
Abstract
Reactive oxygen species (ROS) such as superoxide (O•−2) and hydrogen peroxide (H2O2) are known cerebral vasodilators. A major source of vascular ROS is the flavin-containing enzyme nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase. Activation of NADPH-oxidase leads to dilatation of the basilar artery in vivo via production of H2O2, but the endogenous stimuli for this unique vasodilator mechanism are unknown. Shear stress is known to activate both NADPH-oxidase and phosphatidylinositol-3 kinase (PI3-K) in cultured cells. Hence, this study used a cranial window preparation in anesthetized rats to investigate whether increased intraluminal blood flow could induce cerebral vasodilatation via the activation of NADPH-oxidase and/or PI3-K. Bilateral occlusion of the common carotid arteries to increase basilar artery blood flow caused reproducible, reversible vasodilatation. Topical treatment of the basilar artery with the NADPH-oxidase inhibitor diphenyleneiodonium (DPI) (0.5 and 5 μmol/L) inhibited flow-induced dilatation by up to 50% without affecting dilator responses to acetylcholine. Treatment with the H2O2 scavenger, catalase similarly attenuated flow-induced dilatation, suggesting a role for NADPH-oxidase-derived H2O2 in this response. The nitric oxide synthase inhibitor NG-nitro-
Introduction
Reactive oxygen species (ROS) including superoxide (O•−2), and particularly hydrogen peroxide (H2O2), have been shown to influence vascular function under both physiologic and pathologic conditions. In the cerebral circulation, H2O2 is a powerful vasodilator that acts via the opening of calcium-activated potassium (KCa) channels to relax vascular smooth muscle (Paravicini et al, 2004; Sobey et al, 1997).
Nicotinamide adenine dinucleotide phosphate (NADPH)-oxidases are a major source of vascular ROS, and are expressed throughout the vessel wall. NADPH-oxidases are a family of multimeric enzyme complexes that produce O•−2 by transferring electrons from NADPH to molecular oxygen via a flavin-containing subunit of the enzyme. NADPH-oxidases are expressed in cerebral arteries (Didion and Faraci, 2002; Paravicini et al, 2004), and their activation leads to cerebral vasodilatation and increased cerebral blood flow (Paravicini et al, 2004; Park et al, 2004). The identity of the flavin-containing subunit varies between isoforms of the enzyme, and isoforms containing either gp91phox, Nox1 or Nox4 have all been found in vascular cells (Lassegue and Clempus, 2003). It is well established that NADPH-oxidases can be activated by a number of hormonal stimuli, such as angiotensin II and thrombin. However, mechanical stimuli such as shear stress have also been shown to stimulate NADPH-oxidase activity. In cultured vascular cells, both the activity and expression of NADPH-oxidase is elevated by either increased shear or vascular wall stress (Lassegue and Clempus, 2003).
In cultured endothelial cells, phosphatidylinositol-3 kinase (PI3-K) is known to be involved in various mechanotransduction pathways that are activated in response to shear stress (Fisslthaler et al, 2000; Go et al, 1998). PI3-K may also be involved in the activation of ROS-producing pathways, including NADPH-oxidase. For example, in cultured vascular smooth muscle cells, inhibition of PI3-K attenuates ROS production by NADPH-oxidase due to angiotensin II stimulation (Seshiah et al, 2002). Likewise, there is evidence that activation of PI3-K is linked to the downstream activation of the small G protein, Rac, which is crucial for the activation of NADPH-oxidase (Griendling et al, 2000; Welch et al, 2003).
In blood vessels, the major determinant of shear stress is intraluminal blood flow. In many vascular beds, increases in blood flow result in dilatation of the major conducting vessel(s). Flow-induced dilatation of cerebral arteries in vivo was first described by Fujii et al (1991), who used a cranial window preparation to show that occlusion of one or both common carotid arteries causes proportional increases in the velocity of blood flow in the basilar artery (measured using pulsed Doppler technology), and consequently the artery diameter (Fujii et al, 1991). There was virtually no delay between occlusion of the common carotid arteries and the onset of the increase in blood flow velocity, whereas the artery diameter began to increase after a further ~ 13 secs. These increases in basilar artery blood flow velocity and diameter occurred while changes in arterial pressure were prevented by controlled bleeding, and were sustained throughout the carotid occlusion, only returning to preocclusion levels shortly after removal of the occlusion (Fujii et al, 1991). However, the mechanism(s) underlying this phenomenon of flow-induced cerebral vasodilatation in vivo could not be identified by Fujii et al, using a range of pharmacological inhibitors available at that time, and still remain unclear. It is noteworthy that, although in many vascular beds flow-induced dilatation is known to be mediated predominantly by the production of nitric oxide (NO) from endothelial nitric oxide synthase (eNOS) (Davies, 1995), flow-induced cerebral vasodilatation appeared to occur largely independently of this mechanism (Fujii et al, 1991). Indeed, there remains very limited knowledge about mechanisms underlying flow-dependent cerebral vasodilatation in general, and in vivo in particular.
The present study has therefore used a similar approach as Fujii et al, to test two novel hypotheses. Firstly, we tested if shear-dependent activation of NADPH-oxidase and subsequent production of ROS is involved in mediating flow-induced dilatation of the basilar artery in vivo. Secondly, we also tested if PI3-K activity contributes to the cerebral vasodilator effects of increased flow.
Materials and methods
Animals
All procedures were approved by the University of Melbourne Pharmacology, Physiology and Biochemistry Animal Experimentation Ethics Committee, and performed in accordance with the Australian Code of Practice for the Use of Animals in Scientific Procedures. In total, 92 Sprague–Dawley rats (weight 250 to 400 g, mean arterial pressure 91 ± 4 mm Hg) were used in this study.
Measurement of Cerebral Artery Diameter In vivo
Rats were anesthetized with pentobarbitone sodium (50 mg/kg i.p.), with supplemental anesthetic administered via a femoral vein cannula as required (10 to 20 mg/kg/h i.v.). Femoral arteries were also cannulated for the measurement of arterial blood pressure and the withdrawal of arterial blood, and a tracheostomy was performed to allow mechanical ventilation of the animal. After tracheostomy, the common carotid arteries were exposed and surgical suture was looped loosely around each of them to allow for later manipulation. As described previously (Fujii et al, 1991; Sobey and Faraci, 1999), a craniotomy was then performed over the ventral brain stem to expose the basilar artery, and this cranial window was superfused with artificial cerebrospinal fluid (CSF; composition in mmol/L: 2.95 KCl, 132 NaCl, 3.69 dextrose, 1.7 CaCl2, 0.64 MgCl2, 23.2 NaHCO3) at 2 ml/min. After preparation of the cranial window, the clamps and sutures used to retract the muscles overlying the basioccipital bone were removed to minimize any potential disturbances to carotid blood flow. The PO2, PCO2 and pH of both the arterial blood and CSF were tested regularly, and kept between normal parameters (arterial blood: PO2 = 145 to 155 mm Hg; PCO2 = 37 to 38 mm Hg; pH = 7.35 to 7.45. CSF: PO2 = 115 to 125 mm Hg; PCO2 = 37 to 38 mm Hg; pH = 7.35 to 7.45). Basilar artery diameter was continuously monitored using a microscope coupled to a computer-based tracking program (Diamtrak, Montech, Australia).
In vivo Protocol
Before commencing the experiment the preparation was allowed to stabilize over a 20 min period. Changes in basilar artery diameter were then recorded, as described in detail previously (Fujii et al, 1991), after blood flow through the vessel was increased by bilateral carotid artery occlusion (BCO). Once a stable plateau was reached, the occlusion was removed with care being taken to ensure that the carotid arteries returned to their resting position, thus restoring normal blood flow. Despite the deep barbiturate-induced anesthesia, in some rats BCO caused a small increase in systemic arterial pressure, which was quickly counteracted by the withdrawal of arterial blood from a femoral artery cannula. In some experiments, responses of the basilar artery to the vasodilator agonists acetylcholine (1 and 10 μmol/L), sodium nitroprusside (10 and 100 nmol/L) and H2O2 (100 and 300 μmol/L) were also examined. In all cases, a 20 min period was allowed before a subsequent vasodilator challenge.
Vasodilator responses to BCO, acetylcholine, sodium nitroprusside and H2O2 were examined in the absence and presence of diphenyleneiodonium (DPI; 0.5 and 5 μmol/L), NG-nitro-
Detection of O•−2 by Dihydroethidium
A further 10 rats were anesthetized and surgically prepared as described above except no cranial window was performed. Dihydroethidium (DHE; 10 mmol/L) was then dissolved in DMSO, and 0.15 mL was further diluted in 1 mL saline and injected intravenously (estimated plasma concentrations: DMSO ⩽ 0.5%; DHE ⩽ 50 μmol/L). After a further 30 mins, either a BCO (high flow) or a sham BCO (control) was performed for 10 mins on a pair of rats on five separate experimental days. The rats were then killed by anesthetic overdose, and frozen sections (16 μm) of the basilar arteries were prepared. Endothelial O•−2 production was then localized and measured. In the presence of O•−2, DHE is oxidized to ethidium and oxyethidium, which intercalate between DNA strands. Fluorescent images of arterial sections were acquired as 8-bit (256 intensity levels) and were analyzed with Image-Pro Plus software (version 5.0.1.11, Media Cypernetics. Inc.). At eight randomly selected locations on the endothelium of each arterial section, fluorescent density was measured. Data from pairs of rats were analyzed by paired t-test.
Measurement of O•−2 Production by Basilar Artery Homogenates
Basilar arteries obtained from rats (n = 28) were homogenized in 70 μL of lysis buffer (composition in mmol/L: 100 NaCl, 10 Tris HCl, 2 EDTANa2, 10 NaF, 10 β-glycerophosphate and 0.2 mg/mL benzamidine · HCl) with protease inhibitors (Roche Complete Mini, #1836153) using 0.2 mL glass homogenizers. O•−2 production was measured using lucigenin-enhanced chemiluminescence. Assay solutions consisting of 5 μmol/L lucigenin and one or more of diethyldithiocarbamate (DETCA; 3 mmol/L; to inactivate Cu2+-containing superoxide dismutases), NADPH (100 μmol/L; substrate for NADPH-oxidase), DPI (0.5 and 5 μmol/L), wortmannin (1 μmol/L), and
Drugs
H2O2 was purchased from Merck, DHE from Molecular Probes, and all other drugs were purchased from Sigma. DPI and wortmannin were prepared at 10 and 2 mmol/L respectively in DMSO, and diluted in either saline (in vivo experiments) or Krebs-HEPES buffer (in vitro experiments) such that the final concentration of DMSO applied topically to the basilar artery was ⩽ 0.05%. All other drugs were dissolved and diluted in either saline or Krebs-HEPES buffer as appropriate.
Data Analysis
All results are expressed as mean ± s.e.m. Statistical comparisons were made using Student's t-test or ANOVA (with Student-Neuman-Keuls for post hoc tests) as appropriate, with P < 0.05 being considered significant.
Results
Flow-Induced Dilatation of the Basilar Artery in Response to Bilateral Carotid Occlusion
Occlusion of the common carotid arteries to increase flow through the basilar artery caused dilatation of the basilar artery in vivo (Figure 1A). The delay between BCO and vasodilatation was relatively short (within 30 secs of carotid artery occlusion). The response was also rapidly reversible on removal of the occlusion and restoration of normal cerebral blood flow (Figure 1A). In some animals (~50%), BCO elicited a transient peak response that was followed by a sustained steady-state dilatation. As this peak vasodilator response was not present in all animals, we focused only on the steady-state responses (which were reproducible within the same animal, n = 46; Figure 1B). Overall, mean basilar artery baseline diameter was 254 ± 5 μm, and maximum diameter was 364 ± 5 μm (n = 46).
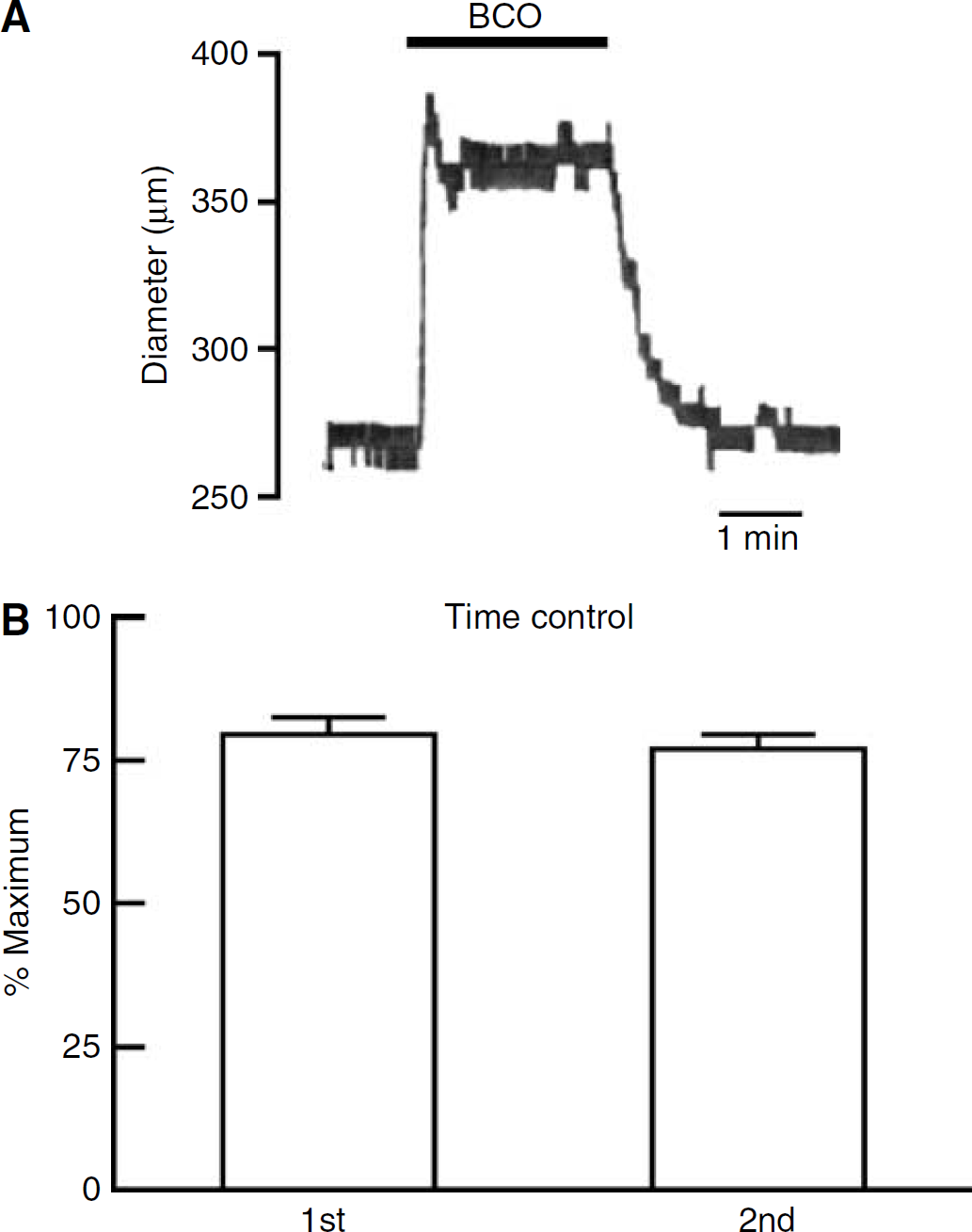
Change in basilar artery diameter in response to bilateral carotid occlusion (BCO). (
Flow-Induced Dilatation of the Basilar Artery is Mediated by NADPH-Oxidase-Derived Reactive Oxygen Species and Nitric Oxide
Treatment of the cranial window with the NOS inhibitor
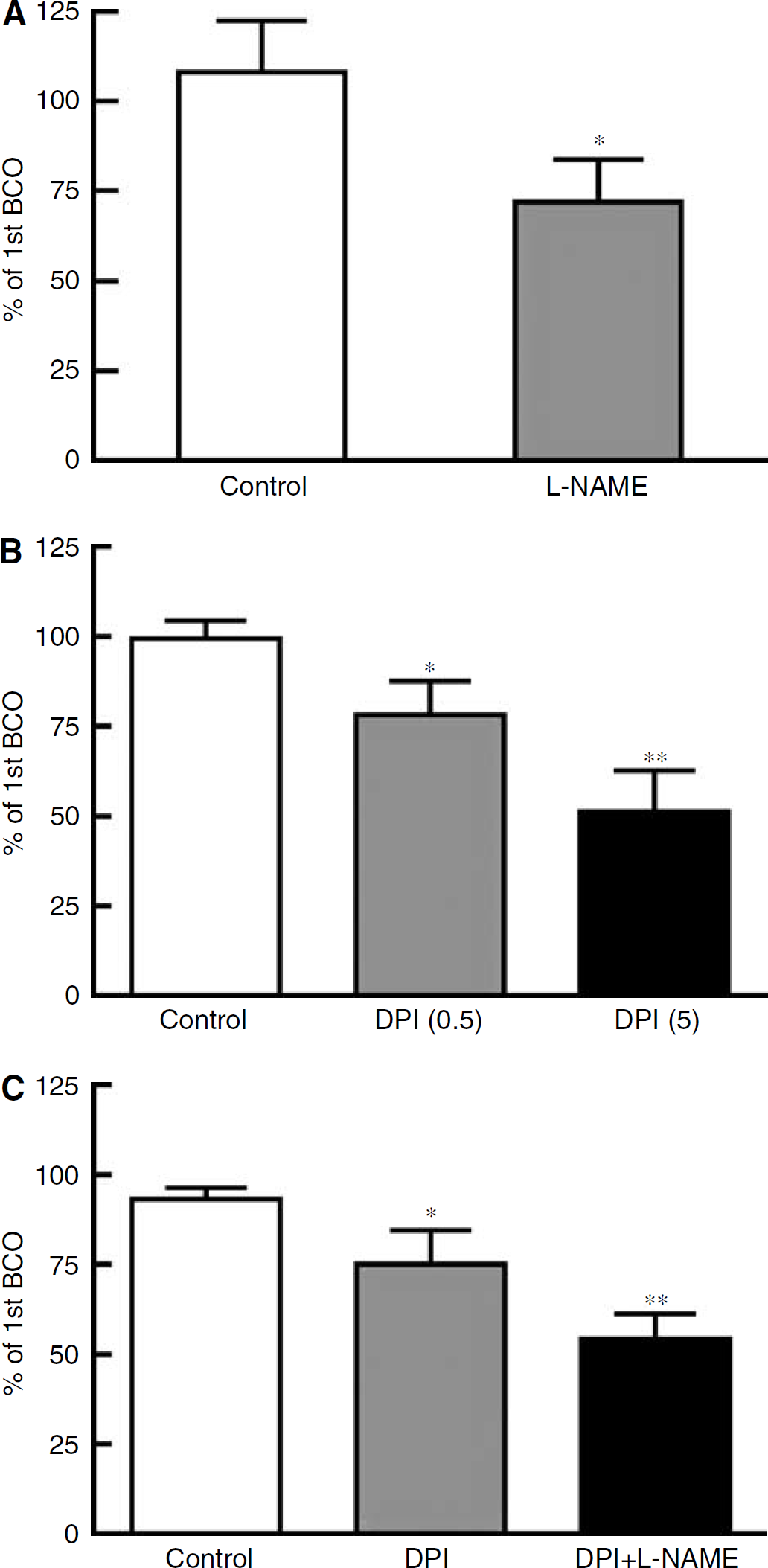
Effect of treatment with (
Effect of treatment with DPI (0.5 μmol/L) or DPI plus
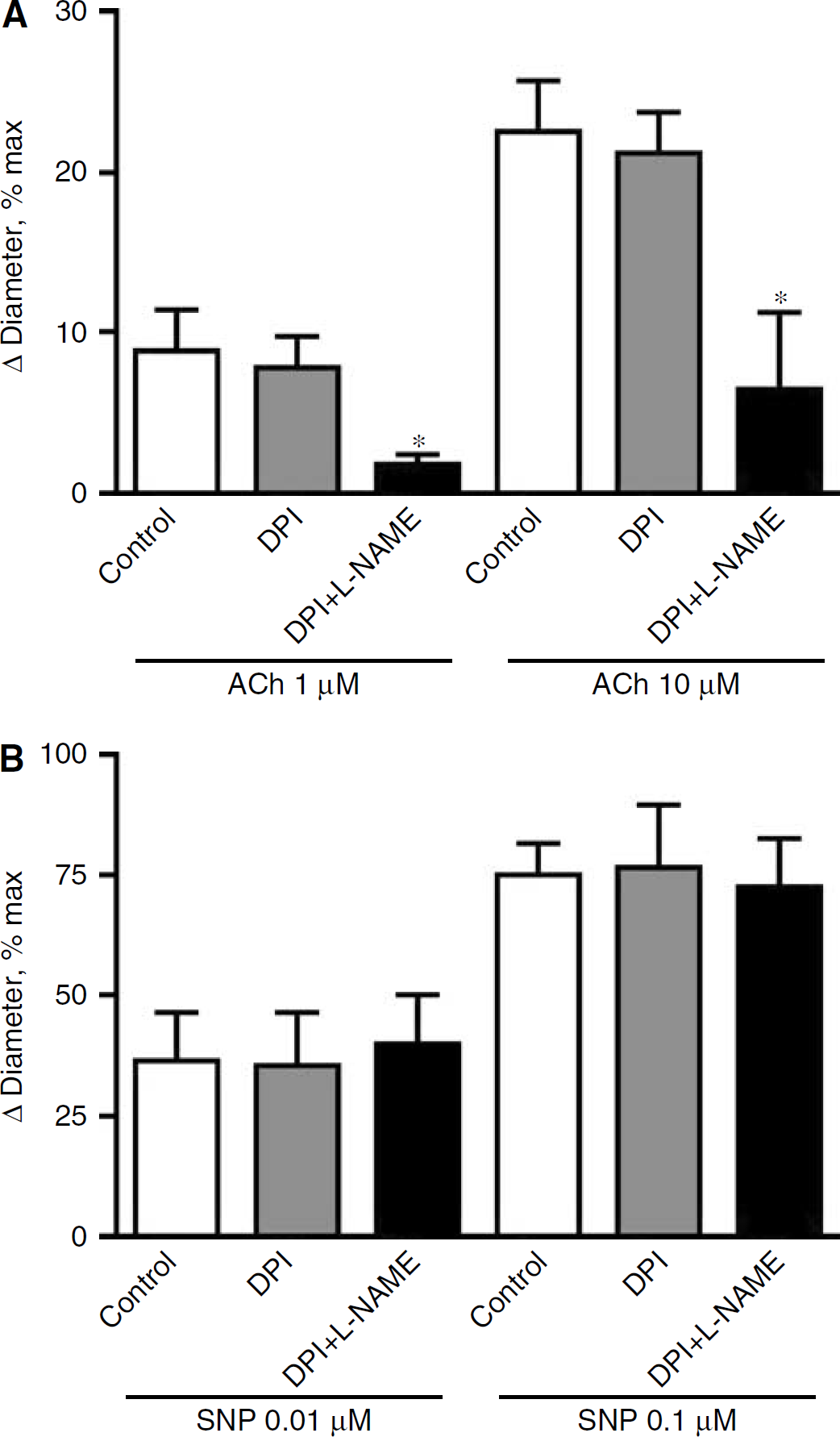
Treatment of the cranial window with DPI (0.5 and 5 μmol/L) had no significant effect on basilar artery diameter, but inhibited flow-induced dilatation by up to ~50% in a concentration-dependent manner (Figure 2B; n = 8 to 16). DPI (0.5 μmol/L) did not alter responses to the NOS-dependent vasodilator acetylcholine (Figure 3A; n = 6), similar to a lack of inhibition by 5 μmol/L DPI found previously (Paravicini et al, 2004), indicating that at these concentrations DPI does not inhibit NOS activity in the basilar artery. Interestingly, combined treatment with
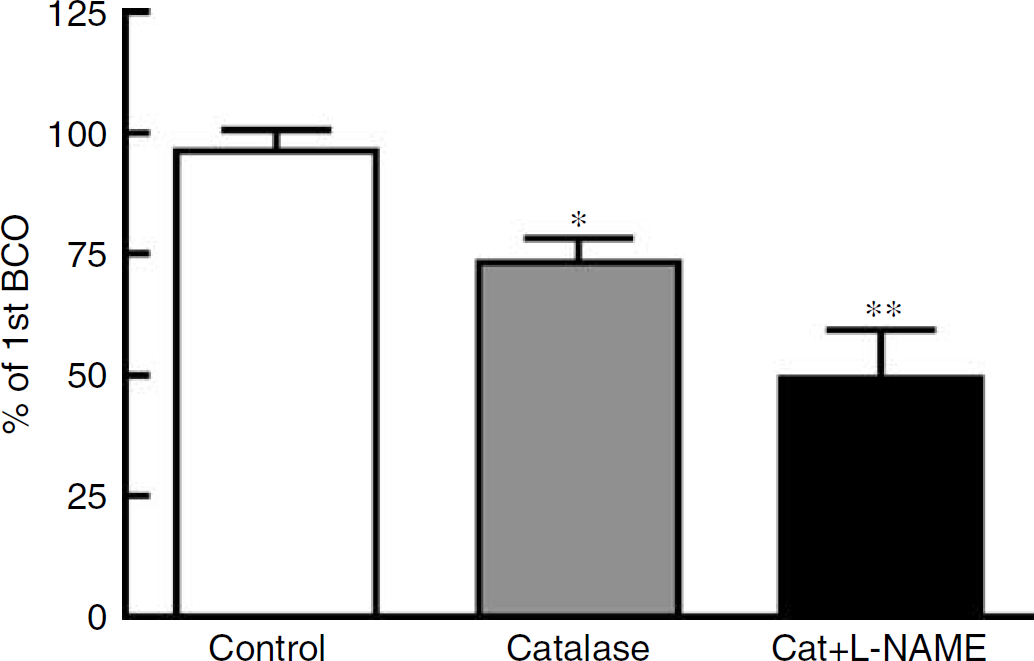
Effect of treatment with catalase alone (1000 U/mL; n = 7) or catalase plus
Flow-Induced Dilatation Involves Activation of Phosphatidylinositol-3 kinase
Given that PI3-K has been implicated as a mediator of mechanotransduction, we tested for a role of PI3-K in the signalling pathway for flow-induced cerebral vasodilatation. Topical treatment of the cranial window with the PI3-K inhibitor, wortmannin (1 μmol/L), had no effect on baseline diameter, but inhibited by ~60% the flow-induced dilator responses of the basilar artery (Figure 5A, n = 15). Combined treatment with wortmannin and
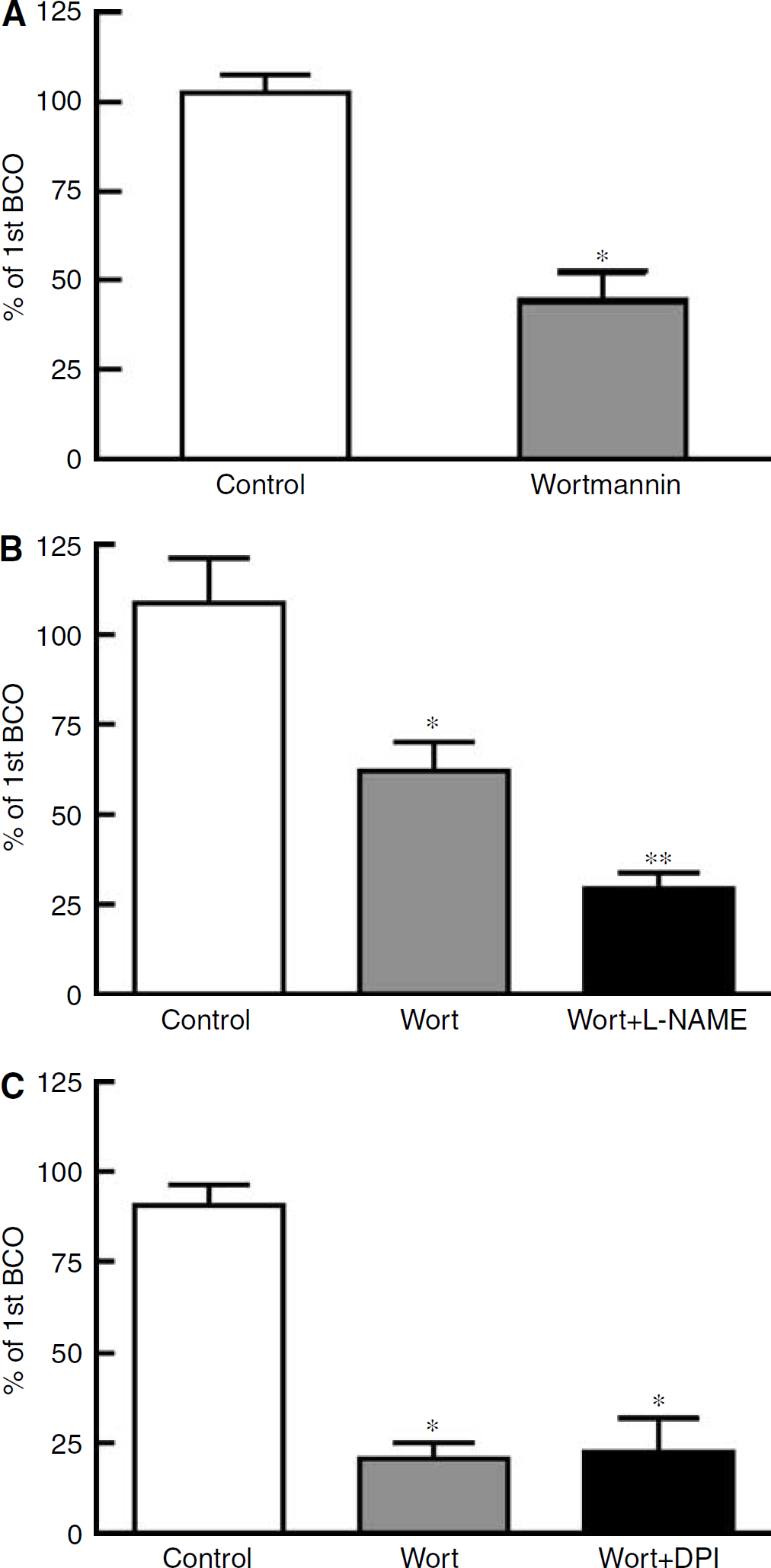
Effect of treatment with (
Effect of Inhibitor Drugs on Superoxide Production
DPI (0.5 and 5 μmol/L) attenuated NADPH-stimulated O•−2 production from basilar artery homogenates by ~45% and ~80%, respectively (Figure 6; n = 6). O•−2 production was abolished by the O•−2 scavenger tiron (1 mmol/L, Figure 6; n = 6). Neither
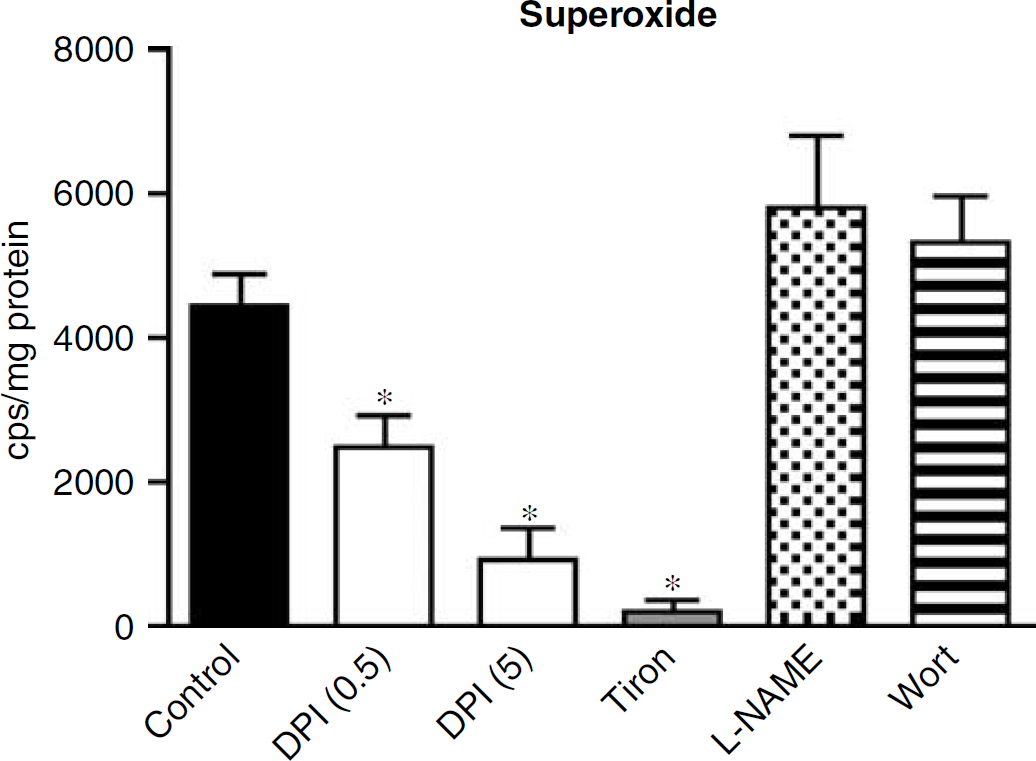
Effect of DPI (0.5 and 5 μmol/L), tiron 10 mmol/L),
Flow-Induced Dilatation Involves Endothelial O•−2 Production
O•−2 production in vivo was localized and measured in sections of basilar arteries after intravenous administration of DHE. Ethidium/oxyethidium fluorescence was observed in endothelial cells of all basilar arteries, but mean intensity was 18 to 125% higher in basilar arteries from rats subjected to high basilar artery blood flow during BCO (n = 5) relative to the specific control rats (n = 5) (Figure 7). Overall, fluorescence intensity was 63 ± 17% higher in endothelium of basilar arteries subjected to high flow in vivo versus paired controls (P < 0.05, Figure 7C).
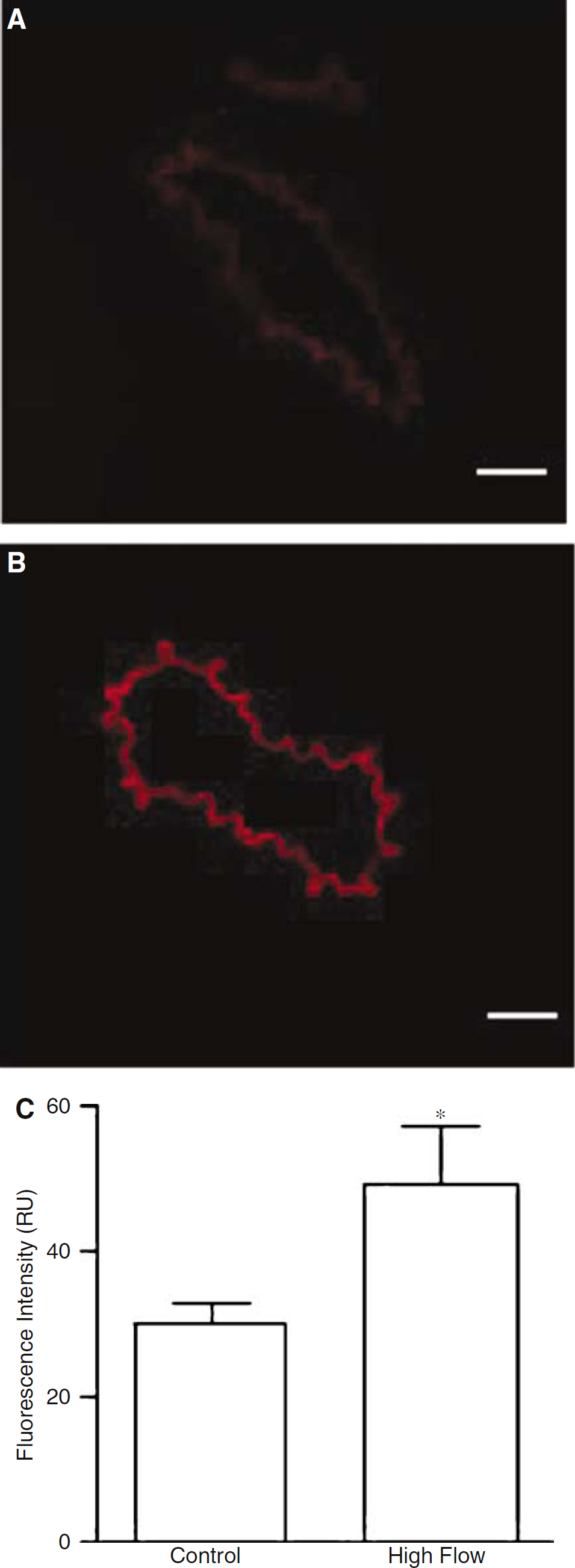
In situ detection of O−2 production by ethidium/oxyethidium fluorescence in endothelium of sections of basilar artery from a control rat (
Discussion
Increased blood flow is a powerful vasodilator stimulus in the cerebral circulation. This study demonstrates that there are multiple mechanisms responsible for flow-induced dilatation of the basilar artery in vivo. One pathway involves the activation of NADPH-oxidase and the subsequent production of superoxide and the vasodilator H2O2. Another pathway involves the activation of NOS and generation of NO. Importantly, activation of PI3-K is a key factor in much of the flow-induced vasodilator response, and may be directly involved in the activation of NADPH-oxidase by increased intraluminal flow. To our knowledge, this is the first study to identify a role for PI3-K or NADPH-oxidase in any flow-induced vasodilator response in vivo.
It is now well established that NADPH-oxidase is a major source of ROS in blood vessels. This enzyme is expressed in cerebral arteries (Didion and Faraci, 2002; Paravicini et al, 2004), and its activation leads to cerebral vasodilatation and increased cerebral blood flow (Paravicini et al, 2004; Park et al, 2004). In cerebral arteries, NADPH-oxidase activity appears to cause vasodilatation by generating O•−2, which is converted by superoxide dismutase to the powerful cerebral vasodilator molecule H2O2 (Paravicini et al, 2004). We have preliminary evidence that NADPH-oxidase activity and function is up to 100-fold higher in cerebral versus systemic arteries (Miller et al, 2005), leading us to consider whether this enzyme might play an important physiologic role in the regulation of cerebrovascular tone. Until now, however, the endogenous stimuli for cerebrovascular NADPH-oxidase activation have not been identified.
In this study, the flavin antagonist and NADPH-oxidase inhibitor, DPI, reduced flow-induced vasodilatation in vivo in a concentration-dependent manner. Further, in vitro studies confirmed that these concentrations of DPI profoundly inhibit NADPH-dependent O•−2 production by the basilar artery. A criticism of the use of DPI as an NADPH-oxidase inhibitor is that its mechanism of action may also lead to inhibition of electron flow through other enzymes, particularly NOS (Wang et al, 1993). However, this seems unlikely in this and in our previous study (Paravicini et al, 2004) as up to 5 μmol/L DPI had no effect on dilator responses of the basilar artery to the NOS-dependent vasodilator acetylcholine. We (and others) have previously found acetylcholine-induced dilatation of the rat basilar artery in vivo to be predominantly, if not exclusively, NO-mediated based on the virtually complete attenuation of the response by NOS inhibitors (Faraci, 1990; Paravicini et al, 2004; Sobey and Cocks, 1998).
We report here evidence that flow-dependent dilatation of the basilar artery in vivo is associated with increased generation of O•−2 in the endothelium. Intraluminal flow was first shown to alter vascular ROS production in a study of rabbit aorta, in which ROS (most likely O•−2) were produced by the endothelium (Laurindo et al, 1994). Although that study did not investigate the molecular source of vascular O•−2, generation of O•−2 by cultured endothelial cells exposed to shear stress is reportedly due to activation of NADPH-oxidase (De Keulenaer et al, 1998). Sustained increases in endothelial cell shear stress also cause alterations in expression of membrane-bound subunits of NADPH-oxidase (Hwang et al, 2003; Silacci et al, 2001); however, until now there have been no studies reporting a functional role of such a response in regulation of vascular tone. Recently, H2O2 generated via mitochondrial respiration, and not NADPH-oxidase, was reported to mediate flow-induced dilatation of isolated cannulated coronary arteries (Liu et al, 2003; Miura et al, 2003), although this did involve activation of KCa channels, as does H2O2-induced cerebral vasodilatation (Iida and Katusic, 2000; Paravicini et al, 2004; Sobey et al, 1997).
Flow-induced vasodilatation has been extensively studied in peripheral arteries, where NO is the major mediator (Davies, 1995). However, there have been very few studies of this phenomenon in cerebral vessels, where the mechanism is clearly more complex. Evidence for both flow-induced dilatation and constriction has been reported in isolated cerebral vessels, and appears to be both species- and vessel-dependent. For example, increases in shear stress and flow are reported to elicit constriction of middle cerebral arteries isolated from cats and rats (Bryan et al, 2001; Madden and Christman, 1999). In contrast, in vivo studies in rats have only demonstrated flow-induced dilatation (not constriction) to occur in the cerebral circulation (Fujii et al, 1991; Fujii et al, 1992). Using the same experimental preparation as Fujii et al (1991), we now report profound flow-dependent dilatation of the rat basilar artery in vivo, and moreover, we have identified key molecular contributors that transduce this response. A difference between the present and previous findings concerning the mechanism(s) of flow-induced dilatation of the basilar artery relates to the role of NO derived from the activation of NOS. Fujii et al (1991) concluded that NO was not involved in the flow-induced dilator response because it was not attenuated by 5 μmol/L NG-monomethyl-
Our data also provide strong support for a role of PI3-K in flow-induced cerebral vasodilatation. We found that the PI3-K inhibitor, wortmannin, markedly reduced flow-induced vasodilatation in vivo. Although a number of studies have identified a role for PI3-K in shear stress-related signalling pathways, to our knowledge this is the first demonstration of a functional role for the activation of PI3-K and regulation of vascular tone in response to shear stress. In cultured vascular cells, inhibition of PI3-K attenuates angiotensin II-induced ROS production by NADPH-oxidase (Seshiah et al, 2002), and activation of PI3-K is linked to the downstream activation of Rac, a small G protein crucial for activation of NADPH-oxidase (Welch et al, 2003). Our data also suggest that PI3-K is upstream of H2O2 in the signalling pathway following increased intraluminal flow, because wortmannin had no inhibitory effect on vasodilator responses to exogenous H2O2. Thus, together these findings suggest that PI3-K is responsive to shear stress caused by increased blood flow, resulting in NADPH-oxidase activity, H2O2 generation, and cerebral vasodilatation in vivo. Hence, it seems likely that this mechanism is of physiologic importance.
The degree of inhibition of flow-induced dilatation by wortmannin was greater than that seen with either DPI or
The lack of any isoform-selective pharmacological inhibitors of NADPH-oxidase makes it difficult to determine which isoform of NADPH-oxidase is involved in the DPI-sensitive part of the flow-induced vasodilator response. However, we have previously shown that Nox4 is expressed at profoundly higher levels than either Nox1 or Nox2 in the basilar artery under physiologic conditions (Paravicini et al, 2004). Moreover, both O•−2 production and vasodilatation to NADPH are augmented in association with elevated Nox4 levels in the basilar artery of hypertensive rats (Paravicini et al, 2004). Hence, it is conceivable that a Nox4-containing isoform of NADPH-oxidase in this artery is also responsible for ROS production and vasodilatation in response to increased flow. Furthermore, Nox4 is localized with p22phox in focal adhesions, which are rich in mechanosensitive proteins such as integrins (Hilenski et al, 2004), and a significant proportion of the NADPH-oxidase complex is reported to exist in a preassembled form associated with the cytoskeleton of endothelial cells (Li and Shah, 2002). Moreover, because Nox4 has been identified as the major NADPH-oxidase catalytic subunit expressed in endothelial cells (Ago et al, 2004), an endothelial Nox4 involvement in the mechanical activation of ROS production seems plausible.
The time course of vasodilatation in response to BCO might be expected to match that of H2O2 if the latter molecule in fact contributes significantly to the response. The time course of the basilar artery dilator response to topically (i.e. abluminally) applied exogenous H2O2 was dependent on the concentration being tested. At 300 μmol/L, H2O2 elicits an increase in diameter in < 1 min, whereas the effect of 100 μmol/L may take 2 to 3 mins to affect artery diameter. The time to reach a full steady-state change in response to exogenous H2O2 may then still take ~5 further min. Thus, the time course of vasodilatation in response to BCO is a little faster than responses purely to topically applied exogenous H2O2. However, it is important to note that our data suggest that H2O2 probably plays only a partial (perhaps minor) role in the flow-dependent vasodilator response, with NO also playing a significant role. Indeed, endogenous NO that is generated in response to acetylcholine causes rapid dilatation of the rat basilar artery in vivo (Faraci, 1990). Moreover, it is noteworthy that in rat cerebral arterioles in vivo, powerful dilator responses that are known to be mediated entirely by endogenous H2O2 are typically complete within 3 mins (Sobey et al, 1997). Importantly, the spatial and temporal distribution of exogenous H2O2, and the signalling pathways consequently activated, may differ substantially from those related to endogenous H2O2 generated by shear-induced activation of the endothelium. Thus, due to the mechanistic complexity of the flow-induced dilator response of the basilar artery, it is difficult to meaningfully correlate its temporal profile with that of the response to exogenous H2O2.
In summary, we have demonstrated that flow-induced vasodilatation in the cerebral circulation in vivo occurs via multiple mechanisms, including H2O2 and NO, generated by activation of NADPH-oxidase and eNOS, respectively. Furthermore, activation of PI3-K appears to play a key role in this response to increased blood flow, especially in the activation of NADPH-oxidase.
Conflict of Interest
CGS and GRD have potential conflicts of interest in that they are consultants for, and have significant ownership interests in, Radical Biotechnology Pty Ltd of Australia.