
Abstract
Spreading depression (SD) is an intense depolarization wave implicated in brain injury. In focal ischemia, recurrent peri-infarct depolarization (PID) waves akin to SD worsen the ischemic injury by exacerbating the blood flow-metabolism mismatch. We recently showed that gabapentin suppresses SD. We, therefore, tested gabapentin on PIDs and stroke outcome. Gabapentin pretreatment (200 mg/kg, intravenously) reduced the infarct volume by 23% after transient focal ischemia in mice. However, the frequency and duration of PIDs were not suppressed when recorded for 2hours during ischemia, suggesting that gabapentin reduces infarct volume independent of PID suppression.
Introduction
Peri-infarct depolarizations (PIDs) are intense neuronal and glial depolarization waves akin to spreading depression (SD) originating within ischemic penumbra in experimental animals, as well as in stroke patients (Dohmen et al, 2008). Although SDs in normal brain do not cause injury, PIDs worsen the ischemic tissue outcome by increasing oxygen and glucose utilization and reducing tissue perfusion, thereby exacerbating the metabolic mismatch in compromised tissue (Nakamura et al, 2010; Shin et al, 2006; Strong et al, 2007). Therefore, suppression of PIDs may afford protection in ischemic brain. However, drugs that are known to suppress SD acutely often have unfavorable neurologic side effects precluding their use in critically ill patients. Gabapentin is an analgesic, adjunct antiepileptic, and migraine prophylactic drug, which targets the auxiliary α2δ subunit of Cav2.1 channels to inhibit the channel and attenuate presynaptic Ca2+ influx and neurotransmitter release. We recently showed that gabapentin can suppress SD susceptibility after a single dose in experimental animals (Hoffmann et al, 2010). We, therefore, tested whether gabapentin, widely used in clinical practice with an excellent safety and tolerability profile, can also suppress PIDs to explain its previously reported protective effect in acute ischemic stroke (Williams et al, 2006).
Materials and methods
Institutional guidelines for animal care and use for research purposes were strictly followed, and study protocol was approved by institutional review board. A total of 59 mice (C57BL/6, male, 25 to 30g, Charles River Laboratories, Kingston, NY, USA) were used. Gabapentin was administered as a single 200 mg/kg intravenous dose (Toronto Research Chemicals, Ontario, Canada) 60minutes before stroke induction; 50 and 400 mg/kg were tested in smaller groups. Intravenous route was used because of the saturable oral absorption kinetics and nonlinear bioavailability (Radulovic et al, 1995). Gabapentin dose and treatment protocol were chosen based on published data (Hoffmann et al, 2010; Radulovic et al, 1995; Traa et al, 2008). Saline was used as vehicle control. Mice were randomly assigned into vehicle and treatment groups. Ischemia induction and outcome assessments were performed in a masked manner.
Tissue and Neurologic Outcome
Middle cerebral artery was occluded (fMCAO) under isoflurane anesthesia (2.5% induction, 1% maintenance, in 70% N2O/30% O2) for 1hour, using an intraluminal silicone-coated 7-0 monofilament inserted into the internal carotid via the external carotid artery. Cerebral blood flow (CBF) reduction and reperfusion were monitored using a laser Doppler probe placed over the ischemic core. Mice were kept in a temperature-controlled warming chamber during and for 2hours after fMCAO. Twenty four hours after stroke onset, mice were neurologically examined and deficit graded based on a 5-point scale: 0, normal; 1, failure to extend the forepaw fully when suspended vertically; 2, circling to the contralateral side; 3, falling or leaning over to the contralateral side; 4, no spontaneous walking and a depressed level of consciousness. Mice were then deeply anesthetized, brains harvested, and infarcts assessed using triphenyltetrazolium chloride (Sigma-Aldrich, St Louis, MO, USA) staining on 1-mm-thick coronal slices. Infarct areas at each slice level were integrated to obtain a final infarct volume. Both direct and indirect (contralateral hemisphere minus ipsilateral non-infarcted volume) infarct volumes were calculated to assess ischemic swelling (direct minus indirect infarct volume). Animals that prematurely died within 24hours were excluded from outcome analysis (see Results). Incomplete ischemia (CBF > 20%) or reperfusion (CBF < 50%), or evidence for subarachnoid hemorrhage directly visualized during brain harvesting at 24hours, were a priori criteria for exclusion, although none of the animals fulfilled these criteria for exclusion in this study.
Electrophysiologic Monitoring of peri-infarct depolarizations
The occurrence and duration of PIDs were studied in a separate group of mice under isoflurane (spontaneously breathing, 5% induction, 1% maintenance, in 70% N2O/ 30% O2) or pentobarbital (50mg/kg, intraperitoneal) anesthesia (mechanically ventilated), and full physiologic monitoring. Blood pressure was continuously recorded (PowerLab; ADInstruments, Colorado Springs, MO, USA) and arterial blood gases and pH were measured every 30 minutes via a femoral artery catheter in spontaneously breathing (n = 17) or mechanically ventilated mice (n = 12). Rectal temperature was kept at 37°C using a thermostatic heating pad (FHC, Bowdoinham, ME, USA). Level of anesthesia was maintained throughout the experiment to eliminate cardiovascular response to tail pinch. In all groups, systemic physiologic parameters were within normal range for anesthetized mice (Table 1).
Systemic physiology and peri-infarct depolarizations
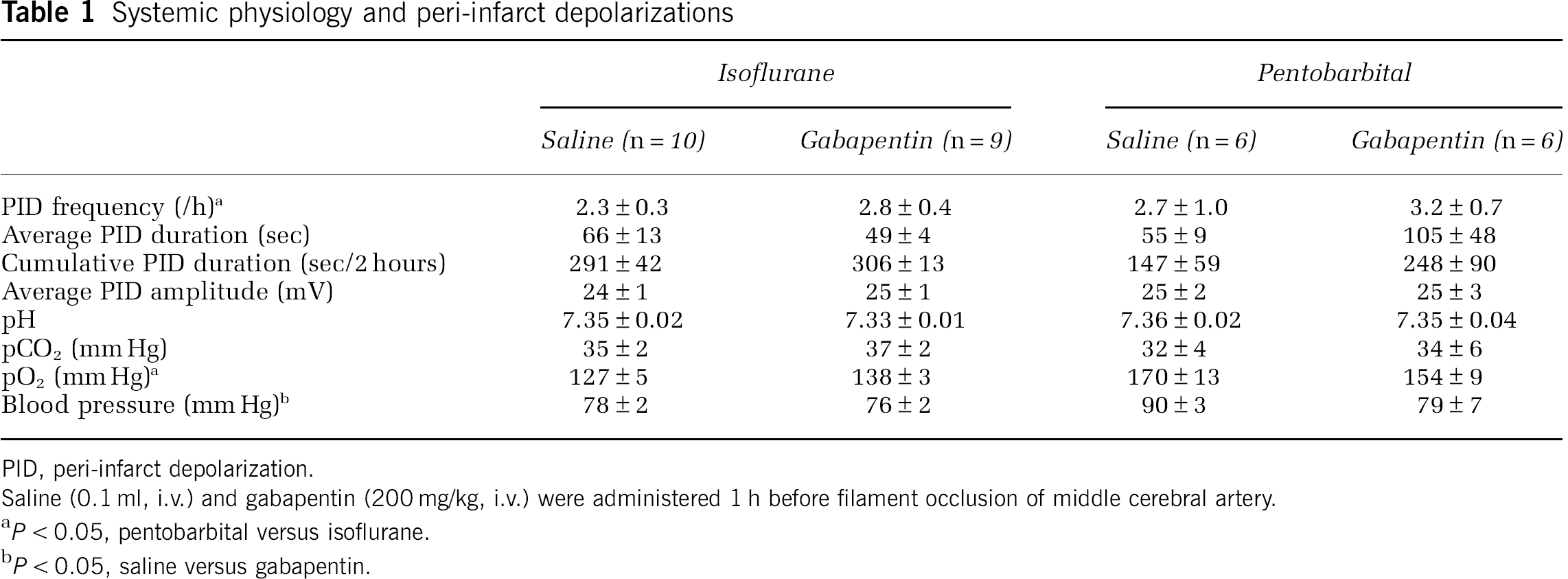
PID, peri-infarct depolarization.
Saline (0.1 ml, i.v.) and gabapentin (200 mg/kg, i.v.) were administered 1 h before filament occlusion of middle cerebral artery.
P < 0.05, pentobarbital versus isoflurane.
P < 0.05, saline versus gabapentin.
After general surgical preparation, fMCAO was performed as described above, mice were quickly placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA), and two burr holes (< 1 mm diameter) were drilled under saline cooling at (posterior and lateral from bregma, respectively): −0.5 and 1.5 mm; 2.5 and 1.5 mm. Dura was kept intact and care was taken to avoid bleeding. The steady (DC) potential and electrocorticogram were recorded with glass micropipettes filled with 200mmol/L NaCl, inserted 300 mm below pia (Axoprobe-1A; Axon Instruments, Burlingame, CA). Ag/AgCl reference electrode was placed subcutaneously in the neck. The data were continuously recorded using a data acquisition system for off-line analysis (ADInstruments, Colorado Springs, MO). All surgical procedures were completed and electrophysiologic recordings started within 30 minutes after fMCAO onset and continued for 2 hours.
Statistical Analysis
The systemic and electrophysiologic data, as well as the stroke outcome, were compared between saline and gabapentin groups using t-test or Mann-Whitney test. Mortality was compared using χ2 test. Data were expressed as mean ± standard error for systemic physiology, electrophysiology and number of PIDs, or as median (25 to 75% range) for neurologic score. P < 0.05 was considered statistically significant.
Results
Gabapentin reduced the infarct volume by 23% compared with saline-treated mice (indirect infarct 56 ± 3 versus 73 ± 4mm3; n = 8 and 9, respectively; P = 0.005; Figures 1A and 1B). Infarct reduction was similar in cortex and subcortical tissues, and present at all coronal levels. the ischemic swelling volume did not differ between groups (8 ± 3 versus 9 ± 2 mm3). Neurologic deficits tended to be milder as well in the gabapentin-treated group (1.5 (1.0 to 2.0) versus 2.0 (2.0 to 2.0), gabapentin and saline, respectively; P = 0.067; Figure 1C), whereas mortality did not significantly differ from the controls (1 out of 9 versus 3 out of 12, in gabapentin and saline groups, respectively; P = 0.42). Cerebral blood flow in the ischemic core also did not differ between the groups during ischemia and reperfusion (Figure 1D).
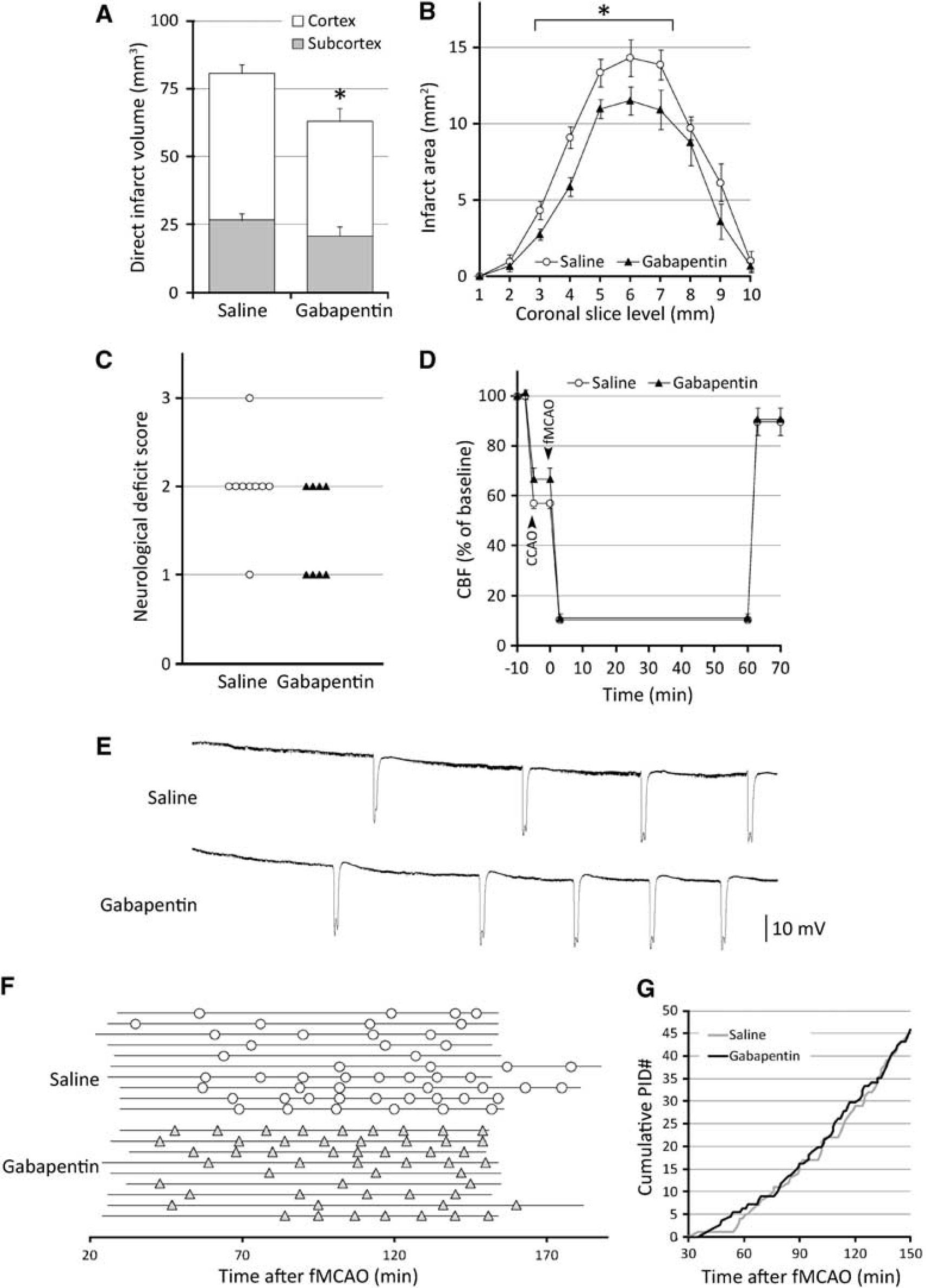
Gabapentin improves ischemic outcome but does not suppress peri-infarct depolarizations (PIDs) after middle cerebral artery occlusion. Gabapentin (200 mg/kg, i.v., 60 minutes before ischemia) reduced infarct volume when assessed 24hours after 60 minutes transient fMCAO (
Despite smaller infarct volumes, the frequency and temporal pattern of PIDs during the 2hours recording period, and their average and cumulative durations were not reduced by 200 mg/kg gabapentin (Figures 1E-G; Table 1). Additional experiments using a lower or a higher dose also did not suppress PIDs (50 and 400 mg/kg, n = 5 and 2, respectively; data not shown). Because isoflurane is known to suppress SD susceptibility, we also tested gabapentin under pentobarbital anesthesia. Although we found higher PID frequencies compared with isoflurane anesthesia, there was again no difference between the treatment groups (Table 1). Electrocorticographic epileptiform discharges were rarely observed under our experimental conditions, and their incidence or duration did not appear to differ between treatment groups (not shown).
Discussion
Our data show that a single pre-ischemic dose of gabapentin reduces infarct volume in an established stroke model in mice, as previously shown in rats (Williams et al, 2006). This neuroprotective effect was not accompanied by electrophysiologic PID suppression in acutely ischemic cortex, suggesting alternative mechanisms by which gabapentin prevents infarct expansion. One such mechanism may be suppression of postischemic seizure activity (Traa et al, 2008; Williams et al, 2006). Although we could not electrophysiologically show this because of general anesthesia, suppression of convulsive or non-convulsive seizures after reperfusion might have contributed to gabapentin's neuroprotective efficacy. Notably, after the initial flurry of PIDs during ischemia, a second phase of PIDs does occur peaking around 12hours after reperfusion and coinciding with infarct growth (Hartings et al, 2003). Therefore, it is also possible that these delayed PIDs are more susceptible to and were suppressed by gabapentin to explain the infarct reduction, although we find this unlikely after a single pre-ischemic dose of gabapentin with a relatively short plasma half-life (Radulovic et al, 1995).
Several factors may have contributed to the lack of efficacy of gabapentin on PIDs. First, elevated extracellular [K+] (e.g., ischemic penumbra) diminishes the efficacy of drugs to inhibit SD (Petzold et al, 2005; Shin et al, 2006). Our recordings were indeed obtained from hypoperfused tissue because the duration of PIDs at the stated coordinates was almost doubled compared with SDs in normal mouse cortex (Eikermann-Haerter et al, 2009). Therefore, under penumbra conditions, gabapentin may have lost its efficacy to suppress SD. Second, inhalational anesthetics are known to suppress SD (Kudo et al, 2008) and may have abrogated the PID suppression by gabapentin. However, this is unlikely because we obtained similar results under barbiturate anesthesia. Third, the dose and timing of gabapentin administration was also unlikely to be a factor, because lower or higher doses of gabapentin also appeared to be ineffective on PIDs in our pilot studies, and because similar treatment protocols have been efficacious in previous rodent studies of pain and epilepsy as well as SD (Hoffmann et al, 2010; Hunter et al, 1997; Welty et al, 1993). A notable shortcoming of single point measurements using glass micropipettes is that they lack spatial information, and might have missed an effect of gabapentin on the duration of PIDs restricted to a specific zone in penumbra. In addition, we cannot rule out an effect of gabapentin on the neurovascular coupling and the hemodynamic response to PIDs, as our CBF measurements using LDF were limited to ischemic core and aimed only to confirm successful occlusion and reperfusion in our study.
It should be noted that gabapentin shows only modest efficacy on SD compared with some other clinically used drugs known to suppress SD, such as migraine prophylactic drugs (Ayata et al, 2006; Hoffmann et al, 2010). Therefore, PID suppression may still be a relevant neuroprotective mechanism that can be targeted by this class of drugs. Nevertheless, gabapentin's rapid onset of action on SD and favorable safety and tolerability profile warrant further investigation of its neuroprotective efficacy and mechanisms in the ischemic stroke, as well as in other brain injury states in neurocritical care.
Footnotes
The authors declare no conflict of interest.