
Abstract
Hyperinsulinemia accompanying insulin resistance (IR) is an independent risk factor for stroke. The objective is to examine the cerebrovascular actions of insulin in Zucker obese (ZO) rats with IR and Zucker lean (ZL) control rats. Diameter measurements of cerebral arteries showed diminished insulin-induced vasodilation in ZO compared with ZL. Endothelial denudation revealed vasoconstriction to insulin that was greater in ZO compared with ZL. Nonspecific inhibition of nitric oxide synthase (NOS) paradoxically improved vasodilation in ZO. Scavenging of reactive oxygen species (ROS), supplementation of tetrahydrobiopterin (BH4) precursor, and inhibition of neuronal NOS or NADPH oxidase or cyclooxygenase (COX) improved insulin-induced vasodilation in ZO. Immunoblot experiments revealed that insulin-induced phosphorylation of Akt, endothelial NOS, and expression of GTP cyclohydrolase-I (GTP-CH) were diminished, but phosphorylation of PKC and ERK was enhanced in ZO arteries. Fluorescence studies showed increased ROS in ZO arteries in response to insulin that was sensitive to NOS inhibition and BH4 supplementation. Thus, a vicious cycle of abnormal insulin-induced ROS generation instigating NOS uncoupling leading to further ROS production underlies the cerebrovascular IR in ZO rats. In addition, decreased bioavailability and impaired synthesis of BH4 by GTP-CH induced by insulin promoted NOS uncoupling.
Introduction
Type 2 diabetes (T2DM) affects over a fifth of people over the age of 65 years in the United States (Center for Disease Control and Prevention. National Diabetes Fact Sheet, 2011). Another 35% of people who are 65 years and older have prediabetes that usually begins as insulin resistance (IR), a disorder in which cells fail to use insulin properly, leading to increased insulin levels to maintain normal glucose levels. Individuals with IR and T2DM exhibit sustained hyperinsulinemia that has been implicated in cerebrovascular dysfunction (Erdos et al, 2004; Phillips et al, 2005) and enhanced stroke injury (Matsumoto et al, 1999).
Insulin, a metabolic hormone, has been known to exert vasodilator (Chen and Messina, 1996; Katakam et al, 2005; Oltman et al, 2000, 2006) as well as vasoconstrictor (Eringa et al, 2002; Katakam et al, 2009a; Miller et al, 2002; van Veen and Chang, 1998) effects on several diverse vascular beds. Recently, we identified insulin receptors in rat cerebral arteries and have reported the cerebrovascular actions of insulin (Katakam et al, 2009b). Cerebral arteries are subjected to hyperinsulinemia in IR and T2DM; however, the impact of insulin on cerebral vasoreactivity in IR has never been examined.
We hypothesized that cerebrovascular IR accompanies metabolic IR in Zucker obese (ZO) rats compared with Zucker lean (ZL) controls. In the present study, we identified the insulin receptors and determined the cerebrovascular actions of insulin in ZO and ZL rats. We also evaluated the endothelium- and vascular smooth muscle-dependent mechanisms involving cyclooxygenase (COX), nitric oxide synthase (NOS), tetrahydrobiopterin (BH4), GTP cyclohydrolase-I (GTP-CH), and reactive oxygen species (ROS) derived from NADPH oxidase. Finally, we determined the intracellular signaling pathways mediating the cerebrovascular actions of insulin in IR.
Materials and methods
The animal protocol was approved by the Institutional Animal Care and Use Committee of Wake Forest University Health Sciences and Tulane University. All experiments complied with the National Institute of Health (NIH)
Zucker Obese Rat Model
The ZO rat with a leptin receptor mutation (
Insulin and Glucose Assays
Blood samples were taken via needle and syringe from the left ventricle after exposure to isofluorane anesthesia before decapitation of fasting rats. Plasma insulin and glucose levels were measured using a rat insulin enzyme-linked immunosorbent assay (ELISA) kit (Crystal Chem, Chicago, IL, USA) and Trinder reagent (Sigma, St Louis, MO, USA), respectively.
Vascular Reactivity
Arterial diameter studies were performed as described previously (Katakam et al, 2009b). Briefly, isolated middle and posterior cerebral arteries (140 to 180 μm) were cannulated in a vessel bath (Chueltech Scientific Design, Houston, TX, USA) filled with warm oxygenated (20%O2/5% CO2/75% N2 at 37°C) physiological salt solution, and intraluminal diameter was measured by a video dimension analyzer (Living Systems, Burlington, VT, USA). Arteries were slowly pressurized to 70 mm Hg with physiological salt solution under no flow conditions until a stable myogenic tone (30% to 45% of passive diameter) developed. Subsequently, drugs were administered abluminally in the bath solution. Endothelium was removed by injecting a bolus of 1 mL of air through the arteries and confirmed by the lack of significant response to bradykinin. Each arterial segment was used only for a single dose—response experiment. Viability of all arteries was verified by contraction to 60 mmol/L potassium chloride (KCl), endothelium-dependent vasodilation to bradykinin- and vascular smooth muscle cell-dependent vasodilation to sodium nitroprusside. Previously, we reported that vascular parameters and responses of the middle and posterior cerebral arteries to insulin were similar to each other (Katakam et al, 2009a); therefore, the data were combined in the present study.
Vascular responses to insulin (0.1 to 100 ng/mL; Humulin R, Eli Lilly Company, Indianapolis, IN, USA) were determined in the presence and absence of indomethacin (nonselective COX inhibitor, 10 μmol/L), Nω-nitro
In-Situ Determination of Reactive Oxygen Species Generation
Dihydroethidium (DHE; Molecular Probes, Eugene, OR, USA) was used to evaluate the
Insulin and Vascular Signaling
Isolated cerebral arteries were treated with and without insulin (40 ng/mL) in low glucose Dulbecco's Modified Eagle Medium medium at 37°C for 15 to 20 minutes. Arteries were washed and snap frozen in liquid nitrogen. Subsequently, homogenates were prepared from the tissues to determine the cellular signaling by western blotting.
RT-PCR
Total RNA was obtained from isolated cerebral arteries using the SV Total RNA Isolation System (Promega, Madison, WI, USA). RT-PCR experiments were performed in an Eppendorf Mastercycler thermocycler (Brinkmann Instruments, Westbury, NY, USA) as previously described (Katakam et al, 2006, 2009a). From each sample, 50 pg of total RNA was reverse transcribed and amplified using QIAGEN OneStep RT-PCR Kit with gene-specific primers targeting rat insulin receptor (sense primer: 5′-GCCATCCCGAAAGCGAAGATC-3′; anti-sense primer: 5′-TCTGGGTCCTGATTGCAT-3′; reference sequence NM_017071). Expected lengths of the RT-PCR product were 224 base pairs. We used 35 cycles for insulin receptor amplification. In control experiments, when reverse transcription was omitted, no amplification was observed.
Western Blot Analysis
Western blot analyses were performed as previously described (Katakam et al, 2009a, b ). Briefly, equal amounts of extracted protein from homogenates of cerebral arteries were separated by 4% to 20% SDS-PAGE and transferred onto a nitrocellulose membrane (Bio-Rad, Hercules, CA, USA). Membranes were blocked with 1% nonfat milk in Tris-buffered saline and 0.05% Tween-20 for 1 hour at room temperature. Subsequently, the membranes were incubated overnight at 4°C with the primary antibodies for human insulin receptor β subunit (1:4,000; BD Transduction Laboratories, San Jose, CA, USA), total and phosphorylated endothelial NOS (eNOS) (1:4,000; BD Transduction Laboratories), total nNOS and inducible NOS (iNOS) (1:2,000; BD Transduction Laboratories), GTP-CH (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA) total Akt (1:4,000; BD Transduction Laboratories), phosphorylated Akt that recognize the phosphorylated Ser473 (1:5,000; Cell Signaling Technology, Danvers, MA, USA), phosphorylated PKC-pan (1:2,500; Cell Signaling Technology), phosphorylated PKC-α/β (1:2,000; BD Transduction Laboratories), total and phosphorylated ERK-1/2 (1:2,500; Promega), and β-actin (1:2,500; Sigma). The membranes were then washed and for 2 hours in the blocking buffer with anti-rabbit IgG (1:50,000 dilution; Jackson ImmunoResearch, West Grove, PA, USA) conjugated to horseradish peroxidase. The final reaction products were visualized using enhanced chemiluminescence (SuperSignal West Pico; Pierce, Rockford, IL, USA) and recorded on X-ray film. Each immunoband intensity was normalized to the corresponding immunoband intensity of β-actin, which was used as a loading control.
Drugs, Chemicals, and Solutions
All chemicals were purchased from Sigma except MnTBAP (Calbiochem, San Diego, CA, USA) and DHE (Molecular Probes). Stock solutions of 7-NI and apocynin were prepared in dimethyl sulfoxide and all the other chemicals were prepared in deionized water. The composition of physiological salt solution (mmol/L) was NaCl (112), KCl (4.8), NaHCO3 (26), KH2PO4H2O (1.2), CaCl2 (1.8), MgSO4·7H2O (1.2), and glucose (10). Physiological salt solution with 60 mmol/L KCl was prepared by replacing NaCl with an equimolar quantity of KCl.
Data Analysis and Statistics
All data are reported as mean ± s.e.m.
Results
Plasma Insulin and Glucose Measurements
Fasting plasma glucose levels (mg/dL) were 143 ± 8 in ZL rats (
Expression of Insulin Receptor
Our previous study was the only report of the presence of insulin receptors in intact rat cerebral arteries. To confirm the presence of insulin receptors, we determined their expression in cerebral arteries of Zucker rats. PCR experiments showed the presence of 224 bp length insulin receptor mRNA in the rat cerebral arteries (Figure 1A). Similarly, the 95-kDa insulin receptor β subunit protein was also identified in the homogenates of rat cerebral arteries (Figure 1B).
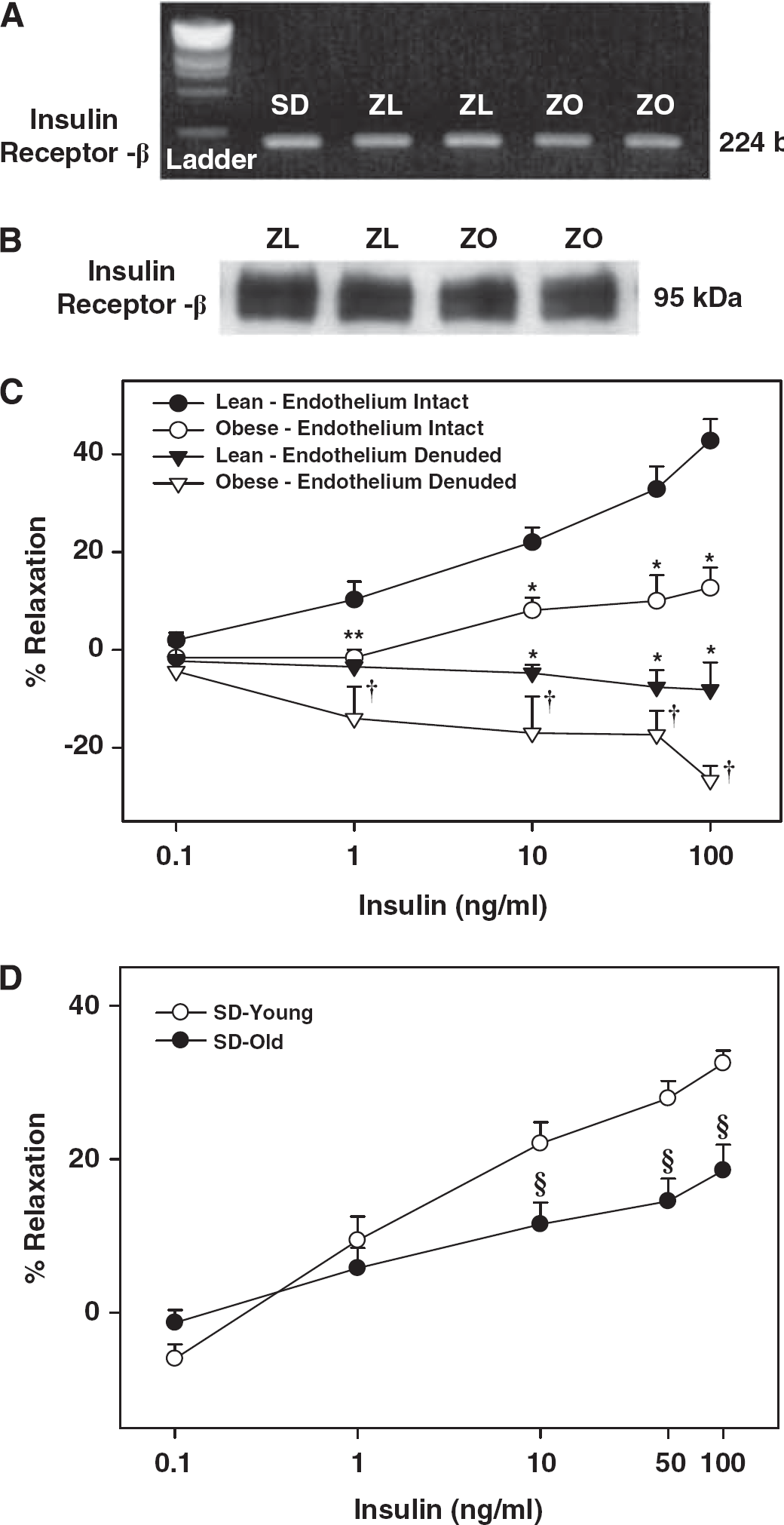
(
Insulin and Vasoreactivity
The average resting intraluminal diameter of cerebral arteries from all rats for each group of experiments was similar and they were preconstricted to a similar extent before the administration of various concentrations of insulin to determine the vasodilation elicited by insulin (online Supplementary Table 1). Insulin elicited a dose-dependent vasodilation in all cerebral arteries; however, significantly reduced vasodilation was observed in ZO compared with ZL rats (Figure 1C). Similarly, decreased insulin-induced vasodilation (% maximal vasodilation) was observed in the cerebral arteries of aged SD rats (19.7 ± 3,
Inhibition of COX by indomethacin enhanced vasodilation in ZL arteries at low insulin concentrations although maximal vasodilation was unchanged (Figure 2A). In contrast, indomethacin enhanced vasodilation to all concentrations of insulin in ZO arteries (
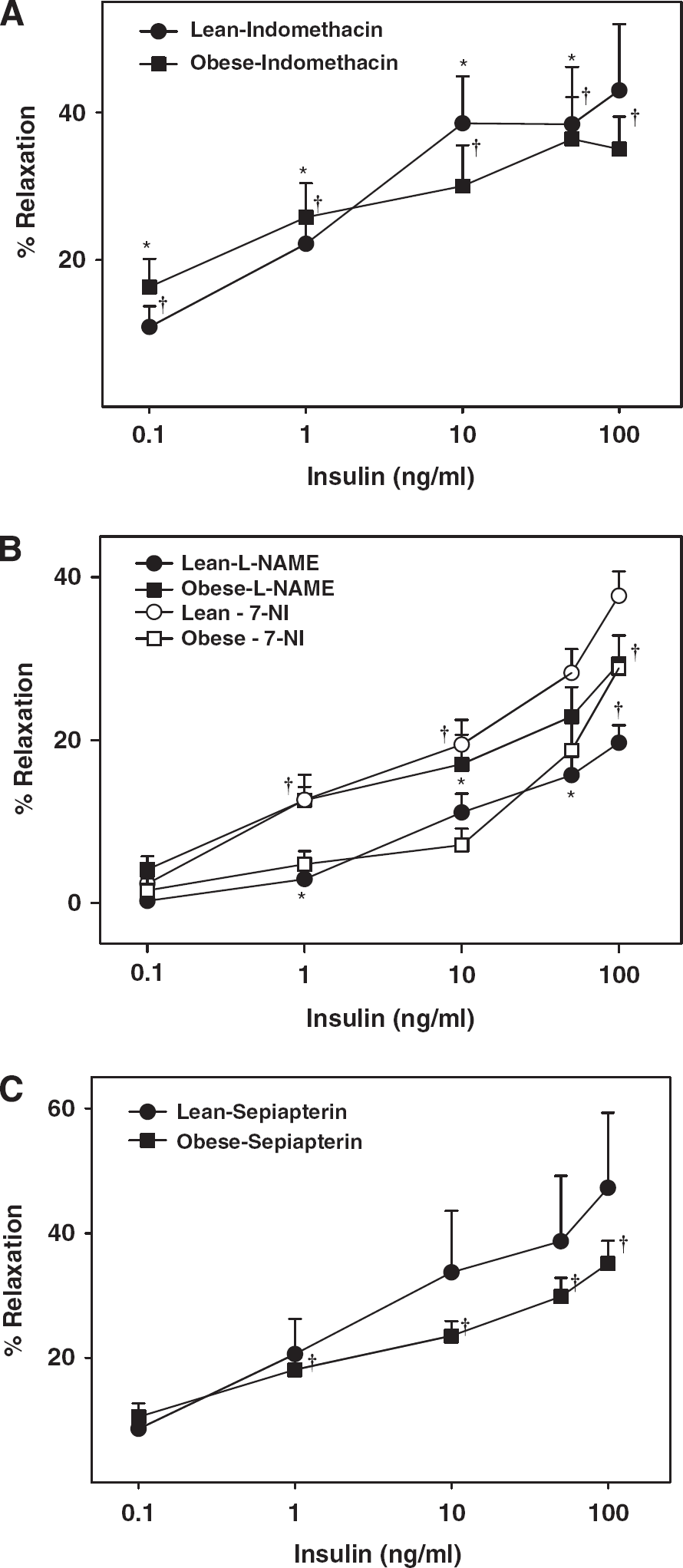
Vascular responses to insulin in cerebral arteries of Zucker obese (ZO) and Zucker lean (ZL) with intact endothelium, in the presence of indomethacin (
Inhibition of ROS generation with apocynin did not affect the maximal vasodilation to insulin in ZL arteries, whereas apocynin improved vasodilation in ZO arteries (Figure 3A). This indicates that insulin activated NADPH oxidase leading to excess ROS generation, which in turn diminished vasodilation to insulin in ZO arteries. However, in ZL arteries, insulin-induced ROS production by NADPH oxidase was not sufficient to alter the vasodilation. Scavenging of the ROS with PEG-SOD improved vasodilation to 1 ng/mL insulin in ZL arteries compared with baseline although maximal relaxation to insulin was unchanged, indicating that insulin promotes ROS generation at basal levels that oppose the vasodilator response to insulin. In ZO arteries, however, PEG-SOD treatment normalized the vasodilation indicating that increased oxidative stress diminished vasodilation in ZO arteries (Figure 3B). In addition, scavenging of the ROS with MnTBAP did affect the vasodilation to insulin in ZL arteries whereas MnTBAP promoted vasodilation in ZO arteries (Figure 3B).
Pretreatment with
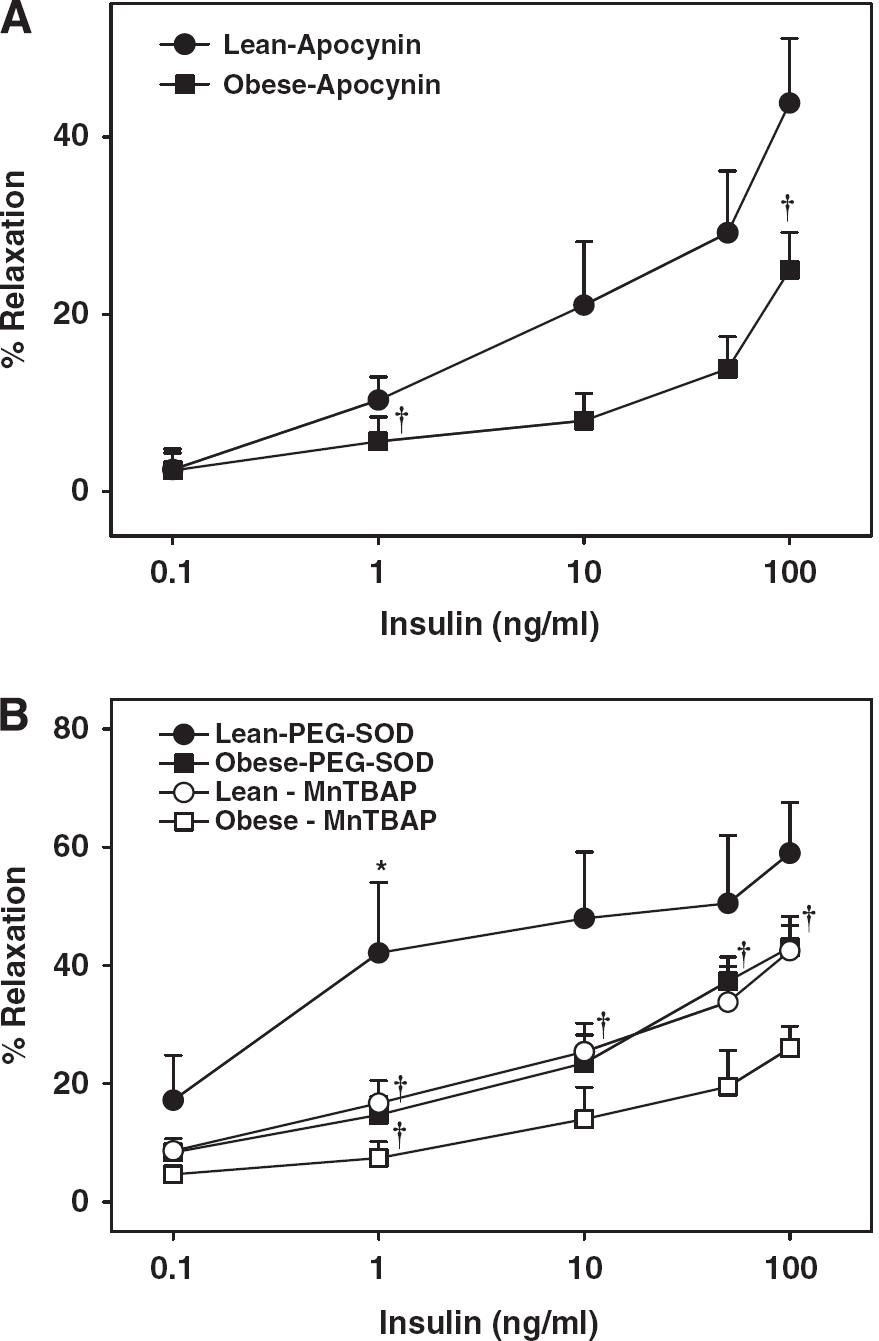
Vascular responses to insulin in cerebral arteries of Zucker obese (ZO) and Zucker lean (ZL) with intact endothelium, in the presence of apocynin (
Vascular Signaling
Cellular kinases mediate vasodilator and vasoconstrictor signaling in arteries. To uncover the signaling mechanisms underlying the impaired vascular actions of insulin in IR, we determined the activation of the key kinases and their targets known to regulate vasoreactivity. Treatment of rat cerebral arteries with insulin
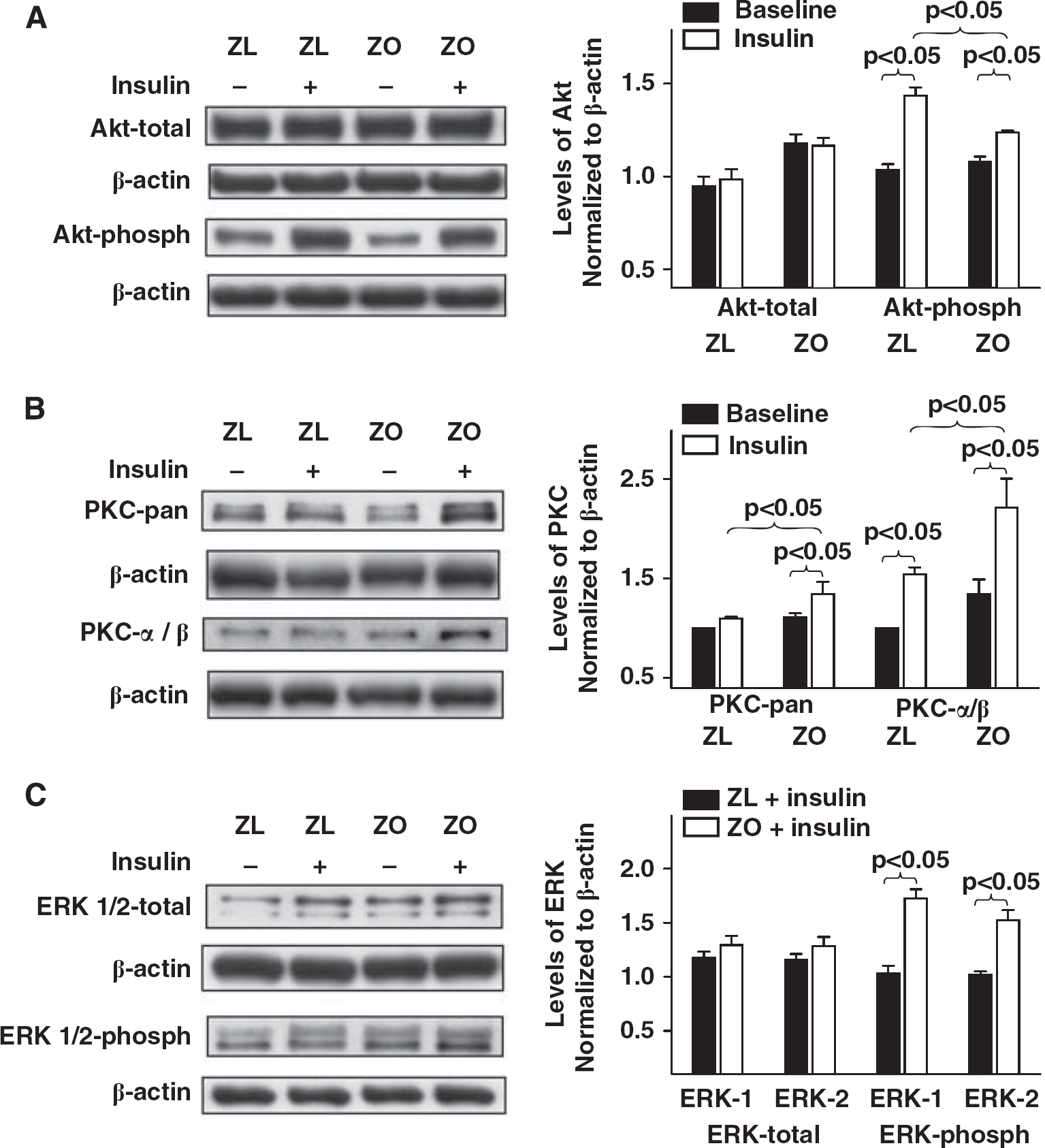
Representative immunoblots of homogenates of Zucker obese (ZO) and Zucker lean (ZL) cerebral arteries treated with or without insulin for 15 to 20 minutes 37°C are shown. Immunoblots of total and phosphorylated Akt (
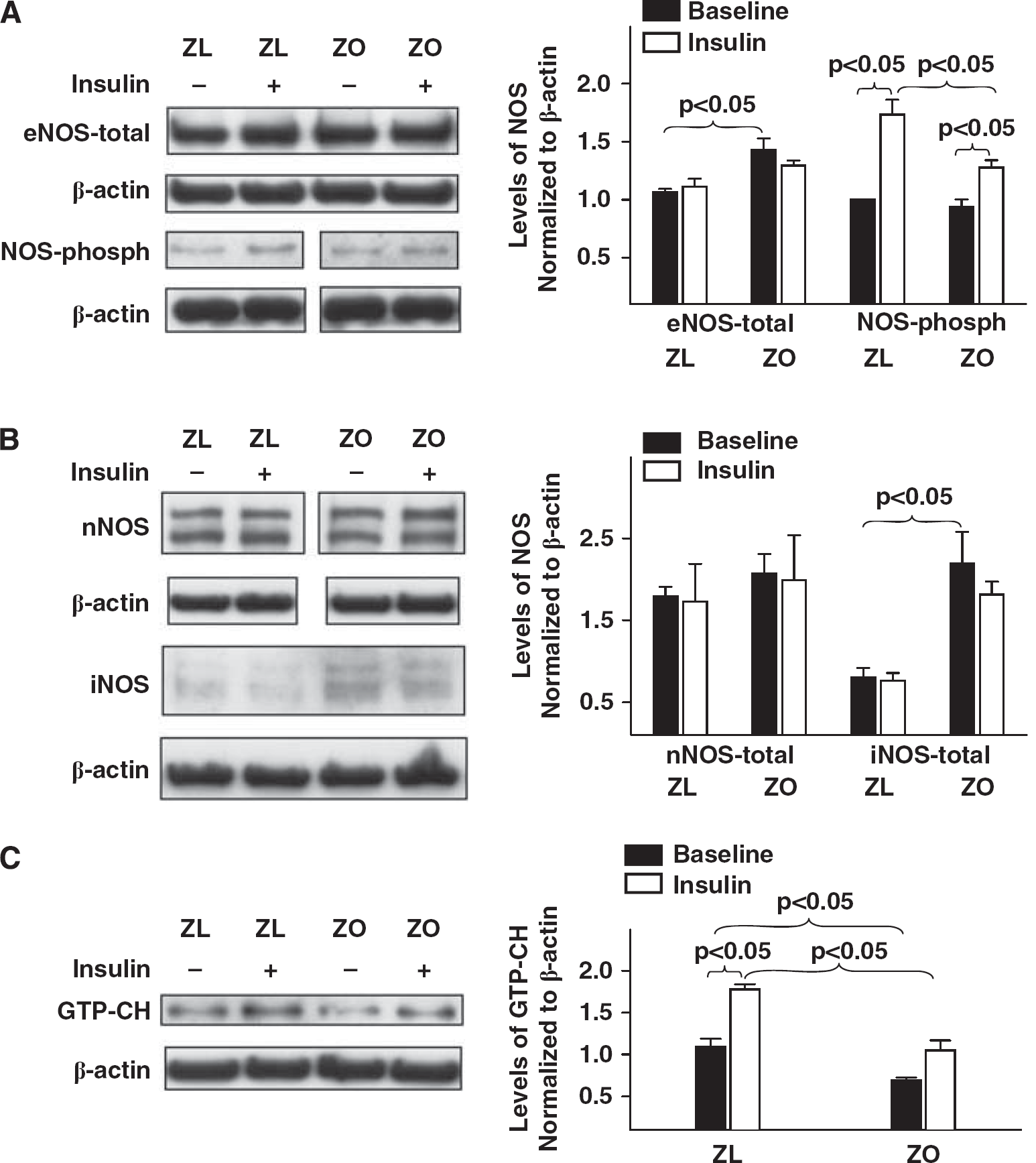
Representative immunoblots of homogenates of Zucker obese (ZO) and Zucker lean (ZL) cerebral arteries treated with or without insulin for 15 to 20 minutes at 37°C are shown. Immunoblots of total and phosphorylated endothelial nitric oxide synthase (eNOS) (
Reactive Oxygen Species Generation
Fluorescence microscopy revealed greater DHE fluorescence in ZO than in ZL arterial sections, indicating increased ROS production under baseline conditions (Figures 6A, 6B, and 6G). In ZL arteries,
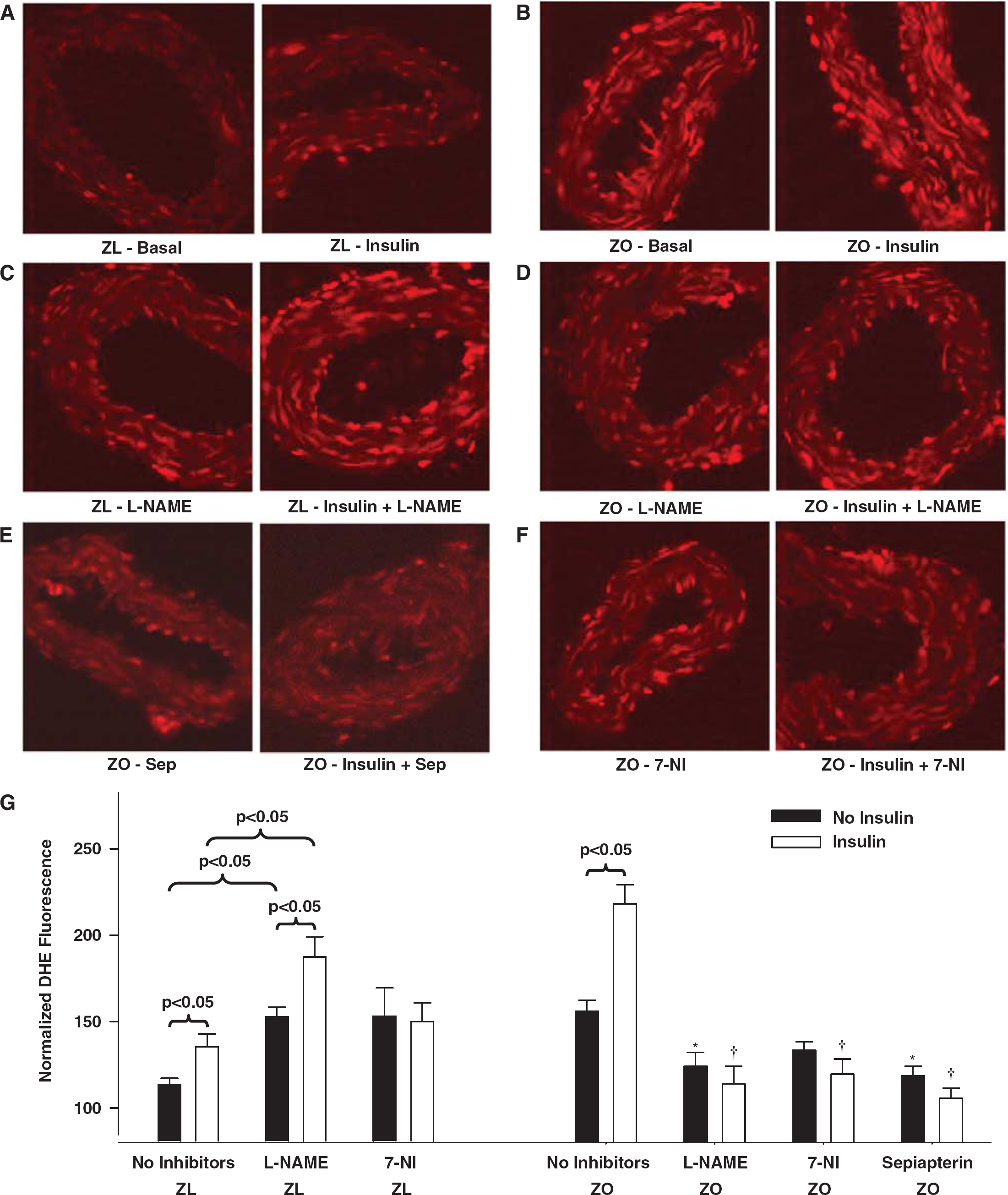
Representative dihydroethidium (DHE) fluorescence images of sections of Zucker obese (ZO) and Zucker lean (ZL) cerebral arteries treated with or without insulin are shown. Fluorescence images of ZL arterial sections acquired at baseline or after insulin treatment in the absence (
Discussion
The present study reports impaired cerebrovascular actions of insulin in diabetes or IR. We show that cerebral vascular responsiveness to insulin is reduced in IR animals associated with obesity due to complex changes in vasodilation and vasoconstriction pathways based on the following original findings: (1) insulin-induced vasodilation in isolated cerebral arteries was significantly diminished in ZO rats compared with ZL rats; (2) eNOS-derived NO mediated insulin-induced vasodilation in ZL arteries whereas in ZO arteries, NO-mediated vasodilation was lost and uncoupling of NOS isoforms resulted in ROS generation contributing to vasoconstriction. Supplementation with the precursor of BH4 diminished the insulin-induced ROS generation and restored vasodilation in ZO arteries implicating NOS uncoupling in cerebrovascular IR; (3) ZO arteries exhibited decreased expression of GTP-CH at baseline and after insulin treatment compared with ZL arteries that contributed to the reduced BH4 bioavailability; (4) insulin promoted abnormal activation of NADPH oxidase in ZO arteries leading to increased generation of ROS and diminished vasodilation. Scavenging of ROS or inhibition of NOS isoforms partially restored insulin-induced vasodilation in ZO arteries confirming the role of oxidative stress in cerebrovascular IR; (5) COX metabolites primarily mediated vasoconstriction in response to insulin and increased production of vasoconstrictor prostanoids reduced vasodilation to insulin in ZO arteries; (6) abnormal insulin-induced phosphorylation of kinases resulted in diminished vasodilatory (eNOS and Akt) and enhanced vasoconstrictor (pan-PKC, PKC-α/β, and ERK-1/2) signaling in ZO arteries. Thus, taken together, these findings show that cerebrovascular IR in ZO rats displays selective resistance to signaling pathways that promote vasodilation compounded by selective promotion of vascular signaling that induce vasoconstriction as summarized in the schematic in Figure 7.
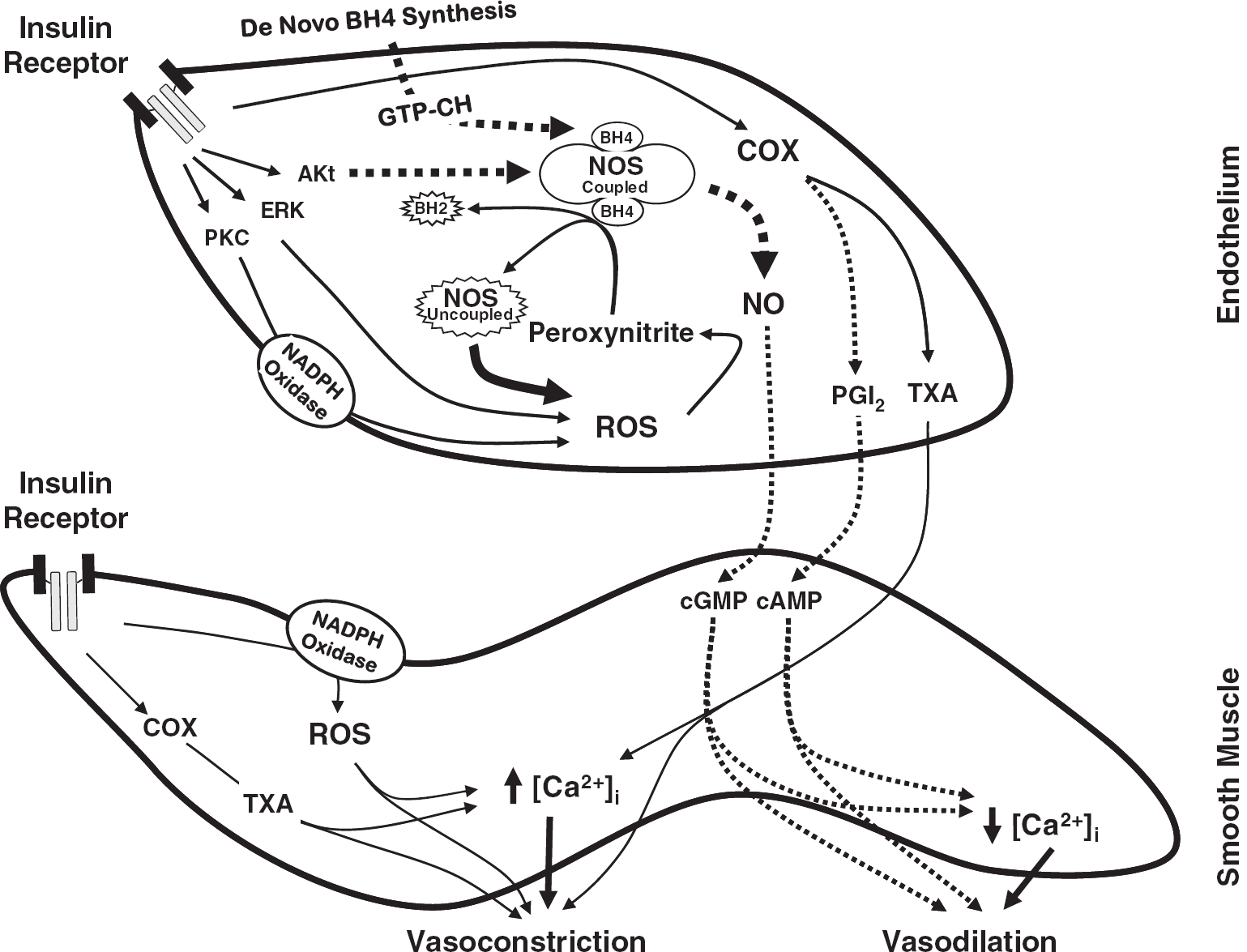
A schematic of the mechanisms underlying cerebrovascular insulin resistance (IR) in Zucker obese (ZO) rats. Insulin binding to insulin receptor leads to activation of kinase signaling by phosphorylation that in turn activates downstream signaling pathways. Zucker obese arteries displayed reduced phosphorylation of Akt and its target, endothelial nitric oxide synthase (eNOS), resulting in diminished nitric oxide (NO)-mediated vasodilation. Insulin binding also resulted in enhanced PKC and ERK 1/2 phosphorylation and activation in ZO arteries that may likely mediate excess superoxide production by NADPH oxidase. Excessive reactive oxygen species (ROS) formation in response to insulin in ZO arteries promoted enhanced vasoconstriction that was reversed by the scavenging of ROS. In addition, oxidative stress in ZO arteries may have induced uncoupling of NOS by oxidation of tetrahydrobiopterin (BH4), an essential cofactor for the activation of NOS that stabilizes the dimeric form of the enzyme, leading to superoxide formation. Zucker obese arteries also displayed decreased expression of GTP cyclohydrolase-I (GTP-CH), a rate-limiting enzyme in the
Our laboratory identified the mRNA and protein of insulin receptors in the cerebral arteries of SD rats (Katakam et al, 2009a). In the present study, we identified mRNA and protein of insulin receptors in the cerebral arteries of Zucker rats comparable in size to the insulin receptors previously reported (Katakam et al, 2009a; Moloney et al, 2010).
Normal fasting plasma insulin levels in humans are <1 ng/mL and they are elevated many times over the basal levels after ingesting a meal; however, obese humans display insulin levels often 3 to 5 times higher at fasting and postprandial states (Polonsky et al, 1988). Moreover, a person with diabetes receiving NovoLog insulin injections (1 unit/kg per day) may display plasma insulin levels of 10 ng/mL or higher (prescribing information of Novo Nordisk, Princeton, NJ, USA), based on the WHO established standard (WHO Expert Committee on Biological Standardization. Thirty-Seventh Report, 1987). In the present study, insulin concentrations ranged from 0.1 to 100 ng/mL and thus covered physiological, pathological, and pharmacological insulin levels.
Cerebrovascular Insulin Resistance Accompanies Metabolic Insulin Resistance
Zucker obese rats and aged SD rats displayed similar plasma glucose levels but hyperinsulinemia when compared with their respective controls, ZL rats and younger SD rats confirming obesity and age-related metabolic IR with normoglycemia. Consistent with our previous observations in SD rats (Katakam et al, 2009a), insulin induced vasodilation in isolated cerebral arteries of Zucker rats. However, the vasodilation to insulin was diminished in ZO compared with ZL rats signifying cerebrovascular IR. Similarly, insulin-induced vasodilation was also reduced in aged SD rats compared with younger SD rats. Thus, cerebrovascular IR accompanied metabolic IR in two diverse models of IR, indicating that vascular IR observed was independent of underlying etiology of IR.
Vasodilation elicited by insulin was entirely endothelium-dependent but endothelial denudation abolished vasodilation, and instead revealed dose-dependent vasoconstriction to insulin. This vasoconstrictor response to insulin was enhanced in ZO arteries compared with ZL arteries. Thus, reduced vasodilation combined with enhanced vasoconstriction underlies the cerebrovascular IR in ZO arteries.
Role of Cyclooxygenase
Consistent with our previous studies (Katakam et al, 2009a), activation of COX by insulin promoted the generation of vasoconstrictor metabolites from the vascular wall in Zucker rats. Cerebral arteries of ZO rats appeared to produce increased vasoconstrictor prostanoids in response to insulin, leading to greater vasoconstriction compared with ZL arteries. Similar abnormal activation of COX in response to insulin has also been implicated in vascular dysfunction in T2DM (Bagi et al, 2005).
Role of Nitric Oxide Synthase Uncoupling
Insulin-induced vasodilation in ZL arteries was mediated primarily by NO originating only from eNOS, unlike SD rat cerebral arteries where both eNOS and nNOS participated in vasodilation (Katakam et al, 2009a). We previously observed that NO-mediated vasodilation was diminished in ZO arteries despite the evidence of increased eNOS and iNOS expression compared with ZL arteries (Erdos et al, 2004; Katakam et al, 2009b). Moreover, a reduced phosphorylated NOS protein in response to insulin, consistent with reduced NO-mediated vasodilation, was observed in ZO arteries compared with ZL arteries. Interestingly, NOS appears to release a vasoconstrictor factor in response to insulin in ZO arteries since inhibition of NOS paradoxically promoted vasodilation. It is known that NOS isoforms in an uncoupled state produce superoxide anion, which promotes vasoconstriction by direct activation of vasoconstrictor pathways or by decreasing the NO bioavailability. Inhibition of all NOS isoforms, or specifically nNOS, partially restored the insulin-induced vasodilation implicating uncoupling of NOS isoforms and subsequent superoxide anion production in the diminished vasodilation. Indeed, direct ROS measurement confirmed enhanced ROS production in ZO arteries in response to insulin that was partially restored by BH4 precursor supplementation or inhibition of NOS in general and nNOS in particular. Western blot studies identified all three isoforms of NOS in cerebral arteries of Zucker rats. Thus, uncoupling of NOS in ZO arteries may likely involve all NOS isoforms.
Reduced Tetrahydrobiopterin Bioavailability and GTP Cyclohydrolase-I Expression
Tetrahydrobiopterin is a cofactor regulating NOS activity and NO synthesis by promoting eNOS dimerization and protein stability. Reduced BH4 levels promote eNOS uncoupling leading to formation of superoxide (Katusic et al, 2009; Tarpey, 2002). In endothelial cells, reduction of BH4 levels occurs due to either increased oxidative conversion to 7,8-dihydrobiopterin (BH2) or decreased biosynthesis by GTP-CH, a rate-limiting enzyme in the
Role of Oxidative Stress
Reduced BH4 levels due to oxidative degradation of BH4 to BH2 has been reported in diabetes (Shinozaki et al, 2000). Furthermore, BH2 competes for binding to NOS at the BH4 site, thus promoting NOS uncoupling and loss of NO production (Katusic et al, 2009). Increased ROS production in ZO arteries is an additional mechanism underlying the uncoupling of NOS by oxidative degradation of BH4 to BH2. Increased vasoconstriction and reduced vasodilation in ZO arteries in response to insulin also appear to be mediated by ROS because scavenging the ROS with PEG-SOD or MnTBAP or inhibition of ROS generation with apocynin,
Consistent with our previous observations in coronary arteries (Katakam et al, 2005), direct measurements of ROS showed the ability of insulin to activate ROS production in rat cerebral arteries. Overall, ROS generation at basal and insulin activated states was greatly increased in ZO arteries. Importantly, ROS generation in ZO arteries was diminished by NOS inhibition or promoting BH4 generation supporting the role of NOS uncoupling in cerebrovascular IR. Surprisingly, NOS inhibition in ZL arteries enhanced vascular ROS. It is not clear whether NO simply quenches ROS or exhibits more complex regulation of ROS generation. In addition to uncoupling of NOS, enhanced insulin-induced activation of NADPH oxidase appears to contribute to the oxidative stress in ZO arteries as inhibition of NADPH oxidase activity partially restored the cerebrovascular vasodilation in ZO rats. We reported similar observations related to coronary vascular dysfunction in ZO rats (Katakam et al, 2005). It has been proposed that in endothelial cells and vascular smooth muscle cells, apocynin reduces ROS not by inhibiting NAPDH oxidase but possibly by scavenging ROS (Heumuller et al, 2008). However, it has been argued that whether apocynin inhibits NADPH oxidase in nonphagocytic cells without myeloperoxidase depends on the cell type studied and the ability of vascular wall to activate apocynin (Touyz, 2008). Insulin-induced vasodilation was improved by apocynin only in ZO arteries, suggesting that insulin enhanced NADPH oxidase activity in ZO arteries. Thus, the present study identified insulin as a novel instigator of NOS uncoupling in cerebrovascular IR via abnormal activation of NADPH oxidase.
Abnormal Vascular Signaling
Insulin elicited phosphorylation of Akt at ser473 in cerebral arteries of Zucker rats consistent with previous observations (Katakam et al, 2009a). Akt phosphorylation has been linked to phosphorylation of eNOS at Ser1177 and NO formation (Mount et al, 2007). Insulin also has been shown to stimulate NO synthesis by Akt-mediated phosphorylation and activation of eNOS (Ritchie et al, 2010). Cerebral arteries of Zucker rats also exhibited insulin-induced eNOS phosphorylation at Ser1177. However, phosphorylation of both Akt and eNOS in response to insulin was diminished in ZO compared with ZL arteries. As previously reported (Bakker et al, 2008b), insulin also induced phosphorylation of PKC (pan and α/β isoforms) in cerebral arteries. Enhanced PKC activation has been shown to promote vasoconstriction in ZO rats (Erdos et al, 2004) and models of T2DM (Bakker et al, 2008a) via activation of NADPH oxidase, promoting endothelin-1 expression and increasing mitochondrial ROS formation (Geraldes and King, 2010). We observed increased insulin-induced phosphorylation of PKC isoforms in ZO that may have contributed to the cerebrovascular IR. Finally, insulin has also been shown to activate ERK 1/2 signaling in vasculature that contributes to vasoconstrictor actions of insulin (Bakker et al, 2008a; Eringa et al, 2004). Cerebral arteries of Zucker rats similarly exhibited activation of ERK 1/2 by phosphorylation, which was greater in ZO arteries compared with ZL arteries. Thus, investigation of kinase signaling in the vascular wall revealed complex activation of pathways leading to impaired vasodilation and enhanced vasoconstriction.
Insulin Versus Other Vasodilators
Cerebrovascular actions of insulin in Zucker rats were unique when compared with other vasodilators. For example, previous studies in our laboratory showed that inhibition of NOS diminished vasodilation in cerebral arteries of ZL arteries in response to two mechanistically different vasodilators, acetylcholine and diazoxide (Erdos et al, 2004; Katakam et al, 2009b). Unlike insulin, neither acetylcholine nor diazoxide improved vasodilation to insulin in ZO arteries in the presence of NOS inhibitors, suggesting that insulin displays unique ability to promote NOS uncoupling in IR (Erdos et al, 2004; Katakam et al, 2009b). In addition, we observed the ability to modulate expression of GTP-CH enzyme and activity of NOS isoforms unique to insulin. Thus, the present study uncovered unique vascular actions of insulin in cerebral arteries of ZO rats that have not been reported in other circulations such as mesenteric (Miller et al, 2002) or coronary (Katakam et al, 2005). It is noted that vasodilation to nitroprusside was unchanged in ZO arteries compared with ZL arteries (Erdos et al, 2004).
Limitations
It is acknowledged that our observations in ZO rats are confounded by the effects of obesity and leptin receptor mutation on the same signaling pathways affected by insulin. However, the significant distinction displayed by insulin in inducing NOS uncoupling in ZO arteries compared with many vasodilators we previously studied (Erdos et al, 2004;, 2005, 2009b) assures the specificity of insulin's action in cerebrovascular IR. Although it has been reported that hyperleptinemia has been associated with increased eNOS expression and uncoupling (Korda et al, 2008); hyperleptinemia has not been shown to directly stimulate NOS uncoupling. Moreover, the demonstration of cerebrovascular IR in aging-associated IR supports the argument that the obesity/leptin receptor mutation is unlikely to directly account for the impaired vascular actions of insulin in ZO rats.
Perspective
In conclusion, we showed that cerebrovascular IR accompanies metabolic IR in ZO rats. Diminished NO generation compounded by the increased oxidative stress resulting from NOS uncoupling and abnormal NADPH oxidase activity underlie the cerebrovascular IR. Inability of insulin to enhance GTP-CH and increased oxidative degradation of BH4 may have contributed to reduced BH4 bioavailability in IR. These observations have contributed to a paradigm whereby excessive ROS production in ZO arteries decreased bioavailability of NO and BH4 leading to eNOS uncoupling and further formation of ROS, thus perpetuating a cycle of vascular oxidative stress in cerebrovascular IR.
Footnotes
Acknowledgements
The authors thank Nancy Busija for the help with editing the manuscript. The authors also acknowledge the help with immunostaining and fluorescence imaging of insulin receptors by Dr Edina A Wappler and Dr Paige S Katz.
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.