
Abstract
Glucagon-like peptide-1 (GLP-1) is an incretin hormone known to stimulate glucose-dependent insulin secretion. The GLP-1 receptor agonist, exendin-4, has similar properties to GLP-1 and is currently in clinical use for type 2 diabetes mellitus. As GLP-1 and exendin-4 confer cardioprotection after myocardial infarction, this study was designed to assess the neuroprotective effects of exendin-4 against cerebral ischemia–reperfusion injury. Mice received a transvenous injection of exendin-4, after a 60-minute focal cerebral ischemia. Exendin-4-treated vehicle and sham groups were evaluated for infarct volume, neurologic deficit score, various physiologic parameters, and immunohistochemical analyses at several time points after ischemia. Exendin-4 treatment significantly reduced infarct volume and improved functional deficit. It also significantly suppressed oxidative stress, inflammatory response, and cell death after reperfusion. Furthermore, intracellular cyclic AMP (cAMP) levels were slightly higher in the exendin-4 group than in the vehicle group. No serial changes were noted in insulin and glucose levels in both groups. This study suggested that exendin-4 provides neuroprotection against ischemic injury and that this action is probably mediated through increased intracellular cAMP levels. Exendin-4 is potentially useful in the treatment of acute ischemic stroke.
Keywords
Introduction
Diabetes mellitus is a major independent risk factor for stroke, and hyperglycemia is associated with poor outcome because of reinforced oxidative stress and inflammation (Kamada et al, 2007). We have recently highlighted the benefits of glucagon-like peptide-1 receptor (GLP-1R) agonists, a new clinically available class of therapeutic agents for diabetes mellitus, in the control of blood glucose levels in the acute phase of ischemic stroke (Bruno et al, 2002).
Oxidative stress and inflammation are responsible for neuronal damage in acute cerebral ischemia. In ischemia–reperfusion injury, reactive oxygen and nitrogen species induce protein oxidation, DNA damage, and lipid peroxidation (Warner et al, 2004). Oxidative damage induced by reactive oxygen radicals causes complex interactions with inflammation and apoptosis-like cell death, and the results expand brain damage (Andrabi et al, 2004). Hence, control of these phenomena is important to achieve neuroprotection.
Glucagon-like peptide-1 is a gut hormone secreted from L cells of the small intestine in response to food ingestion, and facilitates glucose-dependent insulin secretion (Baggio and Drucker, 2007). Exendin-4 is a GLP-1R agonist that shares 53% amino-acid sequence identity to GLP-1. It exhibits biologic actions similar to GLP-1 and has a longer half-life than does GLP-1 by resisting degradation by dipeptidyl peptidase-4 (Perry and Greig, 2003). Furthermore, exendin-4 can also induce pancreatic β-cell proliferation and inhibition of β-cell apoptosis, similar to GLP-1 (Baggio and Drucker, 2007). Consequently, exendin-4 was recently approved for the treatment of type 2 diabetes mellitus. Apart from their effects in diabetes, several studies have shown that GLP-1 and exendin-4 are neuroprotective in brain damage caused by various insults (Perry et al, 2002a, b , 2003; Perry and Greig, 2003) and can induce neurogenesis after a 6-hydroxydopamine insult (Bertilsson et al, 2008; Harkavyi et al, 2008). However, to our knowledge, no report has analyzed the action of exendin-4 in cerebral ischemia, although exendin-4 confers cardioprotection and myocardial functional recovery under myocardial ischemic injury (Bose et al, 2005; Timmers et al, 2009). In this study, we investigated whether exendin-4 can prevent brain damage after ischemia–reperfusion injury.
Materials and methods
Experimental Protocol
Animal procedures were conducted after obtaining the approval of the Animal Care Committee of the Juntendo University. Adult 8-week-old male C57BL/6 mice weighing 20 to 25g were used in this study, and were housed under controlled lighting and provided with food and water ad libitum. Mice were divided at random into three groups. (1) The exendin-4 group (n = 49): these mice were treated with tail vein injection of exendin-4 (Sigma-Aldrich, St Louis, MO, USA) after left middle cerebral artery occlusion (MCAO), which was performed using an intraluminal thread for 60 minutes as described previously (Hara et al, 1996). The selected dose and schedule of exendin-4 treatment were based on preliminary experiments that used exendin-4 at 0.1, 1, 10, or 50μg/100μL per mouse and that administered exendin-4 at 0 (immediately), 1, or 3 hours after cerebral reperfusion. (2) The vehicle group (n = 24) received intravenous infusion of 0.9% saline after MCAO at a rate and volume similar to those applied in the exendin-4 group. (3) The sham-operated group (n = 8) underwent the procedure except for MCAO. During this procedure, body temperature was kept at 37.0°C±0. 5°C using a heating pad. Systolic blood pressure was monitored using a noninvasive tail-cuff system (Softron BP-98A NIBP, Softron Inc., Tokyo, Japan) in conscious mice. Regional cerebral blood flow was measured by laser-Doppler flowmetry before, during, and after MCAO, as well as before killing. At 24, 72 hours, or 7 days after reperfusion, mice of each group were anesthetized by an intraperitoneal injection of 50mg/kg pentobarbital and then decapitated. To evaluate infarct area and volume, brain slices were stained with cresyl violet, scanned using Axio-Vision software (Carl Zeiss, Jena, Germany), and measured by the ImageJ program (NIH, http://rsb.info.nih.gov/nih-image/) (Tureyen et al, 2004).
Neurologic Evaluation
Neurologic function was evaluated by a modified scoring system described previously (Hara et al, 1996): 0, no observable neurologic deficit (normal); 1, failure to extend the right forepaw on lifting the whole body by the tail (mild); 2, circling to the right side (moderate); 3, loss of walking or righting reflex (severe).
Measurement of Insulin and Glucose Levels
Serum insulin level was determined using an insulin enzyme-linked immunosorbent assay (Ultra Sensitive Mouse Insulin ELISA Kit, Morinaga, Yokohama, Japan), and plasma glucose level was measured using a blood glucose meter (Johnson & Johnson, New Brunswick, NJ, USA). In this experiment, blood (200 μL) was collected from the ophthalmic venous plexus before MCAO and at 0 (immediately), 3, 6, 12, and 24 hours after reperfusion.
Immunohistochemistry
Immunohistochemistry was performed on 20-μm-thick free-floating brain sections. After pretreatment as reported previously (Miyamoto et al, 2009), sections were stained overnight using rabbit anti-GLP-1R (dilution 1:50, Medical Biological Laboratories Co., Nagoya, Japan), mouse anti-8-hydroxy deoxyguanosine (8-OHdG, dilution 1:100, Japan Institute for the Control of Aging, Shizuoka, Japan), mouse anti-4-hydroxy 2-hexenal (HHE, dilution 1:100, Japan Institute for the Control of Aging), rabbit antiionized calcium-binding adapter molecule-1 (Iba-1, dilution 1:500, Wako Pure Chemical Industries, Osaka, Japan), and rabbit antiphosphorylated cyclic AMP (cAMP) response element-binding protein (pCREB, dilution 1:100, Upstate Biotechnology, Lake Placid, NY, USA) antibodies. The antigen-retrieval method was used for staining GLP-1R as described earlier (Miyamoto et al, 2009). Sections were then incubated with biotinylated secondary antibodies (dilution 1:300, Vector Laboratories, Burlingame, CA, USA) and subsequently processed using avidin-biotinylated peroxidase (Vectastatin ABC kit, dilution 1:400, Vector Laboratories).
Double Immunofluorescence Histochemistry
Double immunofluorescence staining was performed by simultaneous incubation of the sections with anti-GLP-1R (dilution 1:50), mouse antineuronal nuclei (dilution 1:100, Chemicon International Inc., Temecula, CA, USA), rat anti-CD31 (dilution 1:100, BD Transduction Laboratories, San Jose, CA, USA), mouse antiinducible nitric oxide (iNOS, dilution 1:100, BD Transduction Laboratories), and anti-Iba-1 (dilution 1:500) antibodies. After incubation overnight with the primary antibody, sections were treated with fluoro-chrome-conjugated secondary antibody (Cy3 or fluorescein isothiocyanate, dilution 1:500, Jackson Immunoresearch Laboratories, West Grove, PA, USA), and then mounted with a Vectashield mounting medium (Vector Laboratories).
Terminal Deoxynucleotidyl Transferase (TdT)-Mediated dUTP-Biotin Nick-End Labeling Staining
For detection of in situ DNA fragmentation, staining with TUNEL (terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick-end labeling) was performed using an In Situ Cell Death Detection Kit (TMR Red, Roche, Mannheim, Germany), as described in detail previously (Miyamoto et al, 2008).
SDS-PAGE and Immunoblotting
In each animal, a brain sample was harvested from the ischemic region comprising the cortex and the striatum on the operated side at 24, 72 hours, and 7 days after reperfusion. Protein extraction and electrophoresis were performed as described previously (Miyamoto et al, 2008). Block Ace (Dainichi-Seiyaku, Gifu, Japan) or phosphate-buffered saline containing 0.05% Tween-20 (Sigma-Al-drich) was used for blocking. The transferred membranes were incubated overnight with anti-HHE (dilution 1:1,000), anti-iNOS (dilution 1:1,000), anti-pCREB (dilution 1:1,000), rabbit anti-CREB (dilution 1:1,000, Cell Signaling Technology, Beverly, MA, USA), and mouse anti-α-tubulin (dilution 1:10,000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) antibodies, followed by reaction with the horseradish peroxidase-conjugated secondary antibody (dilution 1:5,000, Amersham, Buckinghamshire, UK). Immunoreactive bands were visualized using enhanced chemiluminescence (ECL kit, Amersham). Equal protein loading was confirmed by measuring α-tubulin (53 kDa).
Cyclic AMP Assay
Each brain sample was taken from the ischemic region comprising the cortex and the striatum on the operated side at 24, 72 hours, and 7 days after MCAO. The samples were lysed in CelLytic reagent (Sigma Chemical Co., St Louis, MO, USA) with protease inhibitor (Calbiochem, La Jolla, CA, USA). The cAMP content in the homogenized brain extracts was measured using an immunoassay kit (R&D Systems Inc., Minneapolis, MN, USA) as described previously (Choi et al, 2002).
Cell Count and Statistical Analysis
In immunohistochemical analysis, positively stained cells in the ischemic boundary zone adjacent to the ischemic core (0.25mm2, Supplementary Figure 1) were counted by an investigator blinded to the experimental groups, using Axio-Vision software (Carl Zeiss). All values in this study are expressed as mean±s.e.m. One-way analysis of variance followed by post hoc Fisher's protected least significant difference test was used to determine the significance of differences in various indexes among the different groups. A P-value <0.05 denoted the presence of a statistically significant difference.
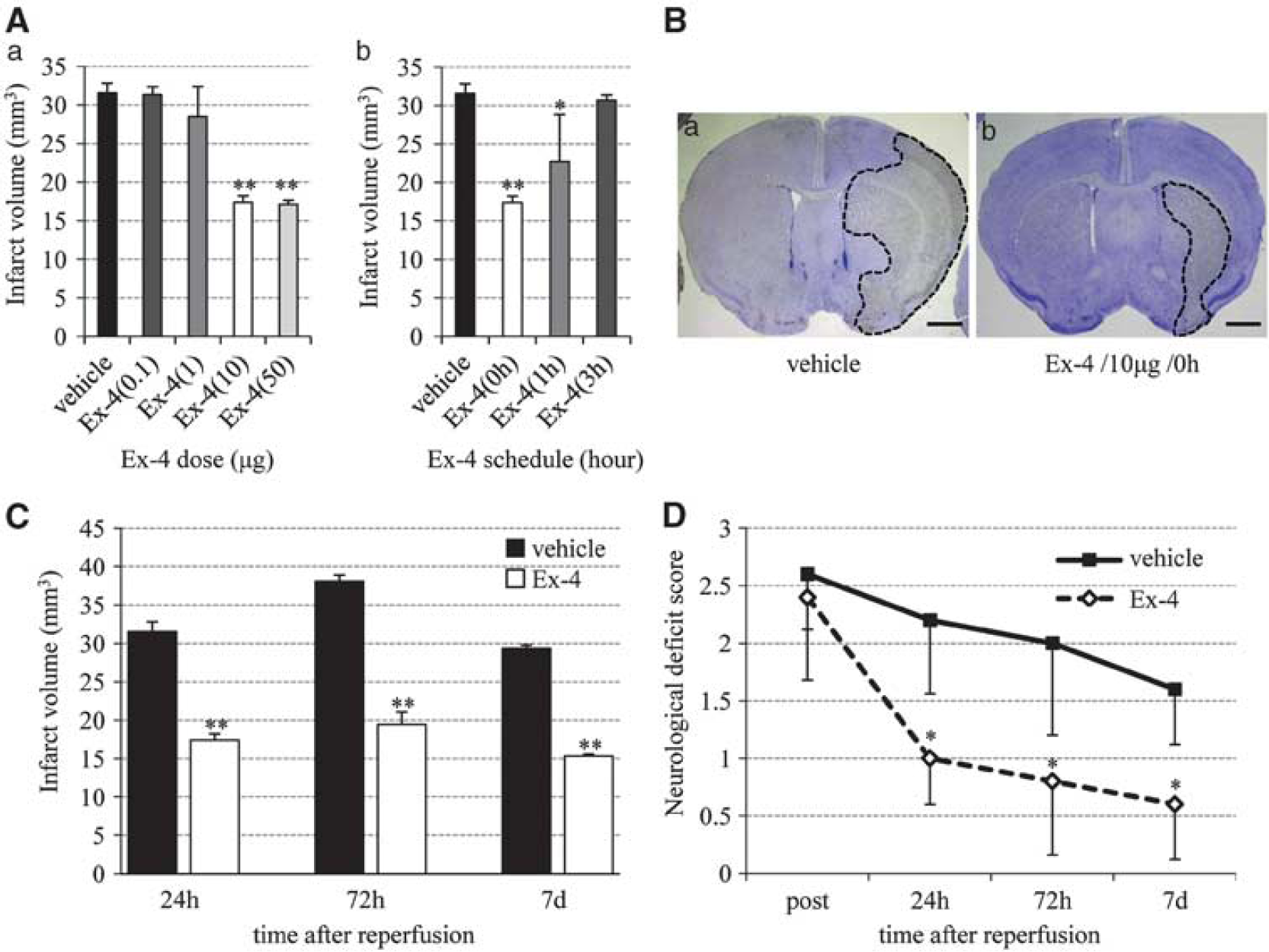
Neuroprotective effects of exendin-4 (Ex-4) against ischemia–reperfusion injury. (
Results
Exendin-4 Reduces Infarct Volume and Improves Neurologic Deficit
The protocol to be used for exendin-4 treatment was determined in a series of preliminary experiments involving the use of different doses and schedules of exendin-4. In these experiments, the infarct volume was clearly smaller in mice treated with exendin-4 at ⩾10 μg than in vehicle mice (Figure 1Aa), and injection of exendin-4 at 0hours after reperfusion produced the best effect with regard to infarct volume (Figure 1Ab). Hence, in the remaining experiments, we used 10 μg exendin-4 at 0 hours. Significant reductions in infarct volume were observed at 24, 72 hours, and 7 days after reperfusion in the exendin-4 group than in the vehicle group (Figures 1B and 1C). Furthermore, mice of the exendin-4 group showed better functional recovery than did those of the vehicle group (Figure 1D).
Physiologic Parameters
The serial changes in serum insulin and plasma glucose levels during the entire experiment until 24 hours after reperfusion were similar in the exendin-4 and vehicle groups (Figure 2A). Similarly, there were no differences in various physiologic parameters including regional cerebral blood flow (Figure 2B) between the two groups.
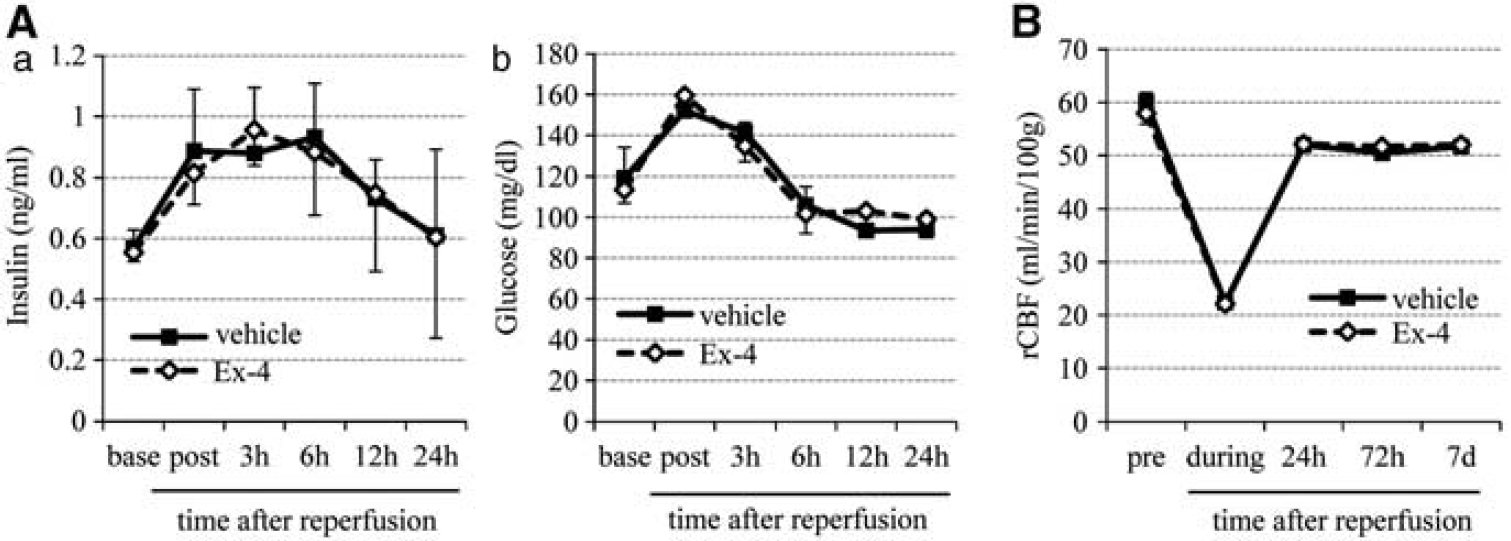
Physiologic parameters. (
Expression of Glucagon-Like Peptide-1 Receptor in the Mouse Brain
Glucagon-like peptide-1 has multiple roles in the central nervous system, and the expression of GLP-1R in the brains of rodents and humans has been established (Perry et al, 2003). First, we confirmed the expression of GLP-1R in the brain. Glucagon-like peptide-1R-immunopositive cells were detected in the brain (normal, untreated), as reported previously (Figure 3A). In addition, double immunostaining showed colocalization of GLP-1R with both neuronal nuclei (a neuronal marker) and CD31 (an endothelial cell marker) (Figure 3B) apart from glial fibrillary acidic protein (which is specifically expressed in astrocytes) and Iba-1 (which is specifically expressed in microglia and cells of monocytic lineage) (data not shown).
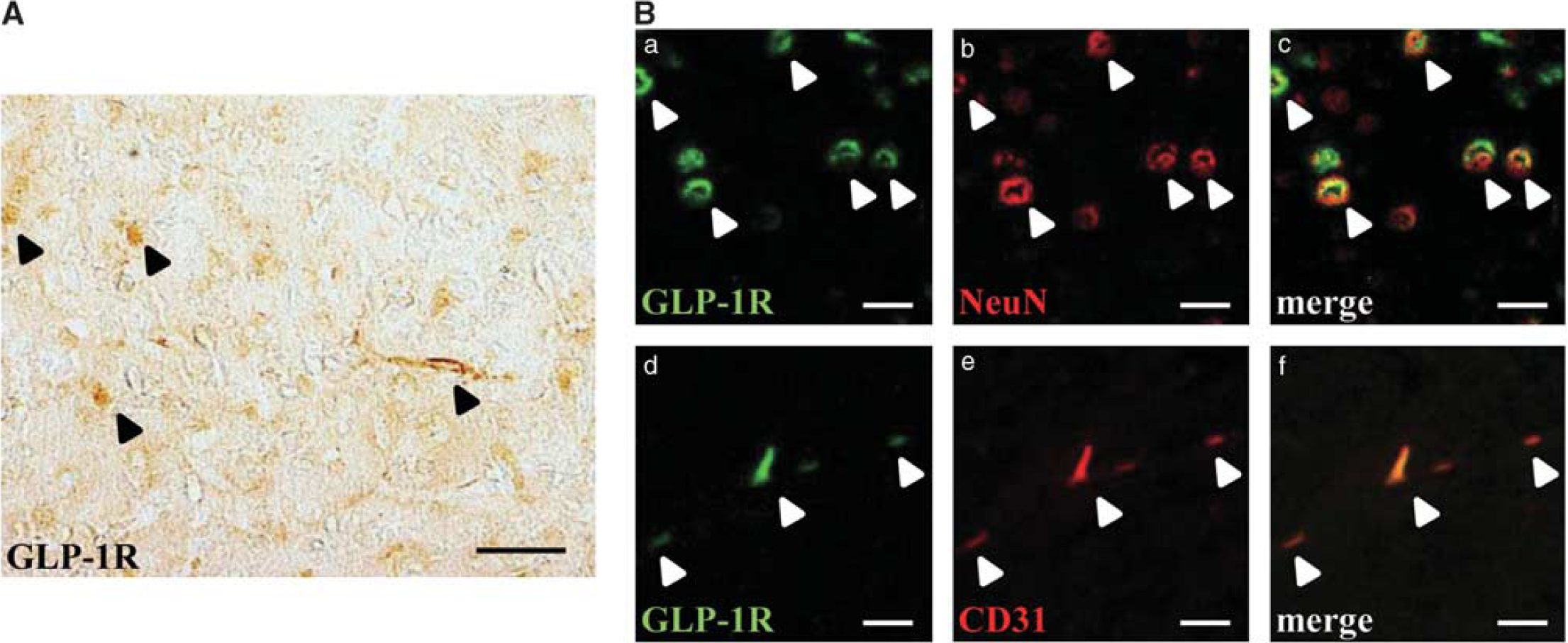
Expression of GLP-1R in the brain. (
Exendin-4 Suppresses Oxidative DNA Damage and Lipid Peroxidation
Next, we investigated whether exendin-4 can control oxidative stress in ischemia–reperfusion injury using 8-OHdG and HHE. 8-Hydroxy deoxyguanosine is a major form of oxidative DNA damage product, and HHE is one of the major lipid peroxidation products that are formed by n-3 polyunsaturated fatty acids in cells exposed to oxidative stress (Yamada et al, 2004). 8-Hydroxy deoxyguanosine- and HHE-positive cells increased until 72 hours after reperfusion, and then tended to decrease. The number of these oxidative stress marker-immunopositive cells was significantly decreased in the exendin-4 group than in vehicle group (Figure 4A). The immunoreactivity of HHE-positive band (75 kDa) at each time point was similar to the results of immunohistochemistry, and the intensity of the band in the exendin-4 group was weaker than that in the vehicle group (Figure 4B).
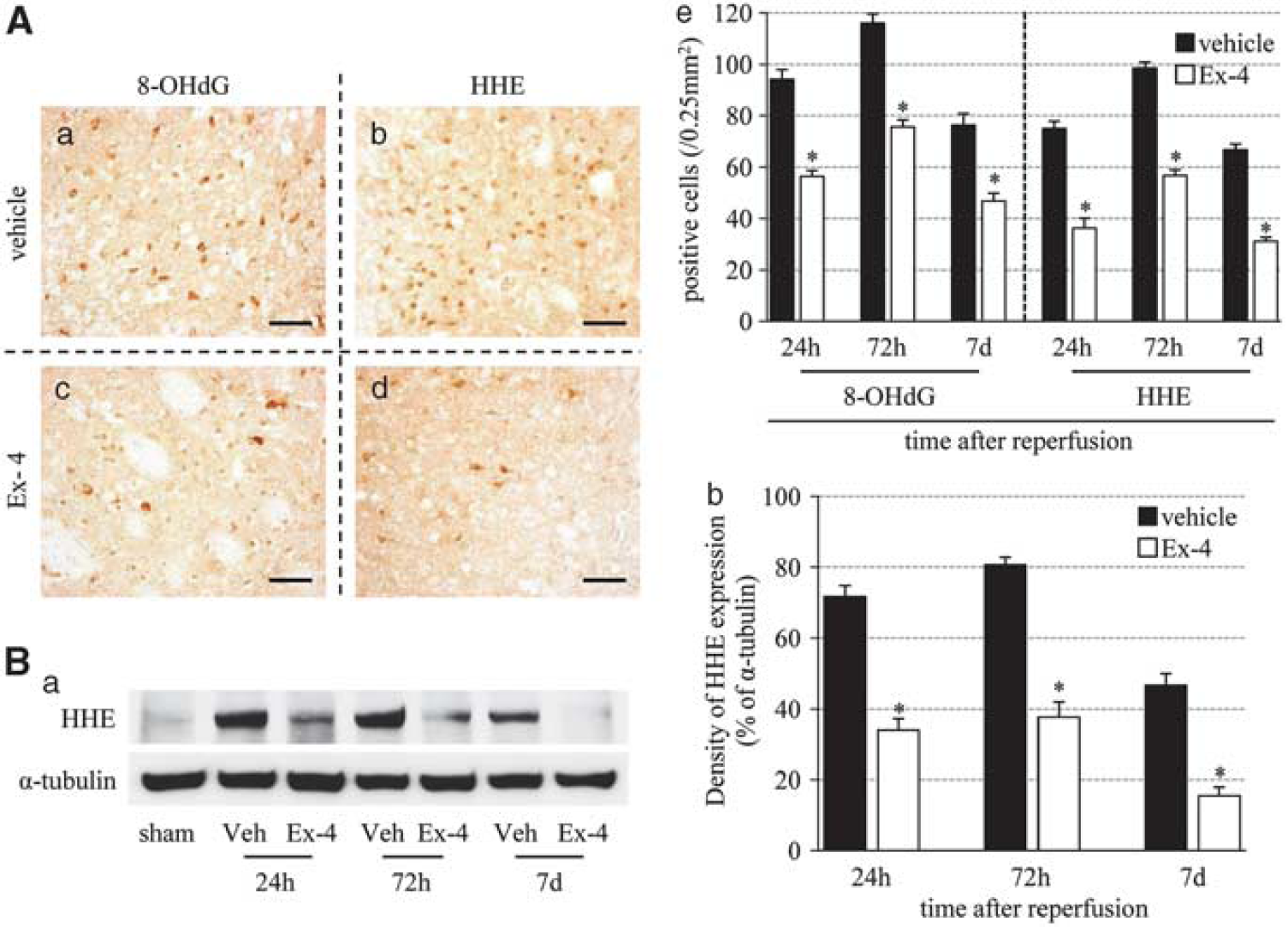
Effects of exendin-4 on oxidative stress. (
Exendin-4 Suppresses Microglial Activation and Inducible Nitric Oxide Expression
Next, we determined the relationship between exendin-4 and reactive oxygen species by examining microglial activation and iNOS formation. The number of ramified Iba-1-positive microglia reached a peak level at 72 hours after reperfusion, and then tended to decrease. The number of Iba-1-positive cells at each time point was significantly lower in the exendin-4 group than in the vehicle group (Figure 5A). The iNOS immunostaining was observed in the microglia from 24 hours after reperfusion, and detected abundantly in the microglia at 72 hours (Figure 5B). The intensity of the iNOS protein band (130 kDa) was stronger until 72 hours after reperfusion, and then tended to decrease. Band density was significantly lower in the exendin-4 group than in the vehicle group (Figure 5C).
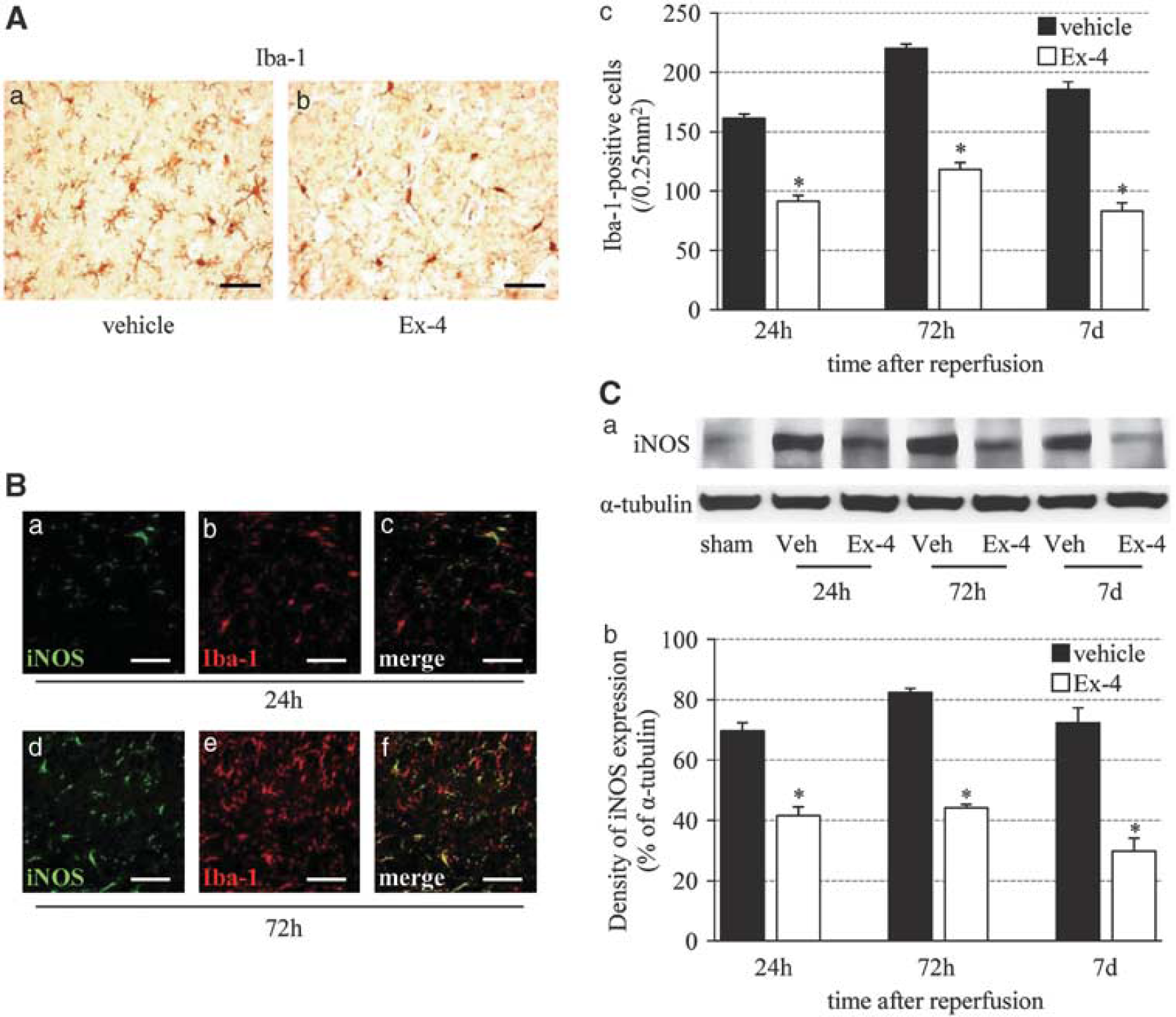
Effect of exendin-4 on the inflammatory response. (
Exendin-4 Prevents Cell Death
Next, we evaluated cell death using TUNEL staining. The number of TUNEL-positive cells increased until 72 hours after reperfusion, and then tended to decrease. Exendin-4 significantly reduced the number of such cells, compared with the vehicle group (Figure 6).
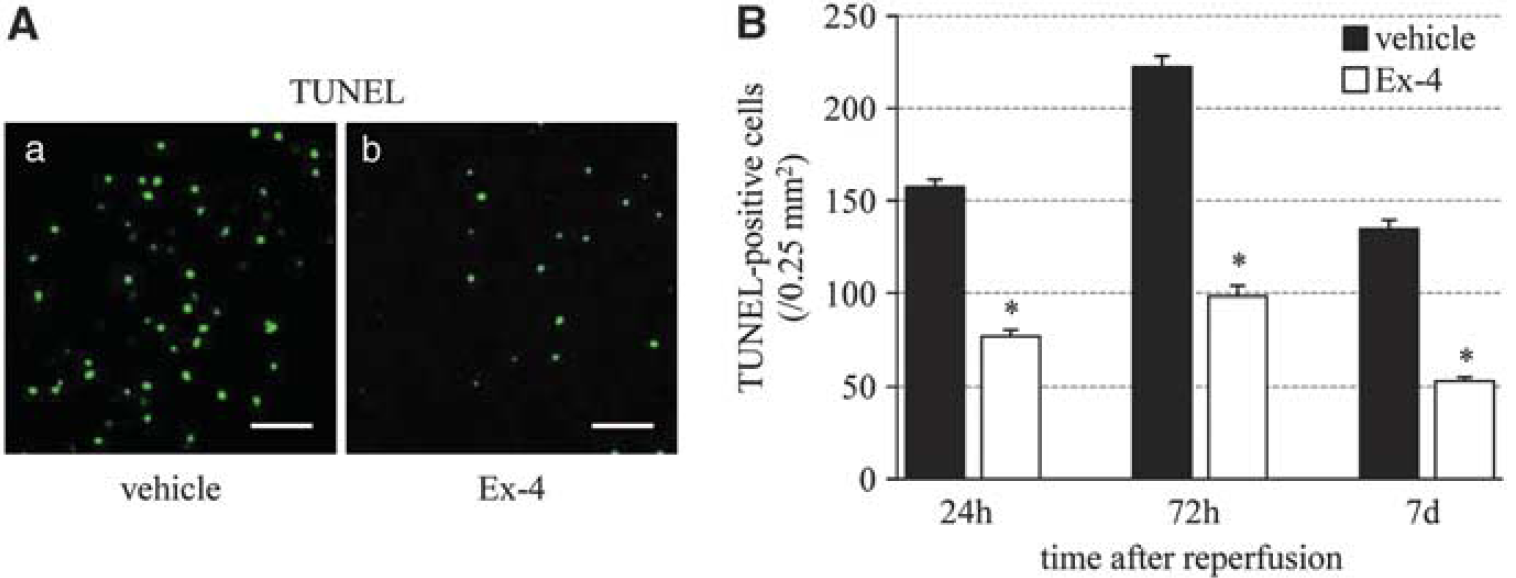
Effect of exendin-4 on cell death. (
Exendin-4 Stimulates Cyclic AMP Response
Finally, we examined the correlation between exendin-4 and cAMP response because activation of GLP-1R is known to increase intracellular levels of cAMP and modulate cell-survival mechanisms in various types of cells (Perry and Greig, 2003). In the exendin-4 group, cAMP levels were somewhat higher than those in the vehicle group at each time point, although the levels decreased in a time-dependent manner in both groups (Figure 7A). The pCREB-positive cells and band (43 kDa) were enhanced mostly at 24 hours after reperfusion, and then gradually decreased in a time-dependent manner.
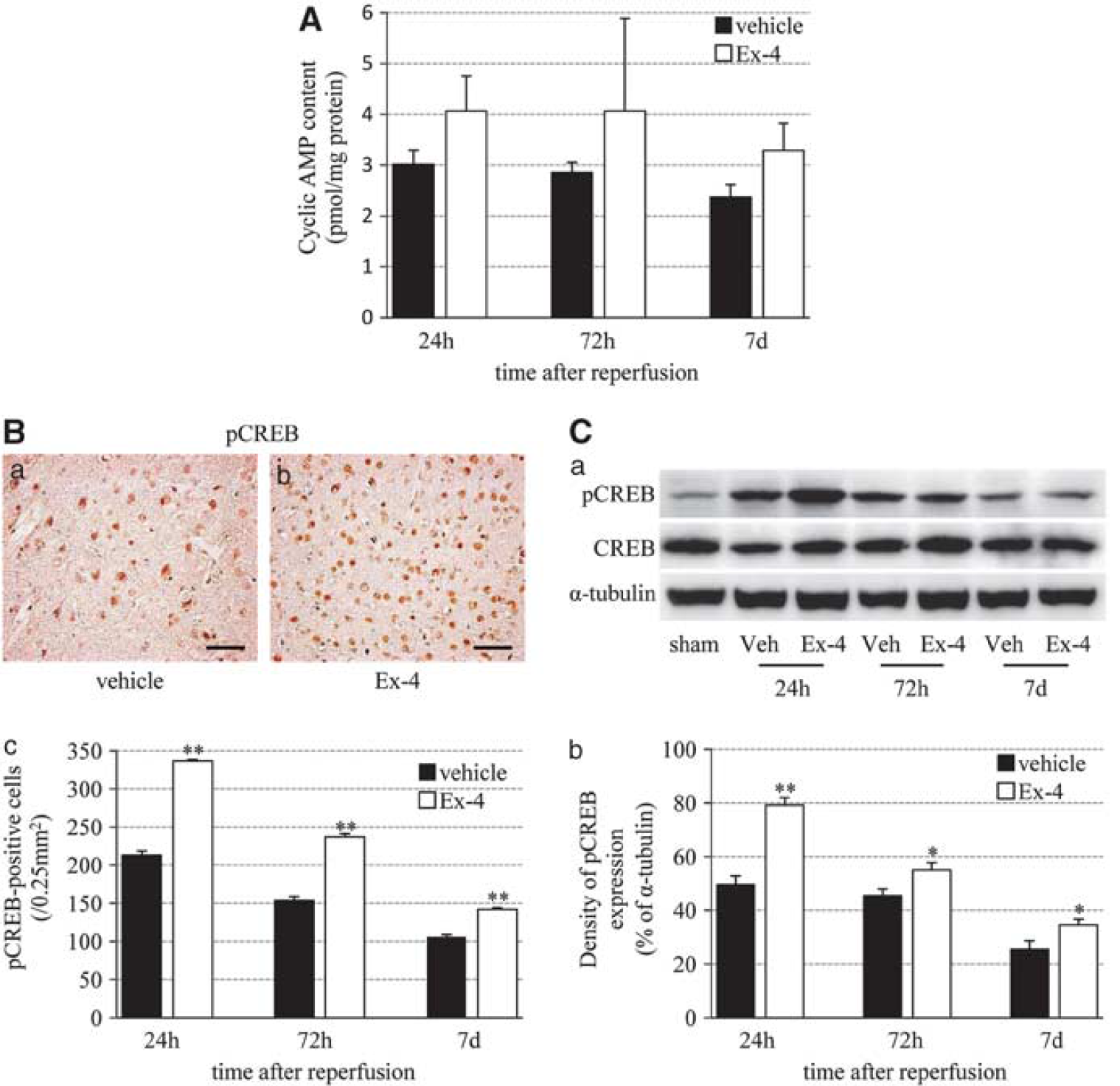
Induction of cAMP upregulation and CREB activation by exendin-4. (
The pCREB immunoreactivity in the exendin-4 group was stronger than that in the vehicle group (Figures 7B and 7C).
Discussion
The major finding of this study was that exendin-4 exhibited neuroprotective effects against ischemia–reperfusion injury. Intravenous injection of exendin-4 after MCAO significantly reduced infarct volume and resulted in recovery of the neurologic deficit. In addition, it provided long-term protection after ischemic injury.
Both GLP-1 and its longer-acting GLP-1R agonist, exendin-4, have multiple physiologic functions, such as the induction of glucose-dependent insulin release, inhibition of glucagon secretion, stimulation of β-cell replication, and antiapoptotic action (Baggio and Drucker, 2007). In the central nervous system, GLP-1 and exendin-4 have relatively smaller molecules, and both can diffuse readily across the blood–brain barrier and access the brain parenchyma directly (Hassan et al, 1999; Kastin et al, 2002; Kastin and Akerstrom, 2003). Peripheral administration of a much larger GLP-1–albumin recombinant fusion protein, which does not cross the blood–brain barrier, also activated neurons coupled to feeding in the central nervous system (Baggio et al, 2004). Glucagon-like peptide-1 receptor is distributed in various tissues including the brain in rodents and humans (Wei and Mojsov, 1995), and is widely expressed throughout the brain (Baggio and Drucker, 2007; Perry and Greig, 2003). The latter was confirmed in this study, which defined the presence of GLP-1R in neurons and endothelial cells of mice. Both central and peripheral administration of GLP-1R agonists decelerated food intake and weight gain in rodents (Meeran et al, 1999; Szayna et al, 2000; Turton et al, 1996). Similarly, in humans, administration of the GLP-1R agonist peripherally inhibited feeding behavior and promoted weight loss (Zander et al, 2002). Glucagon-like peptide-1 receptor stimulation enhanced cognitive functions and preserved hippocampal neurons from kainic acid neurotoxicity (During et al, 2003). Other studies showed that GLP-1 and exendin-4 have neurotrophic actions involving neurite outgrowth in PC12 cells (Bertilsson et al, 2008; Perry et al, 2002b) and provide neuroprotection against excitotoxicity caused by neurodegeneration (Harkavyi et al, 2008; Perry et al, 2002a, 2003). In addition to the above-mentioned effects, exendin-4 has been recently reported to prevent the generation of reactive oxygen species in INS-1 cells (a pancreatic β-cell line) and in the liver (Raab et al, 2009), and to suppress oxidative stress after myocardial infarction (Timmers et al, 2009). Furthermore, GLP-1R agonists were reported to protect neurons from brain injury induced by oxidative damage (Perry and Greig, 2003). Expansion of the brain damage area after ischemia–reperfusion injury is explained by the immediate and direct cytotoxic effects of oxidative DNA damage and lipid peroxidation (Love, 1999), and the subsequent redox-mediated inflammatory insult that generates oxygen-free radicals (Peters et al, 1998). Microglial activation, an inflammatory response, induces various cytotoxic mediators such as NO and inflammatory cytokines, and contributes to infarct progression in the postischemic period. Accordingly, suppression of oxidative damage is a key factor in neuroprotection. Using 8-OHdG and HHE as markers of oxidative stress and Iba-1 and iNOS as markers of inflammatory response, our study showed that exendin-4 reduced accumulation of oxidative DNA damage and lipid peroxidation and inhibited the inflammatory pathway of microglial activation, followed by induction of iNOS. Moreover, exendin-4 treatment significantly reduced cell death. These results indicate that exendin-4 has antioxidant and antiinflammatory properties in cerebral ischemia.
The relationship between GLP-1 and cAMP has been examined in detail previously. Ligand activation of GLP-1R stimulates adenylyl cyclase, leading to an increase in cAMP levels in various cells including β-cells (Perry and Greig, 2003). Elevation of cAMP levels suppresses the generation of superoxide and hydrogen peroxide (Takei et al, 1998). In addition, upregulated cAMP can trigger phosphorylation of CREB at serine 133. The role of CREB phosphorylation in neurons has been studied extensively, and is known to be important in neuronal development, synaptic plasticity, and memory formation (Silva et al, 1998). Furthermore, CREB phosphorylation leads to the expression of neuroprotective genes such as B-cell lymphoma 2 and Brain-derived neurotrophic factor (Kitagawa, 2007), and has a role in neuroprotection against ischemic insults (Tanaka et al, 1999; Walton et al, 1996). Cilostazol, an antiplatelet drug, increases intracellular cAMP levels by blocking its hydrolysis by type III phosphodiesterase, and its use resulted in a decrease in cerebral infarction by reduction of oxidative and apoptosis-like cell death associated with increased cAMP (Choi et al, 2002). One previous study reported that GLP-1 and exendin-4 prevented neurodegeneration through a cascade involving cAMP (Perry et al, 2002a). In this study, exendin-4 resulted in a slight increase in cAMP contents and in a significant activation of CREB compared with the vehicle group. These findings suggest that the neuroprotective actions of exendin-4 are mediated, at least in part, through the cAMP/CREB signaling pathway.
Insulin and glucose levels can influence brain injury. In particular, insulin is reported to reduce brain damage induced by ischemia–reperfusion injury in animal and human studies (Hui et al, 2005; Rizk et al, 2006). However, because we used nondiabetic animals, insulin-dependent neuroprotection was limited. In fact, insulin levels remained unchanged after administration of exendin-4.
In conclusion, this study showed that exendin-4 can protect against oxidative products and neuronal cell death caused by ischemic brain damage. Exendin-4 treatment slightly increased intracellular cAMP levels. Activation of the cAMP response could, at least in part, mediate GLP-1-induced neuroprotection in cerebral ischemia. Our results suggest that GLP-1-related agents, like exendin-4, are potentially useful for the treatment of patients with acute ischemic stroke.
Footnotes
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.