
Abstract
Mitochondria play a central role in the pathophysiological processes of acute ischemic stroke. Disruption of the cerebral blood flow during acute ischemic stroke interrupts oxygen and glucose delivery, leading to the dysfunction of mitochondrial oxidative phosphorylation and cellular bioenergetic stress. Cells can respond to such stress by activating mitochondrial quality control mechanisms, including the mitochondrial unfolded protein response, mitochondrial fission and fusion, mitophagy, mitochondrial biogenesis, and intercellular mitochondrial transfer. Collectively, these adaptive response strategies contribute to retaining the integrity and function of the mitochondrial network, thereby helping to recover the homeostasis of the neurovascular unit. In this review, we focus on mitochondrial quality control mechanisms occurring in acute ischemic stroke. A better understanding of how these regulatory pathways work in maintaining mitochondrial homeostasis will provide a rationale for developing innovative neuroprotectants when these mechanisms fail in acute ischemic stroke.
Keywords
Introduction
The primary therapeutic goal for patients with acute ischemic stroke (AIS) is the timely restoration of blood flow to salvage oxygen-starved tissues. Intravenous thrombolysis with recombinant tissue plasminogen activator (t-PA) has been used as an effective first-line treatment for AIS in numerous studies. However, the narrow time window and the low reperfusion rate have largely hindered its application in clinical practice. 1 In 2015, the publications of five positive clinical trials of endovascular thrombectomy (ET) started the era of highly effective reperfusion treatment in AIS. 2 Later, data from the DAWN 3 and DEFUSE III 4 trials further revealed that the time windows for performing ET treatment in patients with anterior circulation stroke could be extended to 24 h, conditioned upon the existing ischemic penumbra. Still, less than half of the patients benefit from these simple reperfusion strategies. One plausible explanation is that reperfusion beyond the limited time window paradoxically causes irreversible detrimental effects because of the small penumbra. Thus, combining these reperfusion therapies with neuroprotective strategies has been proposed as a future direction for AIS treatment. 5
Since the brain is one of the most energy-demanding organs, it is highly dependent on mitochondria as an efficient source of energy to maintain its normal function. 6 Emerging data suggest that mitochondrial disruption during cerebral ischemia and reperfusion (I/R) is a critical determinant of the final extent of brain injury.7–9 During cerebral ischemia, mitochondrial dysfunction leads to the inhibition of the electron transport chain (ETC), thereby impairing adenosine triphosphate (ATP) production and increasing reactive oxygen species (ROS) generation. Augmented oxidative stress during reperfusion further aggravates mitochondrial damage. 10 In response to this series of perturbations, eukaryotic cells can employ a set of well-organized mitochondrial quality control (MtQC) mechanisms that effectively monitor and maintain the integrity of the intracellular mitochondrial network (Figure 1). These elegant mechanisms include the mitochondrial unfolded protein response (UPRmt), mitochondrial fission and fusion, mitophagy, mitochondrial biogenesis, and intercellular mitochondrial transfer. The UPRmt is an active retrograde signal transduced from the mitochondria to the nucleus, wherein it initiates the targeted transcription of mitochondrial chaperones and proteases, promotes ROS detoxification, and facilitates glycolysis, thereby maintaining the mitochondrial network at the molecular level. 11 At the organelle level, mildly damaged mitochondria can compensate for their loss of function by fusing with other, healthy mitochondria or eliminating deleterious constituents via fission. Damaged mitochondria that cannot be recovered by fission or fusion are subsequently cleared through mitophagy. 12 Once degraded, the mitochondrial components are renewed through protein and lipid biogenesis. 13 Intercellular mitochondrial transfer, a novel type of MtQC, is important for rescuing the mitochondrial network at the cellular level. 14 However, if the primary damage exceeds the capacity of the MtQC, then the mitochondria will translate these danger signals into cell-death decisions. Thus, an in-depth understanding of how mitochondria respond to stressors during cerebral I/R and how MtQC adapts cellular metabolism to environmental changes will help identify novel neuroprotective approaches for AIS treatment.
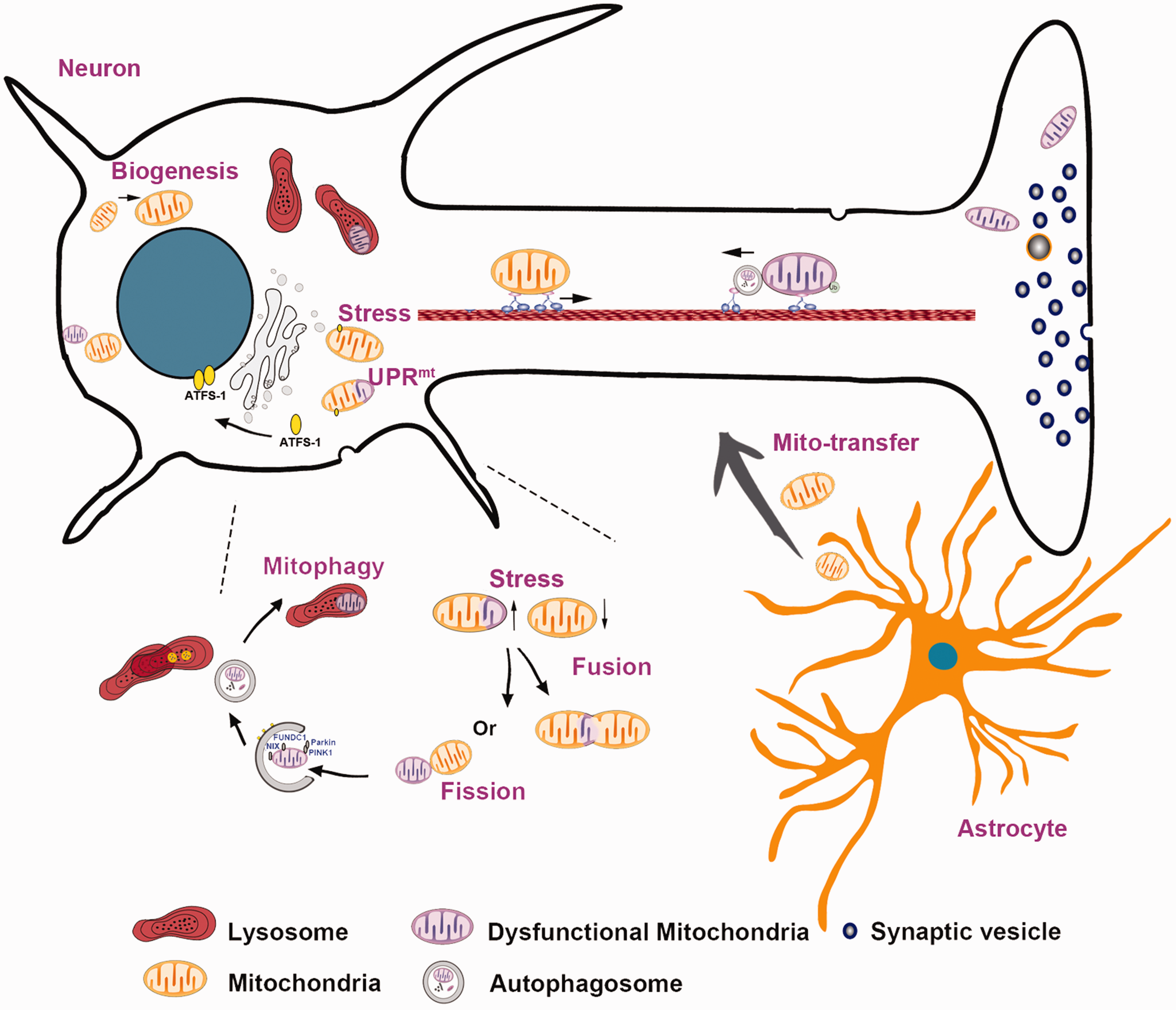
Mitochondrial quality control and intercellular mitochondrial transfer. When exposed to stressors, mitochondria can initiate a set of quality control mechanisms, including the mitochondrial unfolded protein response (UPRmt), mitochondrial fission and fusion, mitophagy, and mitochondrial biogenesis. In addition, mitochondria can be transferred from adjacent astrocytes to injured neurons as a compensatory internal adaptive response, which is a novel type of mitochondrial quality control mechanism.
The aim of this review is to shed light on the pathophysiological mechanisms of mitochondria during cerebral I/R, especially the mechanisms of MtQC, and evaluate mitochondria as therapeutic targets for AIS based on recent findings.
Pathophysiological mechanisms of mitochondria in AIS
The phospholipid bilayer membrane of mitochondria separates their internal environment from the cytosol. Several protein channels in the outer mitochondrial membrane (OMM) allow the permeation of molecules of less than 10 kDa, such as water, ions, nutrient molecules, adenosine diphosphate (ADP), and ATP. The mitochondrial ETC is located on the inner mitochondrial membrane (IMM), and its essential function is to generate ATP from electron donors (such as reduced nicotinamide adenine dinucleotide (NADH)) through different redox reactions, thereby furnishing cells with usable energy. During these redox reactions, the proton pumps (complexes I, III, and IV) transfer protons from the mitochondrial matrix to the intermembrane space (IMS), constituting the proton motive force and membrane potential (ΔΨm) used by complex V for ATP production. However, a small percentage of electrons can escape from the ETC during these processes, especially from complexes I and III, and react with O2 to form superoxide. Under physiological conditions, endogenous antioxidant systems can dispose of ROS and maintain ΔΨm within the normal range, in which ATP production is efficient, but ROS generation is minimal.
When cerebral ischemia occurs, inadequate delivery of oxygen and glucose inhibits mitochondrial respiration. Several studies have revealed that after 2 h of cerebral ischemia, the activity of mitochondrial ETC is reduced by 45–60% in focal tissues and 15–40% in perifocal tissues,15,16 resulting in decreased ATP production and presumably ROS accumulation. The duration of ischemia determines the mitochondrial fate, as mitochondrial respiration can recover after a short period of ischemia, whereas prolonged ischemia leads to irreversible mitochondrial injury. During the reperfusion stage after a prolonged period of cerebral ischemia, oxygen promptly infiltrates and replenishes the brain tissue, but the burst of ROS generation also aggravates oxidative brain damage. A comparative in vivo metabolomics analysis developed by Chouchani et al. first reported that succinate accumulation in ischemic tissues could drive reverse electron transport through complex I and was responsible for mitochondrial ROS production during reperfusion. 17 Excess ROS production directly leads to oxidative damage of lipids, proteins, and DNA. Oxidative DNA damage can activate poly (ADP-ribose) (PAR) polymerase-1, leading to the accumulation of PAR polymers, which bind to the apoptosis-inducing factor (AIF) and subsequently initiate caspase-independent apoptosis. 18 Oxidative stress can also promote the translocation of Bax, a pro-apoptotic member of the Bcl-2 family, from the cytosol to mitochondria and initiate caspase-dependent apoptosis. 19 Interestingly, the oxidative signaling can also remodel the mitochondrial network and mitochondrial function, suggesting that a tight bidirectional interplay exists between the MtQC and pathological stress.20–22
MtQC mechanisms and AIS
Communication between mitochondria and the nucleus: UPRmt
Although mitochondria have their own set of small circular genomes (mtDNA), only 13 oxidative phosphorylation (OXPHOS) components of over 1,300 mitochondrial proteins are encoded by mtDNA, whereas 99% of the mitochondrial proteins are encoded by the nuclear genome and are continuously imported into mitochondria. Thus, there is close communication between mitochondria and the nucleus that ensures normal mitochondrial function and cellular homeostasis.
Consistently, mitochondrial biogenesis is regulated by anterograde signaling from the nucleus to the mitochondria. Most nuclear genome-encoded mitochondrial proteins are synthesized with N-terminal presequences that function as import signals, which can be recognized by the translocase of the outer membrane (TOM) complex and delivered to translocase of the inner membrane 23 (TIM23) complexes. 23 Translocation through the TIM23 complex is ATP-dependent and requires the ΔΨm to be maintained by the functional mitochondrial respiratory chain. 24 These precursor proteins are then transported to the mitochondrial matrix with the assistance of mitochondrial chaperone 70 (mtHsp70), 25 and their presequences are removed by mitochondrial proteases once they enter the matrix. 26 In addition, mtHsp70 cooperates with other mitochondrial chaperones (such as Hsp60 and Hsp10) and proteases (such as the matrix-localized Lon protease) to facilitate the folding of precursor proteins and their assembly into multi-protein complexes.27,28 However, oxidative stress and other detrimental processes that threaten cellular homeostasis will inevitably lead to the accumulation of misfolded or unfolded proteins in the mitochondrial matrix and cytosol. Once the accumulation of unfolded proteins is beyond the steady-state capacity that mitochondrial chaperones and proteases can manage, a retrograde signal is sent to the nucleus, a process known as UPRmt, initiating a series of adaptive responses that help to repair the mitochondrial network (Figure 2).11,29–31
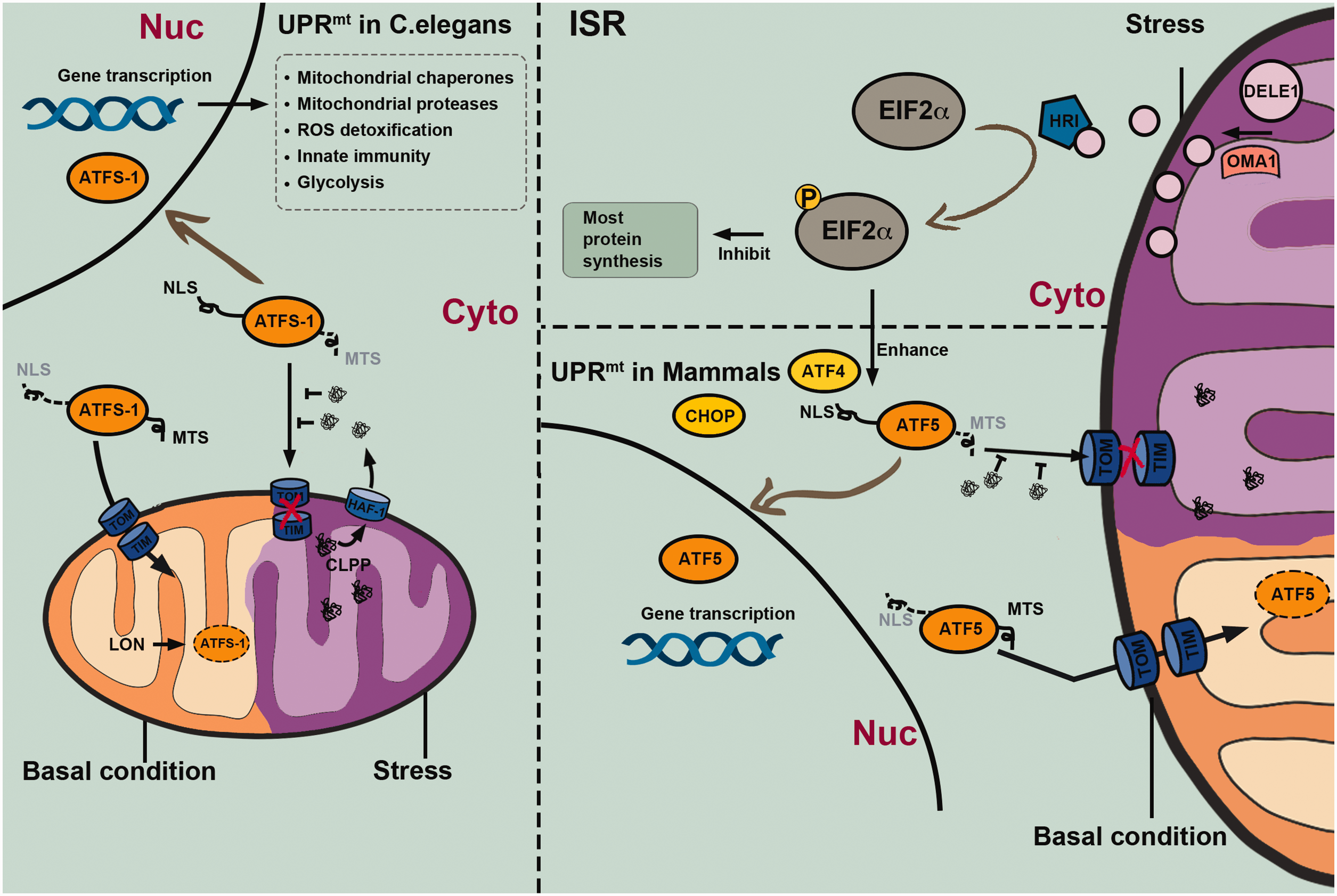
Mechanisms of the mitochondrial unfolded protein response (UPRmt) in Caenorhabditis elegans and mammalian cells. The UPRmt is an adaptive transcriptional response that can be activated by various mitochondrial stressors. In C. elegans, the key regulator of UPRmt is the activation transcription factor associated with stress 1 (ATFS-1), which is continuously transported into mitochondria via mitochondrial targeting sequence (MTS) guidance and cleaved by the Lon protease under physiological conditions. However, the ATFS-1 mitochondria targeting can be suppressed by the accumulation of aberrant peptides in the cytosol, which are unfolded or misfolded proteins that are cleaved by the caseinolytic protease P (CLPP) and extruded into the cytosol via the transporter HAF-1 under stress conditions. Stabilized ATFS-1 translocates through guidance of the nuclear localization sequence (NLS) into the nucleus from where it promotes gene transcription related to mitochondrial chaperones and proteases, detoxification of reactive oxygen species (ROS), innate immunity, and glycolysis. Together, these protective responses facilitate the recovery of mitochondrial homeostasis. UPRmt in mammalian cells may be regulated by basic leucine zipper (bZip) protein homologs including the C/EBP homologous protein (CHOP), activating transcription factor 4 (ATF4), and activating transcription factor 5 (ATF5), which are transcribed under the control of the integrated stress response (ISR) mechanism mediated by phosphorylated eukaryotic initiation factor-2 alpha subunit (eIF2α). The mitochondrial metallopeptidase OMA1 cleaves the DELE1 protein into short peptides, contributing to heme-regulated inhibitor (HRI) activation, eIF2α phosphorylation, and a general inhibition of gene transcription, while promoting transcription of genes encoding bZip proteins.
The UPRmt was first described in 1996 by Hoogenraad et al., 32 who found that mtDNA depletion in cultured rat hepatoma cells through ethidium bromide treatment elicited a nuclear response, promoting the transcription of genes that encode mitochondrial chaperones and proteases. This is presumably an adaptive transcriptional response by cells to maintain proteostasis within mitochondria. The major UPRmt mechanisms have been characterized in Caenorhabditis elegans and require the regulation of activation transcription factor associated with stress 1 (ATFS-1). 31 ATFS-1 has both an N-terminal mitochondrial targeting sequence (MTS) and a C-terminal nuclear localization sequence (NLS). Under resting conditions, ATFS-1 is effectively imported into mitochondria through MTS guidance and continuously degraded by the matrix-localized Lon protease. 33 However, under mitochondrial stress,34–36 the mitochondrial matrix exhibits aberrant accumulation of misfolded or unfolded proteins, which are cleaved into peptides by the mitochondrial caseinolytic protease P (CLPP)30,31 and exported by the HAF-1 transporter into the IMS from where they diffuse into the cytosol. 31 The extruded peptides block the mitochondrial target of ATFS-1, leading to its nuclear re-localization via the NLS. 33 Cells can evaluate the mitochondrial network fitness through the mitochondrial import efficiency of ATFS-1. 33 Once activated, ATFS-1 upregulates many genes involved in mitochondrial protein folding and degradation, ROS detoxification, and innate immunity. 37 More importantly, UPRmt activation can transiently shift the cellular metabolism from mitochondrial OXPHOS to glycolysis, which temporarily decreases the cells’ dependence on mitochondria during stress. However, it is worth noting that glycolysis is less effective for ATP production than OXPHOS. The final recovery of OXPHOS is vital for the brain to meet the challenges of high energy demand, 38 which is consistent with the concept that the UPRmt should be suppressed when short-term stress has been mitigated. Consequently, aberrant or prolonged activation of UPRmt triggers apoptosis. 39 Moreover, reduced expression of the cytosolic pentose phosphate pathway (PPP) enzyme transaldolase has been associated with UPRmt activation. 40 The PPP is essential for cytosolic redox homeostasis. Thus, UPRmt may also act as an important way to mitigate the oxidative stress caused by PPP dysfunction and, therefore, prevent ferroptosis. 41
The regulation of UPRmt in mammals is similar to, but more complicated than that in C. elegans. Three basic leucine zipper (bZip) transcription factors, including the C/EBP homologous protein (CHOP), activating transcription factor 4 (ATF4), and activating transcription factor 5 (ATF5), are known to be responsible for the UPRmt in mammalian cells.42–44 However, exactly how these transcription factors interact during mitochondrial stress remains unclear. ATF5 has been described as the mammalian ortholog of ATFS-1, based on its role in rescuing UPRmt signaling in worms lacking ATFS-1. 42 Similar to ATFS-1, ATF5 trafficking to mitochondria is inhibited by mitochondrial stress, which promotes its translocation to the nucleus and activates UPRmt. 42 However, using multi-omics analyses, Quiros et al. found that ATF4 was activated instead of ATF5 during the mitochondrial stress responses in mammals. 43 Zhao et al. induced the accumulation of protein aggregates within the mitochondrial matrix via mutant ornithine transcarbamylase (delta-OTC) overexpression, revealing aberrant misfolded protein aggregates in mitochondria that led to increased CHOP expression. 36 Although the key regulator of UPRmt remains to be identified in mammals, increasingly clear evidence reveals the stressor and model dependence of the UPRmt. Notably, CHOP and ATF4 are not mitochondria-specific, as they are also activated during the endoplasmic reticulum (ER) UPR (UPRER)36,45 and have been demonstrated to have even stronger activation during UPRER than during UPRmt. 36
The expression of these three bZIP proteins in mammalian cells requires activation of the integrated stress response (ISR), which is mainly mediated by phosphorylated eukaryotic initiation factor-2 alpha subunit (p-eIF2α). 46 eIF2α can be phosphorylated by four kinases, including double-stranded RNA-dependent protein kinase (PKR), PKR-like ER kinase (PERK), general amino acid control nonderepressible 2 (GCN2), and heme-regulated inhibitor (HRI), when responding to different types of cellular stress. eIF2α phosphorylation inhibits the action of eIF2’s guanine nucleotide exchange factor (eIF2B), which controls the formation of the eIF2-GTP-methionyl initiator tRNA ternary complex (TC). 47 ISR-induced reduction of TC formation suppresses the initiating of global protein synthesis to alleviate mitochondrial protein folding load, but selectively triggers the translation of certain mRNAs with open reading frames upstream of the main coding region, such as ATF5, ATF4, and CHOP.48–50 Very recently, the Kampmann and Jae groups elucidated the molecular mechanisms of the mitochondrial axis of ISR, which comprises the OMA1-DELE1-HRI pathway.51,52 OMA1, a mitochondrial stress-activated protease, cleaves DELE1 (a less characterized protein that is associated with the IMM) and leads to the release of truncated DELE1 peptides. The accumulation of truncated DELE1 petides in the cytosol facilitates their interactions with HRI, thereby promoting its activation. ATF4 or CHOP, as part of the ISR, is then activated by p-eIF2α. Both ATF4 and CHOP can directly bind to the ATF5 promoter. 44 It has been reported that ATF5 expression is decreased in Chop-/- mouse embryonic fibroblast cells in both the resting state and during mitochondrial stress, whereas ATF4 expression is increased in stressed CHOP-deficient cells. Thus, ATF4 may act upstream of CHOP, whereas ATF5 seems to be the downstream target of CHOP. 44 Notably, ISR activation must be controlled within a suitable range, and the TC concentration must not fall to zero. If the mRNAs of targeted factors of the UPRmt are not translated, then ISR and UPRmt activation would serve no purpose within the cell. 47
Although mitochondrial dysfunction is a prominent feature of ischemic stroke, it is not clear whether UPRmt has functional implications. Previous studies have shown that UPRmt is closely related to aging, and the modest UPRmt activation has been implicated in lifespan extension.35,53 UPRmt disorders have also been found to be involved in various neurodegenerative diseases, including Alzheimer’s disease, 54 Parkinson’s disease,55,56 and Huntington’s disease, 57 leading to speculations that UPRmt may act as an endogenous compensatory mechanism for mitochondrial perturbations. A recent study proposed that pharmacologic UPRmt activation protects the heart against I/R injury in an ATF5-dependent manner. 58 In this study, oligomycin and doxycycline, used as UPRmt inducers, were administered 6 h prior to ex-vivo heart I/R injury, significantly upregulating many known UPRmt-linked genes and decreasing infarct sizes in wild-type mice, but not in ATF5-/- mice. However, whether basal UPRmt per se is activated after heart I/R remains unclear, and intrinsic UPRmt activation after I/R may not be sufficient for developing resilience to damage. Nevertheless, the organs may be protected from I/R injury by enhancing endogenous UPRmt via exogenous administration of UPRmt activators.
Mitochondrial dynamics: fission and fusion
Mitochondria are highly dynamic organelles that continuously alter their morphology via two opposing processes, namely fission and fusion (Figure 3). 59 Mitochondrial fission facilitates the creation of healthy daughter mitochondria or the elimination of damaged mitochondrial contents. 60 Mitochondrial fission is initiated when ER-localized inverted formin 2 (INF2) mediates interactions between the ER and actin to generate constrictions that are suitable for dynamin-related protein 1 (Drp1) assembly. 61 Subsequently, Drp1 is recruited from the cytosol to the OMM 62 and actively forms a circular oligomer, which further constricts the mitochondria. 63 Although Drp1 has been identified as the primary regulator of mitochondrial fission, Drp1 and its receptors do not independently segregate mitochondrial membranes. Recently, Lee et al. 64 revealed that dynamin-2 (Dyn2) works in concert with Drp1 to organize continuous constriction events leading to mitochondrial fission.
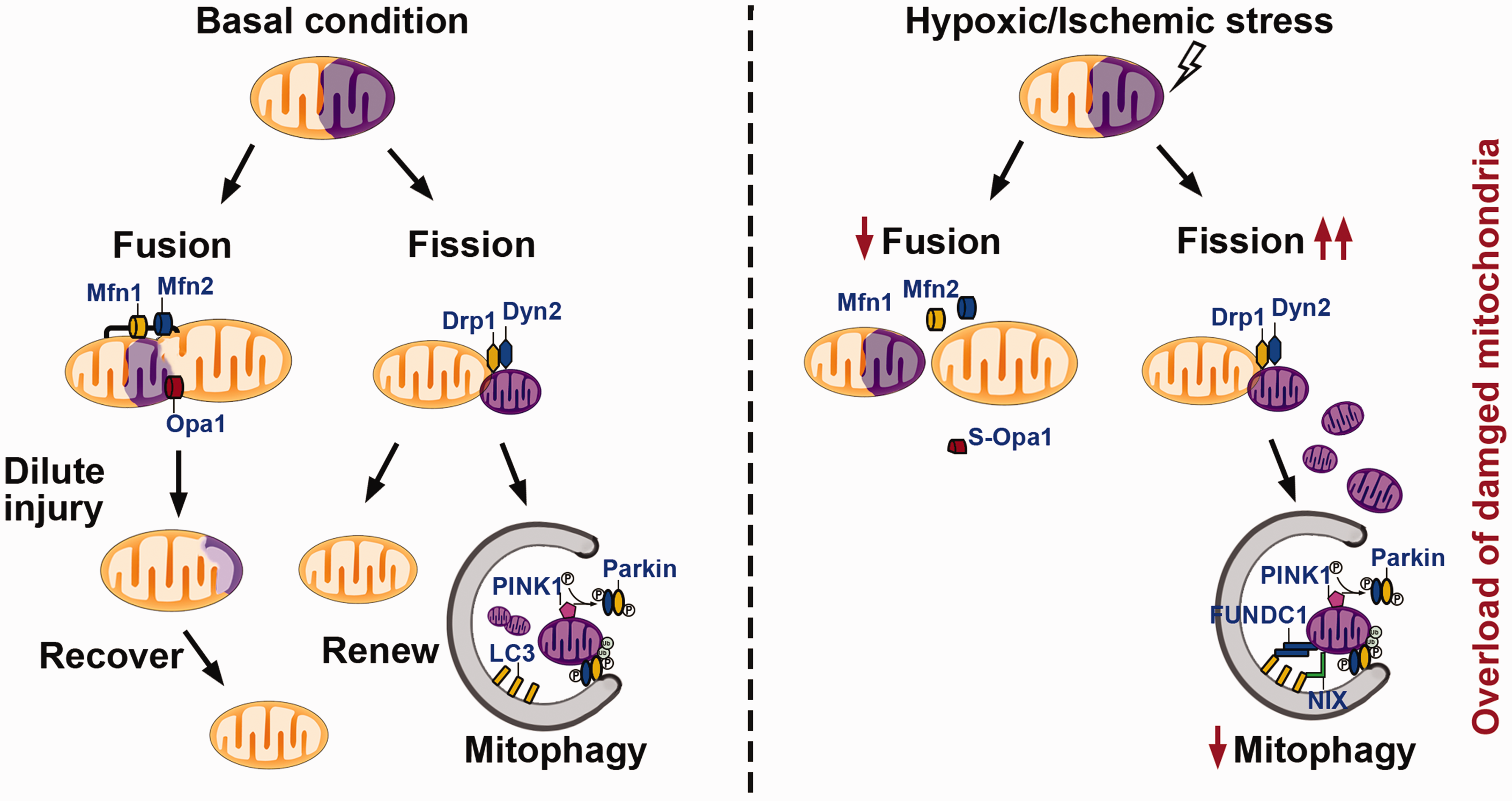
Mechanisms of mitochondrial dynamics and mitophagy under basal conditions and hypoxic/ischemic stress. Typically, fission and fusion machineries balance injured or aged mitochondria by dislodging damaged contents via dynamin-related protein (Drp)1- and dynamin (Dyn)2-mediated fission or merging with other healthy mitochondria via mitofusin (Mfn)1-, Mfn2-, or optic atrophy (Opa)1-mediated fusion. Mitochondria that fail to recover homeostasis will undergo mitophagy in a Parkin-dependent or Parkin-independent (FUNDC1 or NIX/Bnip3L) manner. However, the balance in mitochondrial dynamics shifts to fission under conditions of hypoxic/ischemic stress. Combined with an attenuated mitophagy capacity, these mechanisms collectively lead to the excessive accumulation of damaged mitochondria and irreversible cellular injury.
Mitochondrial fusion involves the merging of mitochondrial membranes and the re-mixing of mitochondrial contents, mediated by several GTPase superfamily members. Mitofusins 1 and 2 (Mfn1 and Mfn2) promote the fusion of OMMs, which is considered the first step of mitochondrial fusion, 65 whereas optic atrophy 1 (Opa1) drives IMM fusion. 66 Classical mitochondrial fusion describes a process in which two mitochondria merge into an elongated tubular mitochondrion, known as complete fusion. However, when Opa1 levels are too low or too high, complete fusion cannot be initiated. Instead, transient fusion occurs via a ‘kiss and run’ encounter, by which mitochondria only exchange partial contents without structural rearrangement. 67 Transient fusion does not seem to be as efficacious as complete fusion, but it is a prominent quick fusion process that occurs within minutes or even seconds. Transient fusion provides an essential recharging mechanism that allows rapid equilibration of the soluble IMS and matrix proteins, as well as smaller molecules (including mRNAs), between organelles. 67 Mitochondrial fusion helps alleviate stress by mixing the contents of partially injured mitochondria with functional components from other mitochondria in the form of complementation, thus enabling the recovery of damaged mitochondria and the maintenance of metabolic homeostasis.60,68
A delicate dynamic balance between fission and fusion is essential for maintaining optimal mitochondrial function and fulfilling specific metabolic and energetic cellular demands under a wide range of conditions. 69 Cerebral I/R induces mitochondrial dynamic imbalance toward fission that favors mitochondrial fragmentation and causes mitochondria to become susceptible to pro-apoptotic proteins. It has been demonstrated that mitochondria disintegrate into multiple smaller units before apoptosis.70–72 Excessively fragmented mitochondria fail to produce sufficient mitochondrial respiratory complexes, resulting in decreased OXPHOS, increased ROS generation, mitochondrial membrane depolarization, and the release of pro-apoptotic factors like cytochrome C.73,74 Recently, many investigators have proposed that inhibiting mitochondrial fission can protect the brain from I/R injury;75,76 hence, Drp1 has received substantial attention as a potential therapeutic target.77,78 The suppression of Drp1 by mitochondrial division inhibitor-1 (Mdivi-1) is protective against cerebral ischemic injury.75,76,79,80 It is noteworthy that mitochondrial fission is not always detrimental; mild and transient fission following brain ischemia may be protective, and fission at a basal level is consistently needed for maintaining mitochondrial homeostasis.
In contrast, mitochondrial fusion is attenuated during I/R. 81 Mitochondrial fusion promotes the generation of networked and elongated mitochondria, which is associated with increased ATP production, increased mtDNA stability, decreased mitochondrial permeability transition pore sensitivity, and reduced cell death.82–84 Interventions that increase mitochondrial fusion have been implicated in alleviating I/R injury. Wei et al. found that melatonin reduces the infarcted area and suppresses neuronal death during reperfusion by enhancing Opa1-related mitochondrial fusion. 85 Zhang et al. showed that exercise training promotes Opa1 expression and ameliorates brain edema in a rat model of middle cerebral artery occlusion (MCAO). 86 In addition to Opa1, Mfn2 is also a potential target for neuroprotection against ischemic brain damage. 87 Mfn2 was shown to translocate to the cytoplasm immediately after transient global brain ischemia, followed by decreased expression during reperfusion. 88 Mfn2 overexpression was found to attenuate mitochondrial dysfunction and revert hypoxia-induced changes in mitochondrial morphology. 87 In addition, some stroke risk factors, such as hyperglycemia, can exacerbate the mitochondrial dynamic imbalance induced by I/R. 89
Mitochondrial turnover: mitophagy and mitochondrial biogenesis
The cell’s metabolic demands are met through two antithetical processes, mitophagy and mitochondrial biogenesis, which control the quality and quantity of mitochondria. Mitophagy (Figure 3) is a selective process that effectively removes damaged or aged mitochondria. In mammalian cells, 85% of mitochondrial fission events lead to the formation of one hyperpolarized and one depolarized daughter mitochondrion. 90 The hyperpolarized daughter re-enters the dynamic mitochondrial network, whereas the depolarized daughter undergoes a ‘grace period’ to recover its ΔΨm. Depolarized mitochondria that fail to regain ΔΨm are recognized by PTEN-induced kinase 1 (PINK1), 91 which is constitutively translocated into polarized mitochondria and rapidly processed by proteolysis. However, upon loss of the ΔΨm, PINK1 cleavage is blocked, 92 leading to its selective accumulation on the OMM, from where it recruits and subsequently activates the E3 ubiquitin ligase Parkin via Ser65 phosphorylation in the ubiquitin-like domain.93,94 PINK1 can also phosphorylate free ubiquitin and poly-ubiquitin chains on resident OMM proteins.95–97 Phosphorylated ubiquitin binds to Parkin, which causes Parkin to release its autoinhibitory domain 98 and converts it to the active form. Activated Parkin then immediately ubiquitinates multiple OMM proteins.99–101 Although the specific roles of these ubiquitinated OMM proteins remain unclear, their poly-phosphorylated ubiquitin chains function as receptors for Parkin. This activity generates a positive feed-forward mechanism, amplifying OMM protein ubiquitination and ubiquitin chain synthesis by recruiting Parkin to damaged mitochondria. 102 Parkin is encoded by the PARK2 gene in humans. 103 PARK2 typically forms ring-like structures surrounding the fragmented mitochondria in the soma and proximal dendritic regions in neurons, where mature lysosomes are predominantly located. This specific spatial distribution enables neurons to efficiently eliminate dysfunctional mitochondria via the autophagy-lysosomal pathway. 104
In addition to the PINK1-Parkin pathway, mitophagy can also be regulated by different mitophagy receptors, including Atg32 in yeast 105 and NIX (BCL2/adenovirus E1B 19-kDa interacting protein 3-like, also known as Bnip3L) 106 and FUNDC1 (FUN14 domain-containing protein 1) 107 in mammalian cells. Both NIX/Bnip3L and FUNDC1 have a typical microtubule-associated protein light chain 3 (LC3)-interacting region motif in their cytosolic domain107,108 through which they recruit autophagosomes to mitochondria via direct binding to LC3 and promote the subsequent clearance of damaged mitochondria.
Accumulating evidence has implicated mitophagy in the pathogenesis of brain I/R injury. However, whether mitophagy is beneficial or detrimental remains a matter of debate. As one of the most important defense mechanisms of the MtQC system, mitophagy is strongly activated at the onset of ischemia, 109 but is attenuated during the reperfusion stage. 110 Thus, it has been proposed that enhanced mitophagy can induce neuroprotective effects in transient ischemia elicited in reperfusion models. Di et al. reported that methylene blue improves neurological function and reduces infarct volumes after acute cerebral ischemia by strengthening mitophagy. 111 Tang et al. revealed that upregulated Parkin expression can protect mouse neuroblastoma Neuro2a (N2a) cells against insults caused by oxygen and glucose deprivation (OGD) and reoxygenation. 112 Chen et al. demonstrated that PINK1 protects transient global ischemia-induced neuronal injuries by promoting Drp1 degradation via phosphorylation. 113 Shen et al. revealed that acidic post-conditioning attenuates ischemic brain injury and extends the reperfusion window by reinforcing I/R-induced mitophagy via a PARK2-dependent pathway. 114 More recently, Cai et al. revealed that t-PA exerts neuroprotective effects by enhancing FUNDC1-mediated mitophagy in ischemic neurons. 115 In their study, the authors found that t-PA upregulated FUNDC1 expression by promoting AMP-activated protein kinase (AMPK) phosphorylation, thereby protecting mitochondria and reducing ischemic injury. Moreover, Bnip3L overexpression can also rescue mitophagy deficiency during cerebral ischemia and attenuate ischemic brain injury, 116 indicating that selective clearance of defective mitochondrial machineries is required for limiting mitochondrial damage. Conversely, the neuroprotective effects of mitophagy may switch to pathological cell death mechanisms under sustained stress caused by long-term brain ischemia. Wen et al. 117 found that excessive activation of autophagy during permanent MCAO contributed to aggravated ischemic injury by inducing autophagic cell death, which could be improved by administering an autophagy inhibitor. Accordingly, Qin et al. 118 demonstrated that autophagy was strongly activated in astrocytes in permanent MCAO in vivo and OGD exposure in vitro, accompanied by reduced astrocyte survival. Pharmacological inhibition of autophagy using 3-methyladenine attenuated OGD-induced astrocyte death and promoted astrocyte survival in the ischemic area following permanent MCAO. These findings suggest that an appropriate level of mitophagy necessitates cellular homeostasis in order to avoid the removal of too many mitochondria.
Once degraded, the cellular mitochondrial pool needs to restore its content through biogenesis to keep pace with its functional demands. As a continuous process, the mitochondrial network is dynamically reshaped by the growth and division of preexisting mitochondria due to the lack of de novo mitochondrial generation. The primary regulator of mitochondrial biogenesis is peroxisome proliferator-activated receptor coactivator 1 (PGC-1α), which initiates mitochondrial biogenesis mainly by stimulating the expression of nuclear respiratory factor 1 (NRF1) and NRF2, 119 which regulate the expression of both nuclear genome and mtDNA-encoded mitochondrial proteins.120–122 Although PGC-1α activation is potentially beneficial to cell stress because enhanced PGC-1α expression can protect against hypoxic/ischemic brain injury, 123 the detailed mechanism whereby mitochondrial biogenesis dysfunction may contribute to ischemic pathology has not been comprehensively studied.
As with most balancing acts, cellular health can be essentially retained at an appropriate level of mitophagy, which can cooperate with the mitochondrial biogenesis machinery to clear dysfunctional mitochondria and maintain the optimal mitochondrial pool. Insufficient or excessive mitophagy will otherwise cause a deficiency in ATP production and failure to meet the energy demands of the cell. Therefore, a proper balance between mitophagy and mitochondrial biogenesis is critical for mitochondrial homeostasis.
Intercellular mitochondrial transfer and exogenous mitochondrial transplantation
In addition to activating internal pro-survival signaling cascades, substantial intercellular communication occurs between different cell types in the neurovascular unit during I/R.124–126 It has been speculated that injured neurons can send “help me” signals to adjacent glial and vascular cells, promoting them to shift into potentially beneficial phenotypes. In turn, non-neuronal cells could transfer protective signals to at-risk neurons and help them remodel the neurovascular unit.125,127 Intriguingly, extracellular mitochondria could serve as a novel type of “help me” signal, enabling metabolic crosstalk among neurons, astrocytes, microglia, and endothelial cells in the neurovascular unit under both healthy and diseased conditions.14,127,128 Hayakawa et al. have shown that astrocytes can release functional mitochondria to injured neurons after stroke mediated by a calcium-dependent mechanism involving CD38 and cyclic ADP ribose (cADPR) signaling. 14 Enhancing astrocytic CD38 signals via clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein 9 constructs or cADPR supplements promoted astrocyte-to-neuron mitochondrial transfer and improved functional recovery after experimental stroke, whereas suppressing CD38 signaling by small interfering RNAs abolished this kind of protective effect. 14 Evaluating these extracellular mitochondria by measuring ATP levels, JC-1 fluorescence ratios, and oxygen consumption revealed that they retained partial respiratory function. 14 Furthermore, the number and quality of extracellular mitochondria may serve as a potential biomarker of disease severity. Chou et al. observed that a higher ΔΨm in cerebrospinal fluid mitochondria correlated with good clinical recovery of patients with subarachnoid hemorrhage at 3 months after disease onset. 129 Additionally, preclinical data have suggested a positive correlation between the function of extracellular mitochondria and neurological outcomes in patients with ischemic stroke. 14
In neurons, mitochondria are enriched within the presynaptic nerve terminals. Due to the time-consuming nature of long-distance transportation of mitochondria from the cell body to the periphery, dysfunctional mitochondria in damaged neurons cannot be replenished quickly after ischemia. Therefore, extracellular mitochondrial transfer from adjacent astrocytes to ischemic neurons may open a new avenue for neuroprotective strategies. One limitation of this endogenous astrocyte-to-neuron transfer of mitochondria is that the effects are transient and may not afford robust and consistent therapeutic benefits against the progressive injuries associated with stroke. 130 Exogenous mitochondrial transplantation, which could enhance ATP generation in recipient cells, has been proposed to be a supplement of endogenous transfer of mitochondria. Previous findings have demonstrated that exogenous mitochondrial transplantation can substantially reduce the infarct volume in a rat model of focal cerebral ischemia and may further augment the neuroprotection of endogenous mitochondrial transfer.131,132 Skeletal muscle-derived mitochondria possess the highest ΔΨm compared to other tissue-derived mitochondria, thus serving as an ideal candidate for mitochondrial isolation. However, the invasive nature of this method and the necessity for freshly isolated mitochondria (ensuring their functional integrity) make this approach inconvenient and probably difficult to implement in clinical practice. It has recently been demonstrated that cryopreserved placenta can be effectively used for mitochondrial isolation and transplantation in ischemic stroke, providing a considerable transformative strategy for the clinical practice of stroke treatment. 132
In addition to the direct transplantation of exogenous mitochondria, stem cells also serve as an effective source of functional mitochondria for ischemic cells, both for neurons and endothelial cells.133–135 Mesenchymal stem cells can transfer mitochondria to neurons and improve neuronal survival after oxidant injury. 133 Moreover, endothelial progenitor cells (EPCs) can donate active extracellular mitochondria with high ΔΨm to ischemic brain endothelial cells. 135 Using an in vitro stroke model, Hayakawa et al. observed that extracellular mitochondria with higher ATP levels exist in EPC-conditioned medium. Moreover, EPC-conditioned medium contains more extracellular mitochondria than medium exposed to other cell types comprising the neurovascular unit, such as astrocytes, endothelial cells, and pericytes. The authors also demonstrated that EPC-derived extracellular mitochondria were transported into brain endothelial cells, helping to maintain brain-endothelial cell energetics, the barrier integrity, and angiogenic functions after OGD. 135 The functions of donor mitochondria determines the effects of stem cell therapies because interference with stem cell-mitochondrial functions reduces their capacity to restore ATP levels in recipient cells and compromises the protective effects of stem cell therapy. 136 Kikuchi-Taura et al. recently found that bone marrow mononuclear cells can promote angiogenesis after cerebral ischemia by transferring vascular endothelial growth factor into endothelial cells. 137 The benefit of this direct cell-cell interaction also depends on mitochondrial transfer. 138 Thus, stem cell-based mitochondrial transfer may be more therapeutically neuroprotective than simple exogenous mitochondrial transplantation, as stem cells serve additional protective effects beyond the transfer of mitochondria per se.
Future perspectives and conclusion
In summary, MtQC is a unique process that monitors the homeostasis of mitochondrial networks. In the past decade, significant progress has been made in identifying the mechanistic details of individual pathways associated with MtQC under both physiological and pathophysiological conditions. Consecutively or simultaneously activated MtQC components cooperate to promote mitochondrial network recovery in a remarkably dynamic manner. Although the complex interplay between elements in the MtQC process still needs to be further investigated, recent findings have increased interest in the possible translational applicability of the MtQC mechanisms for treating stroke. Discovering the neuroprotective effects of various pharmacological and nonpharmacological agents that modulate MtQC has expanded these appealing opportunities.139,140 While chemically induced MtQC still faces challenges such as overt toxicity, 141 noninvasive remote ischemic conditioning (RIC), as a simple and safe therapeutic option, displays efficacy in alleviating hypoxic/ischemic injury and serves as an attractive treatment strategy.142,143 More importantly, RIC also promotes mitophagy and regulates mitochondrial dynamics; thus, it holds promise as a therapeutic strategy for targeting mitochondria-related pathologies.144,145
However, several questions still need to be addressed. First, it is imperative to better understand the time-dependent changes (especially the MtQC regulation) that occur during the I/R stages, which is of central importance in determining the optimal treatment timing. For example, as a pretreatment, Drp1 inhibition can protect cultured cardiomyocytes exposed to OGD, whereas cell damage is paradoxically aggravated when the inhibitor is administered during reoxygenation. 146 Second, although appropriate upregulation of mitophagy is beneficial for eliminating dysfunctional mitochondria during cerebral I/R, excessive mitophagy has detrimental effects. This potential therapeutic strategy will only be effective if a sufficient threshold for mitochondrial removal can be determined. In addition, if receptors associated with selective mitophagy and cargo recognition that do not affect healthy mitochondria could be identified and targeted, the resulting mitochondrial network would provide both sustained elimination of dysfunctional mitochondria and sufficient bioenergy in treated cells. Third, the MtQC is not a neuronal-specific event, studies have also found MtQC in other cell types in the neurovascular unit, such as glial cells and endothelial cells.147,148 As components of the blood-brain barrier, astrocytes and vascular endothelial cells are coupled with neurons to utilize energy substrates. Compartmentalized metabolic cooperation is also particularly affected by resident microglia, whose inflammatory responses during ischemic stroke are governed by MtQC and cellular metabolism. 149 Furthermore, in addition to intracellular MtQC mechanisms, transcellular mechanisms also serve important functions. For example, axonal mitochondria in retinal ganglion cells can undergo lysosomal degradation within adjacent astrocytes through a process termed transcellular mitophagy. 150 However, it remains unclear how these MtQC mechanisms direct crosstalk, what the primary mechanism of each pathological process is, and how cell-type-specific MtQC is regulated. Finally, it would be useful to examine MtQC in animal models with comorbidities associated with mitochondrial dysfunction, such as obesity, diabetes mellitus, and senescence. Success in these avenues of investigation will bring basic research closer to real-life clinical situations.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Beijing Natural Science Foundation (7192103); National Natural Science Foundation of China (81971198); Chinese Ministry of science and Technology (2019YFA0508603), China; and National Natural Science Foundation of China (82027802).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.