatments with such compounds have been shown to increase the expression of UCP2 and be neuroprotective in an experimental rat model of global ischemia (Chen et al, 2006). The clinical usefulness of a similar approach, if any, remains to be investigated.