
Abstract
Effects of insulin on cerebral arteries have never been examined. Therefore, we determined cerebrovascular actions of insulin in rats. Both PCR and immunoblot studies identified insulin receptor expression in cerebral arteries and in cultured cerebral microvascular endothelial cells (CMVECs). Diameter measurements (% change) of isolated rat cerebral arteries showed a biphasic dose response to insulin with an initial vasoconstriction at 0.1 ng/mL (−9.7%±1.6%), followed by vasodilation at 1 to 100 ng/mL (31.9%±1.4%). Insulin also increased cortical blood flow
Keywords
Introduction
Insulin, an important metabolic hormone, has multiple targets in the body including cells comprising the vasculature. Previous studies have shown that insulin exerts potent dilator (Chen and Messina, 1996; Hasdai et al, 1998b; Katakam et al, 2005b; McKay and Hester, 1996; Oltman et al, 2000, 2006; Scherrer et al, 1994; van Veen and Chang, 1997) and constrictor (Eringa et al, 2002; Hasdai et al, 1998a; Miller et al, 2002; Schroeder et al, 1999; van Veen and Chang, 1998) effects on resistance vessels, with the final, integrated vascular response depending on factors, such as dosage, regional circulation, vessel size, endothelial status, and physiologic condition (Mather et al, 2001). Surprisingly, the effects of insulin on the vasoreactivity of cerebral arteries or cerebral blood flow have never been examined directly.
Cerebral microvessels have been shown to bind insulin and insulin-like growth factor (Frank and Pardridge, 1981; Haskell et al, 1985); however, neither insulin receptor protein nor mRNA has been identified in cerebral arteries. Given the sensitivity of the peripheral vasculature to insulin, it seems likely, but untested, that the cerebral vasculature would respond to increases in circulating levels of insulin. Although insulin is not necessary for the uptake of glucose into the brain, insulin binding has been shown in cerebral arteries (Frank and Pardridge, 1981; Haskell et al, 1985). Transient meal-associated elevations in blood insulin levels occur throughout the day in normal people (Polonsky et al, 1988), and even more dramatic increases in blood insulin concentrations occur after meals in obese individuals and after therapeutic injections in patients with diabetes. Furthermore, sustained hyperinsulinemia is present for several months and years in insulin-resistant (IR) individuals, and IR is associated with cerebrovascular dysfunctions (Erdos et al, 2004; Phillips et al, 2005), enhanced stroke injury (Matsumoto et al, 1999), and Alzheimer's disease (Moloney et al, 2008; Razay et al, 2007). Thus, the cerebral resistance blood vessels are subjected to elevated insulin levels in the normal and disease conditions, but vasoactive effects of insulin have not been studied.
In this study, we documented the presence of insulin receptors and determined the vascular actions of insulin in isolated rat cerebral arteries. In addition, we determined the changes in cortical blood flow (CoBF) in response to insulin in anesthetized rats. We also evaluated the function of both the endothelium- and the vascular smooth muscle-dependent mechanisms, such as cyclooxygenase (COX), cytochrome P450 monooxygenase (CP450), nitric oxide synthase (NOS), reactive oxygen species (ROS), various types of K+ channels, and endothelin receptors. Finally, we measured intracellular calcium dynamics, NO synthesis, and ROS production in cultured rat cerebral microvascular endothelial cells (CMVECs).
Materials and methods
The animal protocol was approved by the Institutional Animal Care and Use Committee of Wake Forest University Health Sciences. All experiments complied with the National Institutes of Health
Vascular Reactivity
Videomicroscopy and diameter studies of isolated cerebral arteries were carried out as described previously (Katakam et al, 2009). Briefly, after a 4-h fast, the rats were anesthetized with isofluorane and were decapitated. The brains were removed and placed in an ice-cold oxygenated physiologic saline solution (PSS). Middle and posterior cerebral arteries (140 to 180
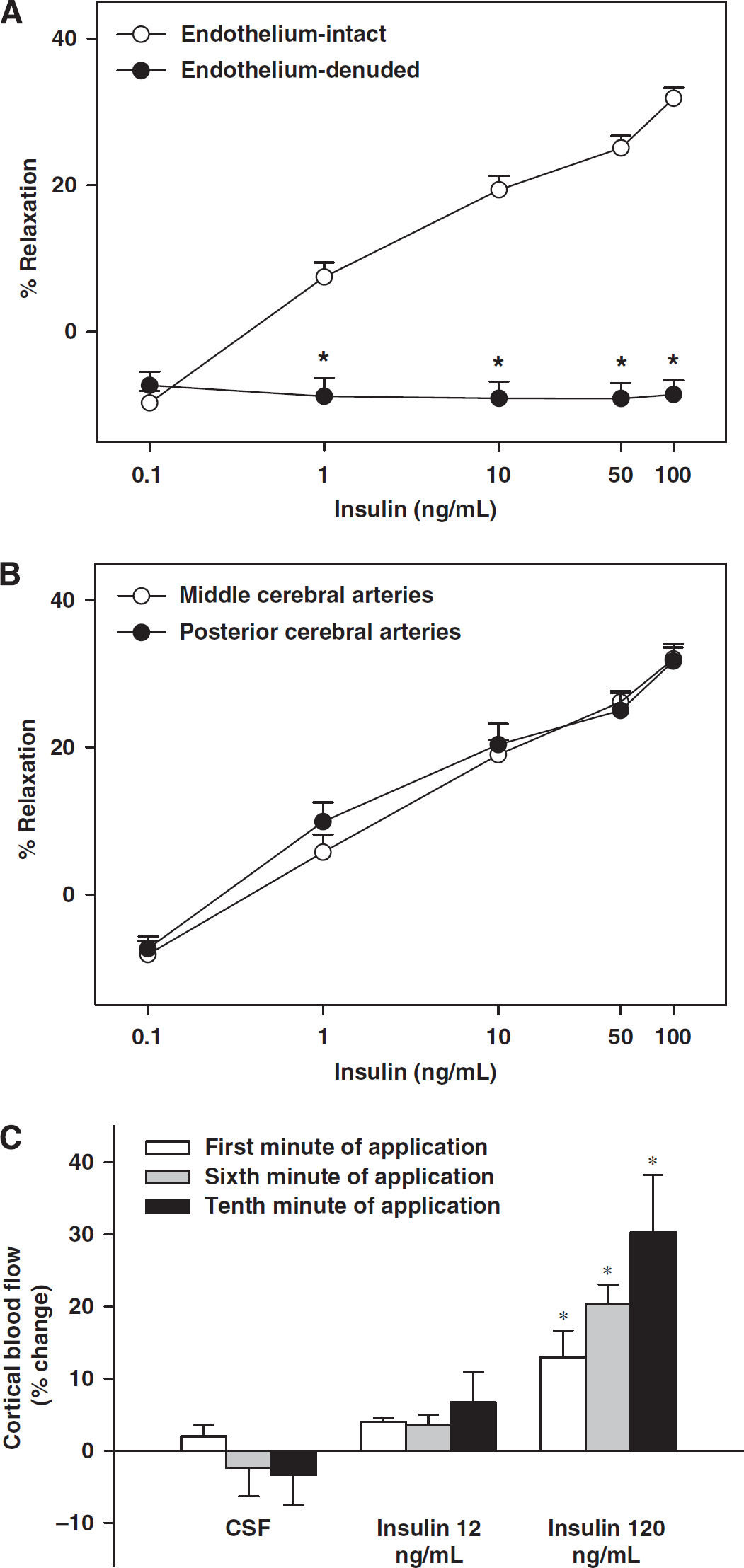
Effects of insulin on the diameter of rat cerebral arteries and cortical blood flow. Vascular responses to insulin in grouped rat cerebral arteries with intact endothelium and endothelium denuded are shown in (
Vascular responses to insulin (0.1, 1, 10, 50, and 100 ng/mL; Humulin R, Eli Lilly company, Indianapolis, IN, USA) were determined in the presence and absence of indomethacin (nonselective inhibitor of COX, 10
Cortical Blood Flow Measurements
Rats were anesthetized by induction with pentobarbital (100 mg/kg, intraperitoneally) and maintained by intermittent intravenous administration of pentobarbital (25 mg/kg), as required. An adequate level of anesthesia was assessed by testing motor and hemodynamic responses to paw pinch. Rats were intubated through tracheotomy and were mechanically ventilated with room air supplemented with oxygen (SAR-830 Ventilator, CWE, Ardmore, PA, USA). Ventilation parameters were adjusted to maintain normal end-tidal CO2 levels (∼40 mm Hg) continuously monitored using a capnometer (Micro-Capnometer, Columbus Instruments, Columbus, OH, USA). Body temperature was maintained at ∼37°C using a heated mat and was monitored by rectal probe. The right femoral artery and vein were cannulated to measure arterial blood pressure and to administer drugs and fluids, respectively. The heads of the animals were placed in a stereotaxic frame and a circular craniotomy (4 mm in diameter, with the center of the window 3 mm lateral and 4 mm caudal to the bregma) was made over the right parietal cortex. The dura was carefully removed and a 2-mm high rim was created around the craniotomy using bone wax and superglue. Tubes were inserted into the rim as input and output ports to enable drug administration and flushing of the brain surface, respectively. Cortical blood flow was measured using a laser Doppler flowmeter (Perimed, Järfälla, Sweden) mounted on a micromanipulator and positioned on the brain surface. The window was sealed with Parafilm (American National Can, Greenwich, CT, USA), and filled with warm (∼37°C) artificial cerebrospinal fluid (aCSF) containing (in mmol/L) the following: 2.95 KCl, 132 NaCl, 3.69 dextrose, 1.7 CaCl2, 0.64 MgCl2, and 23.2 NaHCO3, and was bubbled with 5% CO2 in N2.
The exposed brain surface was superfused with aCSF at 0.5 mL/min for 10 mins, followed by the superfusion of insulin (Humulin R, 12 and 120 ng/mL in aCSF) for 10 mins each. Cortical blood flow, blood pressure, and end-tidal pCO2 were recorded continuously. The venous blood glucose level was also checked before and after insulin application. At the end of the experiments, the animals were euthanized by an intravenous injection of saturated KCl solution after which the biologic zero was determined. Relative changes in CoBF were calculated as a percentage of the average flow during 1 min before the drug/aCSF application. Average flow changes during each minute were used for further statistical analysis.
Endothelial Cell Cultures
To assess the ability of insulin to induce elevation of cytosolic calcium and to increase the production of NO and ROS, we determined calcium, NO, and ROS levels in primary rat CMVECs. Two-week-old Sprague–Dawley rats were decapitated under deep anesthesia, and the brain cortices were freed from meninges, homogenized, and digested. The homogenate was redistributed in 20% bovine serum albumin and was centrifuged at 1,000
Insulin and Akt Pathway
Isolated cerebral arteries and CMVECs were treated with and without insulin (100 ng/mL) in low-glucose DMEM medium at 37°C for 15 mins. Subsequently, homogenates were prepared from cells to determine the total and phosphorylated Akt by western blotting.
Reverse Transcription-PCR
Total RNA was obtained from isolated cerebral arteries and CMVECs using the SV Total RNA Isolation System (Promega, Madison, WI, USA). Reverse transcription-PCR experiments were carried out in an Eppendorf Mastercycler thermocycler (Brinkmann Instruments, Westbury, NY, USA) as described previously (Katakam et al, 2005a). From each sample, 50 pg of total RNA was reverse transcribed and amplified using the Qiagen OneStep RT-PCR Kit (Qiagen, Valencia, CA, USA) with gene-specific primers targeting rat insulin receptor (sense primer: 5′-GCCATCCCGAAAGCGAAGATC-3′; anti-sense primer: 5′-TCTGGGTCCTGATTGCAT-3′; reference sequence NM_017071). Expected lengths of the RT-PCR product were 224 base pairs. We used 35 cycles for insulin receptor amplification. In control experiments, when RT was omitted, no amplification was observed.
Western Blot Analysis
Western blot analyses were carried out as described previously (Katakam et al, 2005a,2005b). Briefly, equal amounts of extracted protein from homogenates of cerebral arteries or CMVECs were separated by 4% to 20% SDS-PAGE and transferred into a nitrocellulose membrane (BioRad, Hercules, CA, USA). Membranes were blocked with 1% nonfat milk in Tris-buffered saline and 0.05% Tween 20 for 1 h at room temperature. Subsequently, the membranes were incubated overnight at 4°C with the primary antibodies for human insulin receptor-
Insulin and Glucose Assays
Blood samples were collected using needle and syringe from the left ventricle after exposure to isofluorane anesthesia before decapitation. Plasma insulin and glucose levels were measured using a rat insulin ELISA (enzyme-linked immunosorbent assay) (Crystal Chem, Chicago, IL, USA) and Trinder reagent (Sigma), respectively.
Drugs, Chemicals, and Solutions
All chemicals were purchased from Sigma except MnTBAP (Calbiochem, San Diego, CA, USA), TMRE, HEt, DAF-FM, Pluronic F-127 (Molecular Probes, Eugene, OR, USA), DMEM (Gibco BRL, Grand Island, NY, USA), fetal bovine plasma-derived serum (Animal Technologies, Tyler, TX, USA), and Percoll (Amersham Biosciences, Uppsala, Sweden). Stock solutions (10 mmol/L) of 7-NI and apocynin were prepared in dimethyl sulfoxide, and all the other chemicals were prepared in deionized water. The composition of PSS (expressed in mmol/L) was NaCl (112), KCl (4.8), NaHCO3 (26), KH2PO4·H2O (1.2), CaCl2 (1.8), MgSO4·7H2O (1.2), and glucose (10). Physiologic saline solution with 60 mmol/L of KCl was prepared by replacing NaCl with an equimolar quantity of KCl. Calcium-free PSS was prepared by replacing CaCl2 with an equimolar quantity of MgSO4·7H2O with 2 mmol/L of ethylene glycol tetraacetic acid. The cell culture medium consisted of DMEM supplemented with 20% fetal bovine plasma-derived serum, 2 mmol/L glutamine, 1 ng/mL basic fibrobast growth factor (bFGF), 50
Data Analysis and Statistics
All data are reported as mean±s.e.m. ‘
Results
Vascular Responses in Isolated Cerebral Arteries
The resting diameters of cerebral arteries were similar for each group of experiments, and they were preconstricted to a similar degree (Table 1). Insulin elicited a biphasic response in cerebral arteries with vasoconstriction at low concentrations (9.7%±1.6% at 0.1 ng/mL) and with vasodilation at high concentrations (31.9%±1.4%,
Changes in arterial diameter after administration of various agents

Diameter changes in isolated rat cerebral arteries. Maximal relaxation was calculated as a % decrease in preconstriction in response to the highest dose (100 ng/mL) of insulin. % Preconstriction was calculated by determining the decrease in diameter and expressing it as a percentage of passive diameter of the arteries. ‘% dilation’ and ‘% constriction’ were expressed as mean±s.e.m. Insulin response in endothelium-intact arteries in the absence of any drugs was designated as baseline response.
∗ Indicates significant difference with regard to baseline insulin response in endothelium-intact arteries (
∗∗Indicates significant difference with regard to insulin response in endothelium-denuded arteries (
Cortical Blood Flow Responses In Vivo
Superfusion of blood vessels with aCSF alone did not alter the CoBF. Topically applied insulin significantly increased the CoBF at the higher (3 mU/mL) concentration; however, the lower concentration (12 ng/mL) failed to evoke significant changes in blood flow (Figure 1C). During the administration of high-dose insulin (120 ng/mL), blood flow increased gradually and reached a plateau near the eighth minute of application. Cortical blood flow changes (% increase) in the 8 to 10 mins were significantly higher than in the first minute (26%±6%, 29%±7%, and 30%±8%, respectively versus 13%±3%;
Mechanisms of Cerebrovascular Actions
Denudation of endothelium abolished vasodilation; instead insulin elicited vasoconstriction at 1 to 100 ng/mL (Figure 1A, Table 1). Pretreatment with indomethacin not only abolished vasoconstriction (15.5.0%±5% at 0.1 ng/mL;
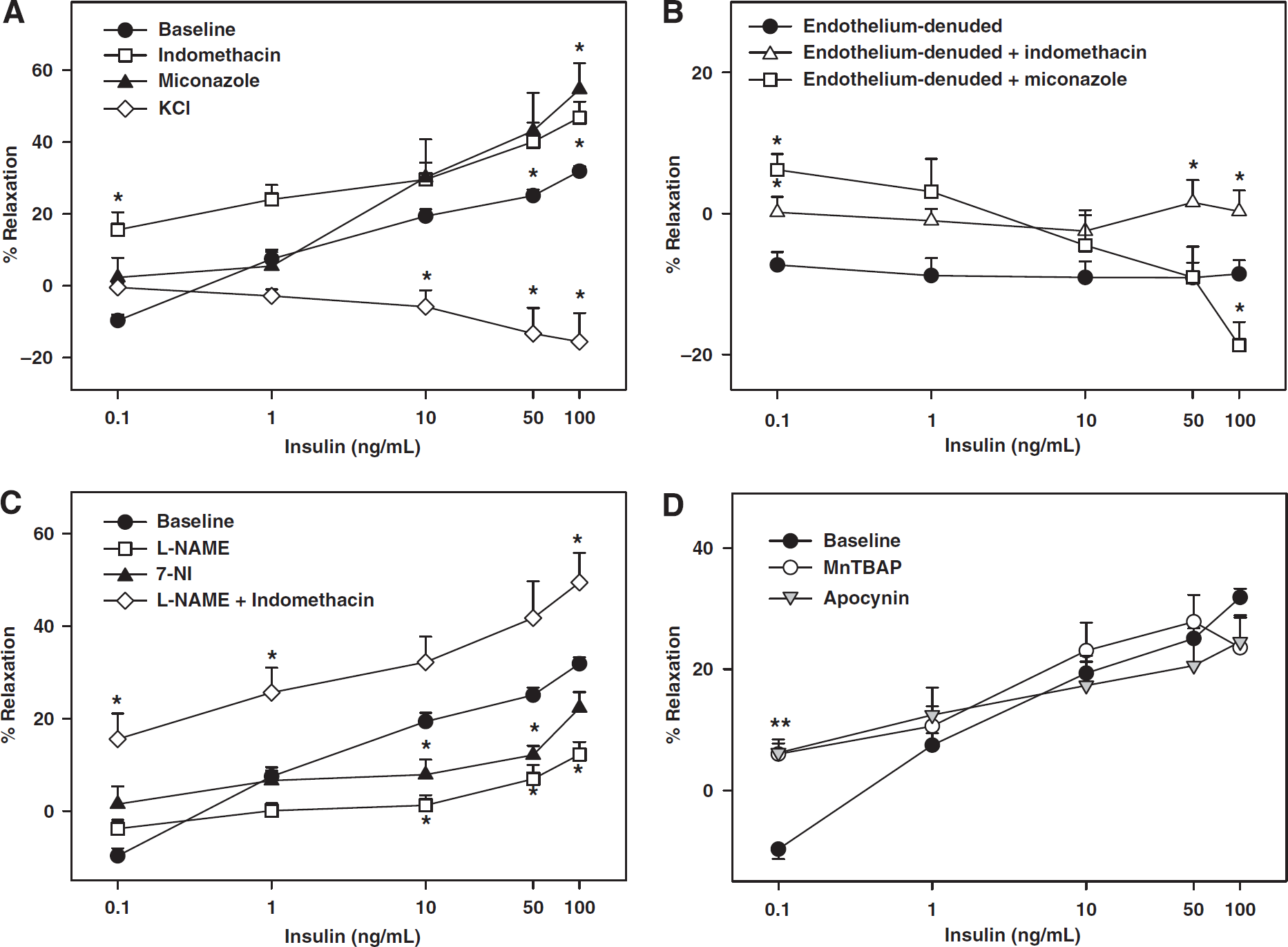
Role of NOS, COX, cytochrome P450, K+ channels, endothelium and ROS in cerebrovascular responses to insulin. Vascular responses to insulin in rat cerebral arteries with intact endothelium, in the presence and absence of indomethacin, miconazole, and KCl are shown in (
In contrast, L-NAME treatment resulted in greatly diminished maximal relaxation to insulin implicating NO in vasodilation (Figure 2C, Table 1). In addition, inhibition of nNOS with 7-NI also resulted in reduced vasodilation, suggesting that nNOS contributes part of the NO to vasodilation. Coapplication of indomethacin and L-NAME abolished vasoconstriction and increased insulin-induced vasodilation. This insulin response was identical to the insulin-induced vasodilation in the presence of indomethacin alone (Figure 2C, Table 1).
Inhibition of ROS generation with apocynin abolished vasoconstriction at 0.1 ng/mL and instead promoted vasodilation to insulin (6.0%±2% at 0.1 ng/mL;
Inhibition of K+ channels with KCl abolished vasodilation in response to insulin leading, instead, to a dose-dependent vasoconstriction (Figure 2A, Table 1). This finding suggests that K+ channels are the primary targets of vasodilation induced by insulin. Similarly, iberiotoxin abolished insulin-induced vasodilation and elicited dose-dependent vasoconstriction to insulin (Figure 3A, Table 1), suggesting that BKCa was the predominant K+ channel target of insulin-induced vasodilation. In contrast, pretreatment with apamin, TRAM-34, Barium, 4-AP, and glybenclamide resulted in diminished maximal dilation in response to insulin thereby implicating SKCa, IKCa, Kir, Kv, and KATP in vasodilation (Figures 3A and 3B, Table 1). BQ-123 abolished insulin-induced vasoconstriction (10.3%±6.2%,
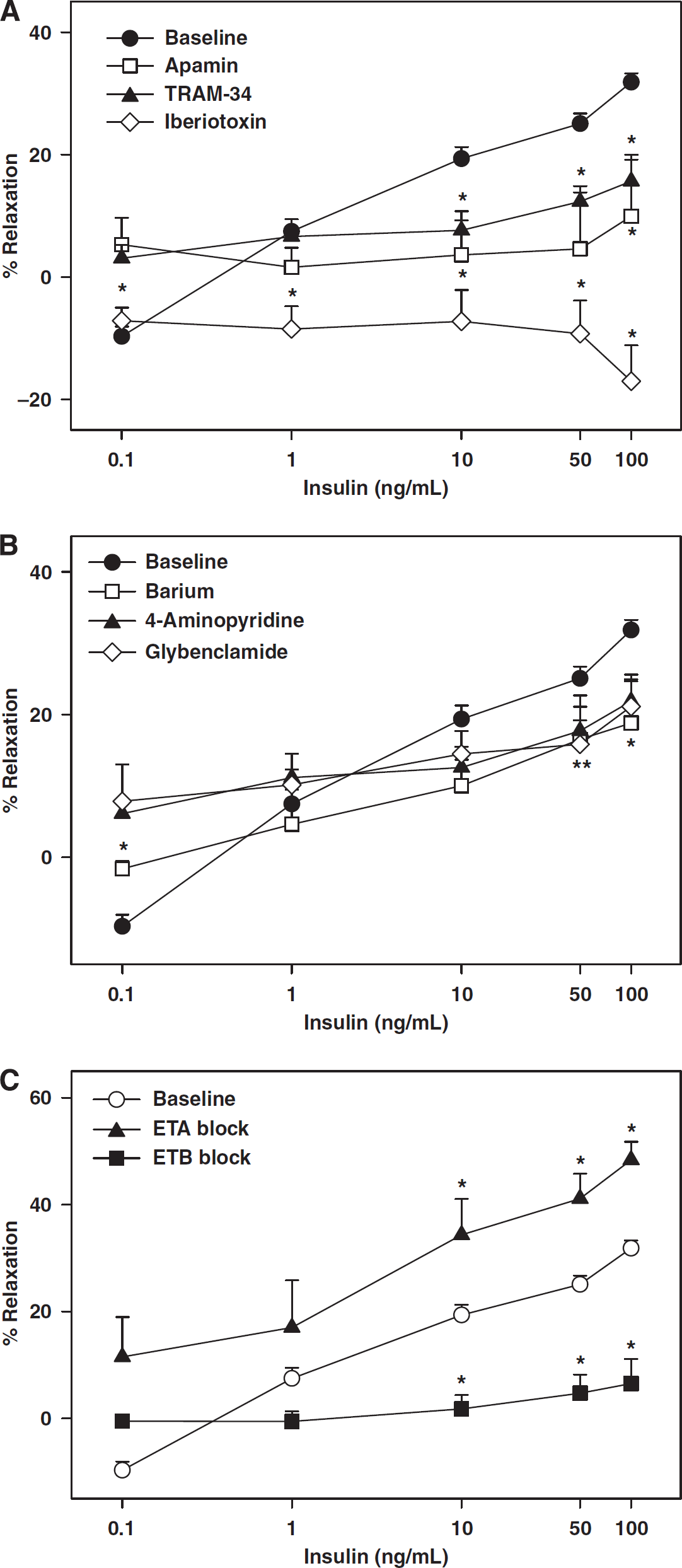
Contribution of K+ channels and endothelin receptors to cerebrovascular responses to insulin. Vascular responses to insulin in rat cerebral arteries in the presence and absence of iberiotoxin, apamin, and TRAM-34 are shown in (
Arteries took ∼20 to 30 mins to attain a stable myogenic tone and maintained a relatively stable diameter for more than 1 h. When compared with vehicle-treated arteries (1.9%±0.5%,
Calcium, Nitric Oxide, and Reactive Oxygen Species Measurements
Fluorescence measurements of Fluo-4 AM showed an increase in fluorescence in response to insulin, indicating an elevation of cytosolic calcium in CMVECs (Figures 4A and 4B). In addition, fluorescence measurements of DAF-FM in CMVECs showed increased fluorescence intensity in response to insulin compared with vehicle, indicating generation of NO. Pretreatment with L-NAME abolished enhanced DAF-FM fluorescence in response to insulin, confirming that DAF-FM fluorescence correlated with NOS-derived NO (Figures 4C and 4D). Measurements of HEt fluorescence showed that insulin increased HEt fluorescence compared with vehicle, indicating enhanced ROS production (Figures 5E and 5F).
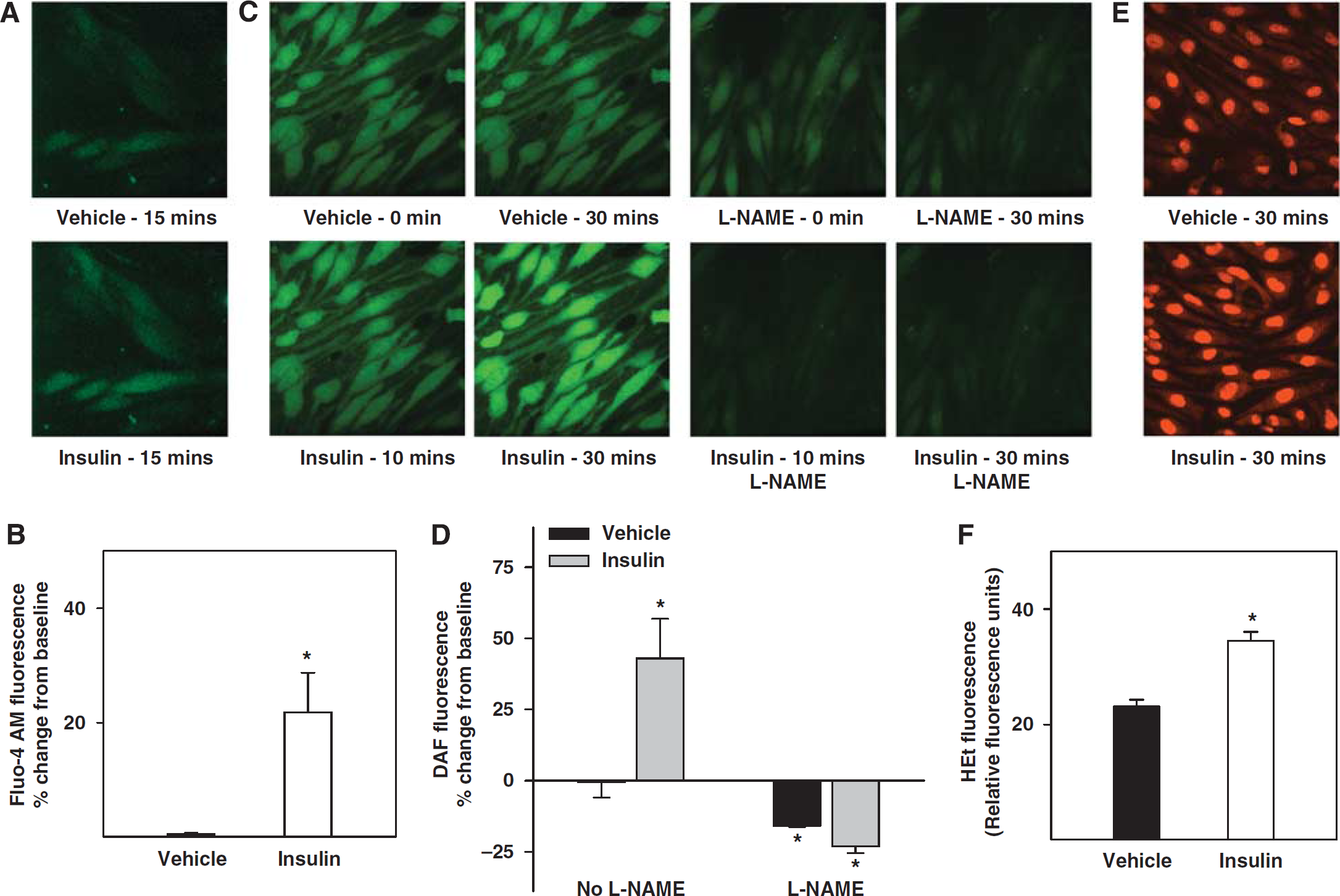
Effect of insulin on intracellular calcium dynamics and generation of NO and ROS in cerebral microvascular endothelial cells. Fluorescence measurements of calcium influx and generation of NO and ROS in the cultured rat CMVECs. (
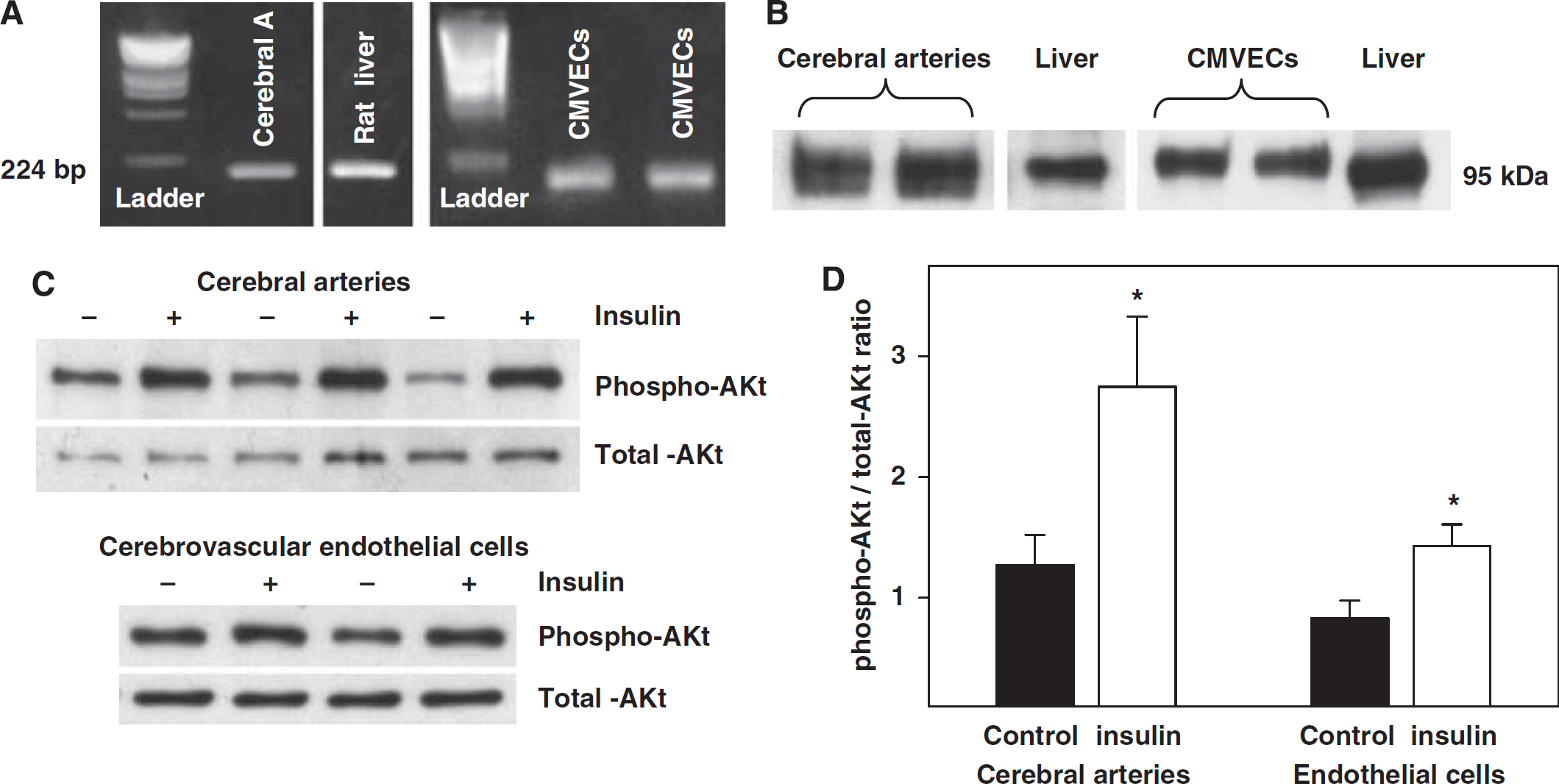
Expression of insulin receptor
Expression of Insulin Receptor
The PCR experiments showed the presence of 224-bp length insulin receptor mRNA in the rat cerebral arteries and CMVECs (Figure 5A). Similarly, the 95 kDa insulin receptor-
Insulin and Akt Phosphorylation
Treatment of rat cerebral arteries and CMVECs with insulin elicited increased phosphorylated Akt compared with untreated arteries and CMVECs, whereas the expression of total Akt was unchanged. The ratio of phosphorylated to total Akt was increased in insulin treated (2.8±0.58 and 2.1±0.27, respectively;
Plasma Insulin and Glucose Measurements
Plasma insulin levels were.07±0.08 ng/mL and fasting plasma glucose levels were 148±8 mg/dL in young Sprague–Dawley rats (
Discussion
The new and original findings of this study are as follows: (1) insulin receptor mRNA and protein were detected in rat cerebral arteries and in CMVECs; (2) insulin elicited a biphasic response in isolated cerebral arteries, including both vasoconstriction and vasodilation; (3) insulin-induced vasodilation seems to be primarily dependent on the endothelium, whereas insulin-induced vasoconstriction was mediated by both endothelial- and VSMC-dependent mechanisms; (4) both endothelial NOS- and nNOS-derived NO contributed to the vasodilation, whereas COX and CP450 metabolites primarily mediated vasoconstriction in response to insulin; (5) K+ channels were the primary targets of insulin-induced vasodilation with BKCa having the predominant role; (6) endothelin receptors mediated cerebrovascular responses to insulin with ETA activation causing vasoconstriction and ETB activation leading to vasodilation; (7) ROS contributed to insulin-induced vasoconstriction in cerebral arteries; and (8) insulin treatment induced enhanced Akt phosphorylation, endothelial [Ca2+]i and NO production in CMVECs, and isolated cerebral arteries. Thus, insulin exhibits concentration-dependent complex and competing effects on rat cerebral arteries as summarized in the schematic in Figure 6.
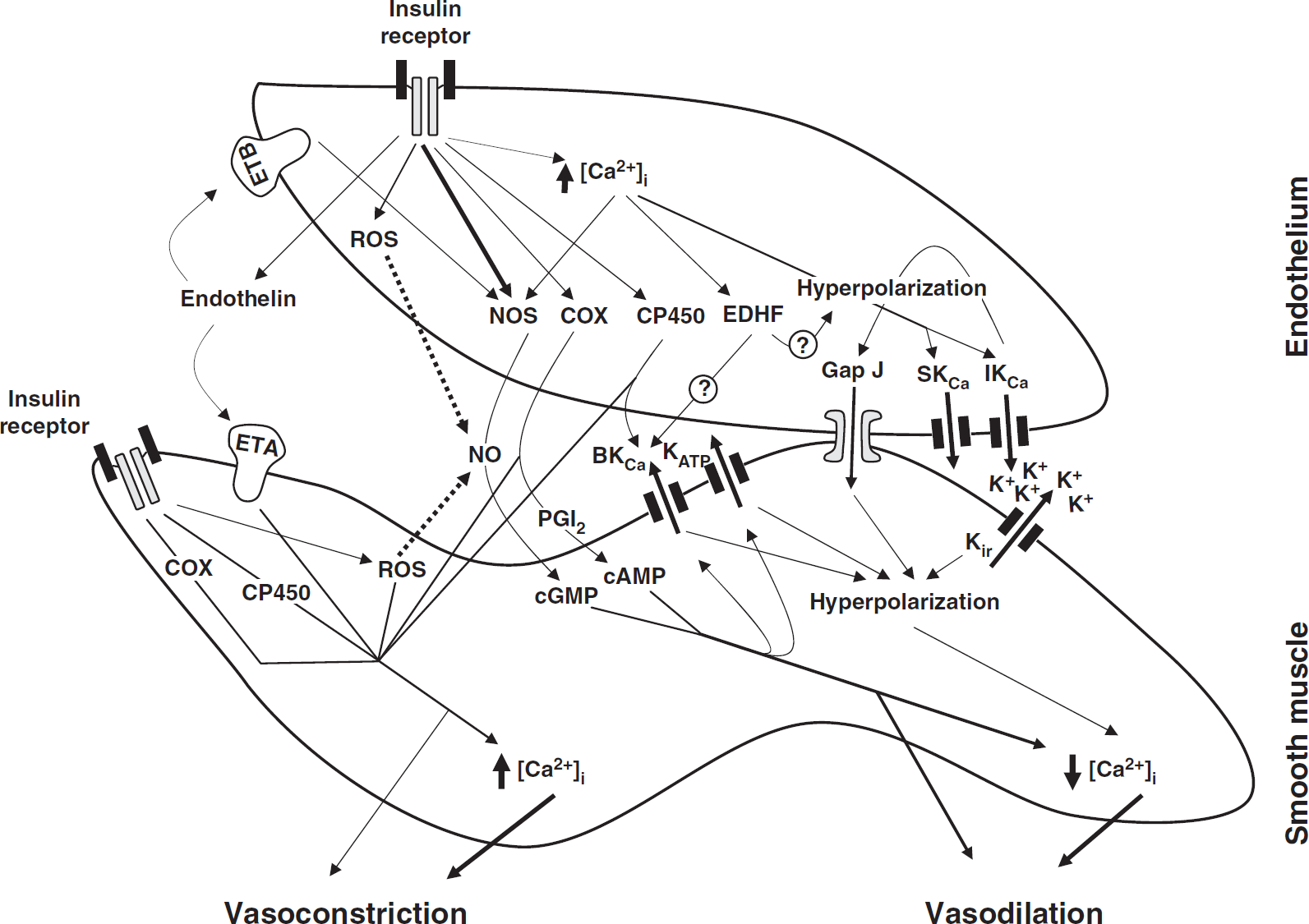
A schematic of the mechanisms underlying cerebrovascular actions of insulin in rats. Insulin-induced vasodilation is primarily endothelium-dependent, mediated by prostacyclin (PGI2), nitric oxide (NO), and endothelium-derived hyperpolarizing factor (EDHF). Elevation of endothelial calcium [Ca2+]i leads to activation of endothelial NO synthase (eNOS), cyclooxygenase, and EDHF pathway, although insulin-induced activation of eNOS is primarily calcium-independent. Calcium-activated K+ channels (SKCa, small; IKCa, intermediate; BKCa, large-conductance), myoendothelial gap junctions and inwardly-rectifying K+ channels (Kir) have all been implicated in the vascular hyperpolarization-mediated vasodilation (EDHF mechanism). In addition, NO and PGI2 activate guanylate and adenylate cyclase to produce cGMP and cAMP in vascular smooth muscle (VSM) cells, respectively, leading to activation of ATP-dependent K+ channels and BKCa. The subsequent K+ efflux and hyperpolarization promotes decrease in [Ca2+]i of VSM cells leading to vasodilation. In addition, cGMP and cAMP induce vasodilation by calcium-independent mechanism. Insulin-induced vasoconstriction results from the production of endothelin, vasoconstrictor prostanoids by COX, arachidonic acid metabolites by CP450 or reactive oxygen species (ROS) by the entire vascular wall. ROS induce vasoconstriction by either decreasing the NO bioavailability or promoting direct vasoconstriction. Endothelin causes predominantly vasoconstriction by activation of type A receptors (ETA) in VSM cells, whereas type B receptor (ETB) activation contributes to endothelium-dependent vasodilation. The vasoconstrictor factors promote vasoconstriction by both calcium-dependent (increased [Ca2+]i) and independent mechanisms. Dotted arrows in the figure represent inhibition mechanisms.
Insulin receptors have been identified in isolated arteries in several vascular beds (King et al, 1985) as well as in the homogenates of the brain (Havrankova et al, 1983). In addition, radioligand-binding studies have also established the existence of insulin receptors in the microvessels of the brain (Frank and Pardridge, 1981; Haskell et al, 1985). However, to our knowledge, identification of mRNA or immunoreactive protein of insulin receptors in cerebral arteries of any species has not been reported previously. For the first time, we have identified the mRNA of insulin receptors using primers designed on the basis of the sequence of rat insulin receptors. Similarly, we have identified the insulin receptor-
Normal fasting plasma insulin levels in humans are below 6
Insulin has been shown to activate both vasoconstrictor and vasodilation pathways in the vascular wall. Vasodilation in response to insulin has been shown to be primarily dependent on the endothelium, although insulin has also been shown to induce vasodilation by the direct activation of VSMC-dependent mechanisms (Oltman et al, 2000). Endothelium-derived relaxing factors such as NO, prostanoids, CP450 metabolites of arachidonic acid, and endothelium-derived hyperpolarizing factor (EDHF) have all been implicated in the vasodilation to insulin (Hasdai et al, 1998b; Katakam et al, 2005b; McKay and Hester, 1996; Oltman et al, 2000; van Veen and Chang, 1997; Verma et al, 1997). The contribution of each of these factors to vasodilation varies with the vascular bed and species studied. In rat cerebral arteries, NO was the primary endothelium-derived relaxing factor mediating the vasodilation to insulin. Interestingly, both endothelial NOS and nNOS appear to contribute NO to the vasodilation in response to insulin. Although the specificity of 7-NI to inhibit nNOS has been doubted, the low concentrations of 7-NI used was insufficient to affect other NOS isoforms (Meng et al, 1998). The contribution of nNOS to insulin-induced vasodilation is a novel observation of this study. Insulin also induced phosphorylation at ser473 of Akt in cerebral arteries and CMVECs consistent with a previous report of insulin action (Muniyappa et al, 2008), leading to the activation of endothelial NOS. The ability of insulin to enhance Akt phosphorylation also validates the vascular actions of insulin observed in cerebral arteries. Furthermore, fluorescence measurements of NO in CMVECs confirm the NO generation by insulin. Notably, increased intracellular calcium in CMVECs in response to insulin implicates calcium as a possible signaling mechanism of NOS activation and NO production, as well as activation of COX and EDHF pathways.
Cylooxygenase metabolites derived from both the endothelium and the VSMCs appear to mediate vasoconstriction and oppose insulin-induced vasodilation. This finding is unique to cerebral arteries in contrast to other vascular beds in which COX metabolites including prostacyclins, induce endothelium-dependent vasodilation (van Veen and Chang, 1997). The interactions between the COX and NOS systems in mediating cerebrovascular responses to insulin are complex and show a dominant effect of constrictor prostanoids. We have found similar interactions in a previous study (Meng et al, 1995). Thus, indomethacin administration enhanced cerebral blood flow responses during cortical spreading depression in rabbits, whereas L-NAME treatment reduced the CBF response. However, dual administration of L-NAME with indomethacin resulted in an enhanced CBF not different than with indomethacin administration alone. As indomethacin treatment did not affect NOS activity, we speculate that the dominant effect of constrictor prostanoids is at one or more signaling steps after the production of prostanoids or NO. For example, it has been shown that many prostanoids or NO-mediated responses in the cerebral circulation are influenced by permissive factors the presence of which is dependent on other signaling pathways (Wang et al, 1999).
CP450 products derived from the endothelium seem to contribute to the vasoconstriction and oppose the insulin-induced vasodilation in cerebral arteries, whereas they cause vasodilation in other vascular beds (Okubo et al, 1999). Interestingly, insulin at a high concentration seems to promote vasodilator CP450 products from VSMCs in endothelium-denuded arteries. Vasoconstriction to insulin also seems to be mediated by ROS because scavenging the ROS or inhibition of ROS generation abolished vasoconstriction. This was supported by the direct measurements of increased ROS generation in CMVECs in response to insulin. The source of ROS produced in response to insulin appears to be nicotinamide adenine dinucleotide phosphate oxidase although other ROS-generating systems, particularly COX and mitochondria, probably also contribute.
The target of insulin-induced vasodilation seems to be primarily K+ channels. In particular, BKCa seems to have the dominant role because inhibition of BKCa totally abolished vasodilation leading to a dose-dependent vasoconstriction in response to insulin. In contrast, inhibition of other K+ channels, including that of Kir, Kv, KATP, IKCa, and SKCa, modestly diminished vasodilation without abolishing vasodilation completely. This observation indicates that these K+ channels have a minor role in the overall vasodilation to insulin. Interestingly, IKCa and SKCa have been implicated in the vasodilation linked to the EDHF (Brahler et al, 2009; Eichler et al, 2003), a yet unknown mechanism of vasodilation dependent on K+ channels. Thus, insulin-induced vasodilation also seems to involve EDHF in cerebral arteries. Previously, EDHF has been shown to mediate insulin-induced hyperpolarization and vasodilation in resistance arteries (Iida et al, 2001; Okubo et al, 1999). However, this study is the first to link IKCa and SKCa to the vasodilation caused by insulin. The underlying basis for the reversal of constriction to 0.1 ng/mL insulin in the presence of several potassium channel blockers, but not to iberiotoxin and barium, is unclear at this time. The similar effect of ETA blockade or ROS scavenging, which occurs only at this dose of insulin, suggests an interaction with some, but not all types of potassium channels. Nonetheless, the precise mode of this interaction in intact arteries is not known at this time.
Insulin also has been shown to cause vasoconstriction (Eringa et al, 2002; Hasdai et al, 1998a; Schroeder et al, 1999; van Veen and Chang, 1998). A major mechanism of vasoconstriction seems to be the promotion of the release of endothelin-1 (ET-1) from the endothelium (Miller et al, 2002; Verma et al, 1997). Endothelin-1 is known to cause potent vasoconstriction by activating ET-receptors, mainly ET type A receptors, on the smooth muscle. Endothelin-1 also causes endothelium-dependent vasodilation through ET type B receptors in the endothelium. Consistent with previous observations, ETA receptors mediated vasoconstriction and ETB receptors mediated vasodilation in response to insulin. Importantly, previous studies have shown that increased endothelin activity results in attenuation of insulin signaling leading to IR at a cellular level (Ishibashi et al, 2001). Furthermore, enhanced endothelin activity leading to vasoconstriction, also promotes IR by reducing the delivery of insulin and glucose to tissues. Moreover, the endothelin system has also been implicated in the vascular spasm associated with traumatic brain injury, stroke, and subarachnoid hemorrhage (Sobey and Faraci, 1998). Thus, the interaction between insulin and endothelin signaling has a critical role in cerebrovascular insulin sensitivity as well as in the pathogenesis of cerebrovascular disease.
There are experimental limitations that should be considered in the evaluation of the results of our study. Pharmacological inhibition is often used to confirm the role of vasodilator/vasoconstrictor pathways mediating the vascular response; however, it is not sensitive enough to make assumptions about the relative contribution of each pathway to overall vascular response. This may be due to the cross-activation or cross-inhibition of various pathways as reported previously. For example, it has been shown that NO inhibits EDHF response (Kessler et al, 1999) and in conditions in which NO production is compromised, upregulation of EDHF response occurred in endothelial NOS knockout mice (Waldron et al, 1999). Thus, the inhibition of NO production may be necessary to uncover the EDHF response in cerebral arteries. In this study, almost complete inhibition of the insulin response by the inhibition of one pathway/mediator does not rule out the contribution of others to the vasodilation.
In conclusion, our study shows the ability of insulin to regulate cerebral vasoreactivity. Insulin-induced vasodilation is primarily by the endothelium-dependent mechanisms, NO, and EDHF, targeting predominantly VSMC K+ channels, whereas vasoconstriction was mediated by metabolites of COX and CP450, endothelin, and ROS. Thus, the multiple mechanisms by which insulin regulates the tone of the cerebral arteries underscore the dynamic role of insulin in cerebrovascular regulation and the potential for cerebrovascular dysfunction in IR.
Footnotes
Acknowledgements
The authors thank Nancy Busija for the help with editing this paper.
The authors declare no conflict of interest.