
Abstract
The invasion of inflammatory cells occurring after ischemic or traumatic brain injury (TBI) has a detrimental effect on neuronal survival and functional recovery after injury. We have recently demonstrated that not only the blood-brain barrier, but also the blood-cerebrospinal fluid (CSF) barrier (BCSFB), has a role in posttraumatic recruitment of neutrophils. Here, we show that TBI results in a rapid increase in synthesis and release into the CSF of a major chemoattractant for monocytes, CCL2, by the choroid plexus epithelium, a site of the BCSFB. Using an
Introduction
Brain injury, whether it is ischemic, hemorrhagic, or traumatic, is accompanied by pathological neuroinflammation, which is thought to significantly contribute to the formation of cerebral edema and loss of neural tissue, and to have an impact on functional recovery after injury. An important part of this brain inflammatory response to injury is the influx of neutrophils and monocytes. Neutrophils are highly toxic to vulnerable neurons (Neumann et al, 2008) and these inflammatory cells have been shown to exacerbate cerebral edema and brain tissue damage in rodent models of traumatic brain injury (TBI) (Schoettle et al, 1990; Semple et al, 2010b). Similarly, invading monocytes have been reported to substantially contribute to the formation of cerebral edema and loss of neural tissue, and to have an adverse effect on neurologic outcome in rodent models of stroke and TBI (Chen et al, 2003; Dimitrijevic et al, 2007; Semple et al, 2010a). Using the Brattleboro rats deficient in vasopressin, a neuropeptide found to substantially augment the posttraumatic synthesis of proinflammatory mediators, we have recently demonstrated that there is an association between the cortical production of neutrophil and monocyte chemoattractants, the magnitude of influx of inflammatory cells, and the extent of loss of neural tissue occurring after TBI (Szmydynger-Chodobska et al, 2010). These results are consistent with previous observations that treatments directed to counter the influx of inflammatory cells into the injured brain are therapeutically beneficial (Beech et al, 2001; Yamasaki et al, 1997).
Although the blood—brain barrier is the major route for inflammatory cells to invade the injured brain, increasing evidence suggests that the blood-cerebrospinal fluid (CSF) barrier (BCSFB) also has an important role in this pathophysiological process (Szmydynger-Chodobska et al, 2009). The BCSFB is formed by a single layer of epithelial cells enclosing blood microvessels in the highly vascularized choroid plexus (CP), a tissue located in all four cerebral ventricles (Strazielle and Ghersi-Egea, 2000). We have previously shown that the choroidal epithelium has the ability to produce neutrophil chemoattractants and provided electron microscopic evidence for neutrophil trafficking across the BCSFB (Szmydynger-Chodobska et al, 2009). However, investigations have not determined whether the BCSFB also has a role in the recruitment of monocytes to the injured brain.
The major chemoattractant for monocytes is monocyte chemotactic protein 1 (MCP-1) or CCL2, which belongs to the CC family of chemokines (Rollins, 1997). This chemokine has previously been shown to be synthesized by variety of epithelial cells in response to proinflammatory cytokines interleukin-1β (IL-1β) and tumor necrosis factor-α (Paine et al, 1993; Prodjosudjadi et al, 1995). Although other members of the MCP subfamily of CC chemokines, such as MCP-2/CCL8, MCP-3/CCL7, MCP-4/CCL13, and MCP-5/CCL12 (mouse only), have been identified, they seem to be weaker chemoattractants for monocytes compared with MCP-1/CCL2 (Sozzani et al, 1994; Takahashi et al, 2009). CCL2 binds to CCR2, a G-protein-coupled receptor (Rollins, 1997). This chemokine also binds to CCR10, a placental chemokine receptor (Bonini et al, 1997); however, CCR10 is not expressed on leukocytes.
The aim of this study was to define a role of the CP in monocyte recruitment to the injured brain. Accordingly, we characterized the ability of the CP to produce CCL2 in response to injury using a rat model of TBI and determined the direction of cytokine-induced release of this chemokine (across the apical versus basolateral membrane of choroidal epithelium) using an
Materials and methods
Reagents and Antibodies
ThermoScript RNase H− reverse transcriptase and RNase inhibitor, RNaseOut, were obtained from Invitrogen (Carlsbad, CA, USA). HotStart
Rabbit polyclonal antibody to rat CCL2 (1 μg/mL for Western blotting and 2 μg/mL for immunohistochemistry) was obtained from Antigenix America (Huntington Station, NY, USA). The following mouse monoclonal antibodies were used: anti-mouse β-catenin (clone 14; 2.5 μg/mL) from BD-Transduction Labs (Lexington, KY, USA), anti-rat
The Rat Model of Traumatic Brain Injury and Cerebrospinal Fluid Sampling
Adult male Long-Evans rats weighing 250 to 350 g (Harlan, Indianapolis, IN, USA) were used. They were kept at 22°C with a 12-hour light cycle and maintained on standard pelleted rat chow and water
At 6 hours after TBI or sham injury, rats (8 to 11 animals per group) were reanesthetized with pentobarbital sodium and the samples of CSF were collected from the cisterna magna. The concentration of CCL2 in these samples was measured by ELISA (enzyme-linked immunosorbent assay) as described below.
Real-Time Reverse-Transcriptase Polymerase Chain Reaction
At 2, 4, and 6 hours, and 1, 2, and 4 days post-TBI, rats (9 to 10 animals per time point) were reanesthetized with pentobarbital sodium and were perfused transcardially with ice-cold 0.9% NaCl. The lateral ventricle CPs, both ipsilateral and contralateral to injury, were collected separately and pooled into three subgroups (3 to 4 rats per subgroup) for each time point. The lateral ventricle CPs were also collected from sham-injured rats. Total RNA was isolated using NucleoSpin RNA II kit (Macherey-Nagel, Düren, Germany). First-strand cDNAs were synthesized using oligo(dT)20 primer (50 pmol) and 15 U of ThermoScript RNase H− reverse transcriptase. Forty units of RNase inhibitor, RNaseOut, were also added to the reverse transcription reactions. For each reaction, 0.5 μg of total RNA was used and the reactions were performed for 1 hour at 50°C.
Real-time polymerase chain reaction (PCR) was performed as previously described (Szmydynger-Chodobska et al, 2009, 2010). Cyclophilin A was used for the normalization of the data obtained. The following primers and TaqMan probes were used: 5'-TGTCTCAGCCAGATGCAGT TA-3' (forward primer for CCL2), 5'-CATTCCTTATTGGG GTCAGC-3' (reverse primer for CCL2), 5'-ATGCCCCACTC ACCTGCTGCTA-3' (probe for CCL2), 5'-GGTGAAAGA AGGCATGAGCA-3' (forward primer for cyclophilin A), 5'-GCTACAGAAGGAATGGTTTGATG-3' (reverse primer for cyclophilin A), and 5'-TTTGGGTCCAGGAATGGCAAG AC-3' (probe for cyclophilin A). The predicted sizes of PCR products were 181 and 152bp for CCL2 and cyclophilin A, respectively. The 50-μL PCR reaction mixtures contained 0.2 mmol/L mixed dNTPs, 0.2 μmol/L each primer, 0.1 μmol/L TaqMan probe, 5 mmol/L MgCl2, 1 U of HotStart
Western Blotting
At 6 hours post-TBI, rats divided into two separate groups (four animals per group) were reanesthetized with pentobarbital sodium and were perfused transcardially with ice-cold 0.9% NaCl. The lateral ventricle CPs were collected and pooled separately for the ipsilateral and contralateral side. The lateral ventricle CPs were also collected from four sham-injured rats. Proteins were extracted using isotonic lysis buffer (150 mmol/L NaCl, 50 mmol/L Tris-HCl, pH 7.4, 2 mmol/L EDTA, 1% Triton X-100) containing protease inhibitors (1 mmol/L benzamidine, 100U/mL aprotinin, 20 μg/mL antipain, 20 μg/mL leupeptin, 1 μg/mL pepstatin A, 1 mmol/L PMSF).
Immunoblotting procedures were performed as previously described (Szmydynger-Chodobska et al, 2010). In brief, proteins were resolved via sodium dodecyl sulfate-polyacrylamide gel electrophoresis (4% to 12%) under reducing conditions and proteins were transferred onto 0.2 μm nitrocellulose membranes (Invitrogen). After blocking with 5% ECL Advance blocking agent (GE Healthcare, Little Chalfont, UK) for 1 hour at room temperature, the membranes were incubated with primary antibodies overnight at 4°C. Membranes were subsequently incubated with horseradish peroxidase-conjugated anti-rabbit or antimouse antibody for 1 hour at room temperature. For detection, SuperSignal West Dura extended duration (Pierce; Thermo Fisher Scientific Inc., Rockford, IL, USA) or ECL Advance (GE Healthcare) chemiluminescence substrate and the Bio Imaging System Chemi Genius2 (Syngene, Frederick, MD, USA) were used.
Immunohistochemistry
Immunohistochemical procedures were performed as previously described (Szmydynger-Chodobska et al, 2009, 2010). Rats (2 to 4 animals per group) were reanesthetized with pentobarbital sodium and were perfused transcardially with ice-cold 0.9% NaCl, followed by ice-cold 4% paraformaldehyde in 0.05 mol/L PBS (phosphate-buffered saline), pH 7.4. Brains were removed and postfixed for additional 4 hours in the paraformaldehyde/PBS solution at 4°C. They were then incubated overnight in 20% sucrose in PBS and embedded in Tissue-Tek OCT compound (Sakura Finetek, Torrance, CA, USA). Coronal brain sections were cut on a cryostat at 10 μm.
All procedures were performed at room temperature, except for the incubation with primary antibodies that was completed at 4°C. To minimize nonspecific staining, the brain sections were incubated for 30 minutes with 10% normal goat serum. Four percent of normal goat serum was also added when the sections were incubated with primary and secondary antibodies. After the initial blocking step, the sections were incubated overnight with primary antibodies and then were incubated for 1 hour with secondary antibodies. The sections were mounted with Vectashield mounting medium (Vector Labs, Burlingame, CA, USA).
Transmission Electron Microscopic Analysis
At 1 day after TBI or sham injury, rats (2 to 10 animals per group) were reanesthetized with pentobarbital sodium and the lateral ventricle CPs were removed and fixed overnight in 2.5% glutaraldehyde in 0.15 mol/L sodium cacodylate buffer, pH 7.4, at 4°C. Following fixation, the specimens were treated with 1% OsO4 in cacodylate buffer for 0 hour at 4°C. They were then dehydrated in a graded acetone series and embedded in Epox 812 resin (Fullam, Latham, NY, USA). Ultrathin sections (50 to 60 nm) were prepared, retrieved onto 300 mesh copper grids, and contrasted with uranyl acetate and lead citrate. Sections were examined using a Morgagni 268 transmission electron microscope (FEI Electron Optics, Eindhoven, Netherlands).
Primary Cell Cultures of Rat Choroidal Epithelial Cells
The procedures were conducted according to the guidelines approved by the French Ethical Committee (decree 87-848) and by the European Community directive 86-609-EEC. Animals were obtained from Harlan (Gannat, France). Choroidal epithelial cells were isolated as previously described (Strazielle and Ghersi-Egea, 1999) and were seeded on Transwell Clear inserts with the pore size of 0.4 μm (Costar, Cambridge, MA, USA) precoated with laminin. The cells were kept at 37°C in a humidified atmosphere of 5% CO2/95% air and were fed every other day with medium containing 10% fetal bovine serum. The experiments were performed 5 days after the cells reached confluence.
Secretion of CCL2
The cells were serum starved in SFM (serum-free medium) supplemented with 0.1% bovine serum albumin for 24 hours before experimentation. On the day of experiment, bovine serum albumin-containing SFM was renewed in both chambers and the apical surface of epithelial monolayers was exposed for 6 hours to recombinant rat IL-
Permeability of Epithelial Monolayers to CCL2
The epithelial cells were serum starved as described above and were then exposed for 6 hours to recombinant rat CCL2, which was added to either the apical (100 ng/mL) or basolateral (50 ng/mL) chamber. After this incubation, media were sampled from both compartments to determine the concentration of CCL2. Recombinant human CCL2 was used to measure the epithelial permeability to this chemokine in IL-1β-treated cells. In these experiments, the cells were pretreated for 3 hours by adding 100 pg/mL of IL-1β to the apical chamber. Control monolayers (incubated in SFM without IL-1β) were processed simultaneously. The concentrations of human CCL2 in the apical and basolateral compartments were determined using the Human MCP-1 ELISA Development kit (PeproTech). This assay showed no crossreactivity with rat CCL2 at a concentration as high as 50 ng/mL. No degradation of the recombinant chemokines during the incubation periods was observed.
Measurement of Paracellular Permeability of Epithelial Monolayers
The integrity of epithelial monolayers was examined by measuring the permeability of each filter to the paracellular permeability marker 14C-sucrose. The permeability × surface area product, PSt, was calculated from the 14C-sucrose clearance rates as previously described (Strazielle and Preston, 2003). The permeability coefficient, Pt, was calculated by dividing PSt by the surface area of the cell monolayer.
Statistical Analysis
For statistical evaluation of data, analysis of variance was used, followed by the tests for multiple comparisons among means, as described in figure 1 and 3 legends.
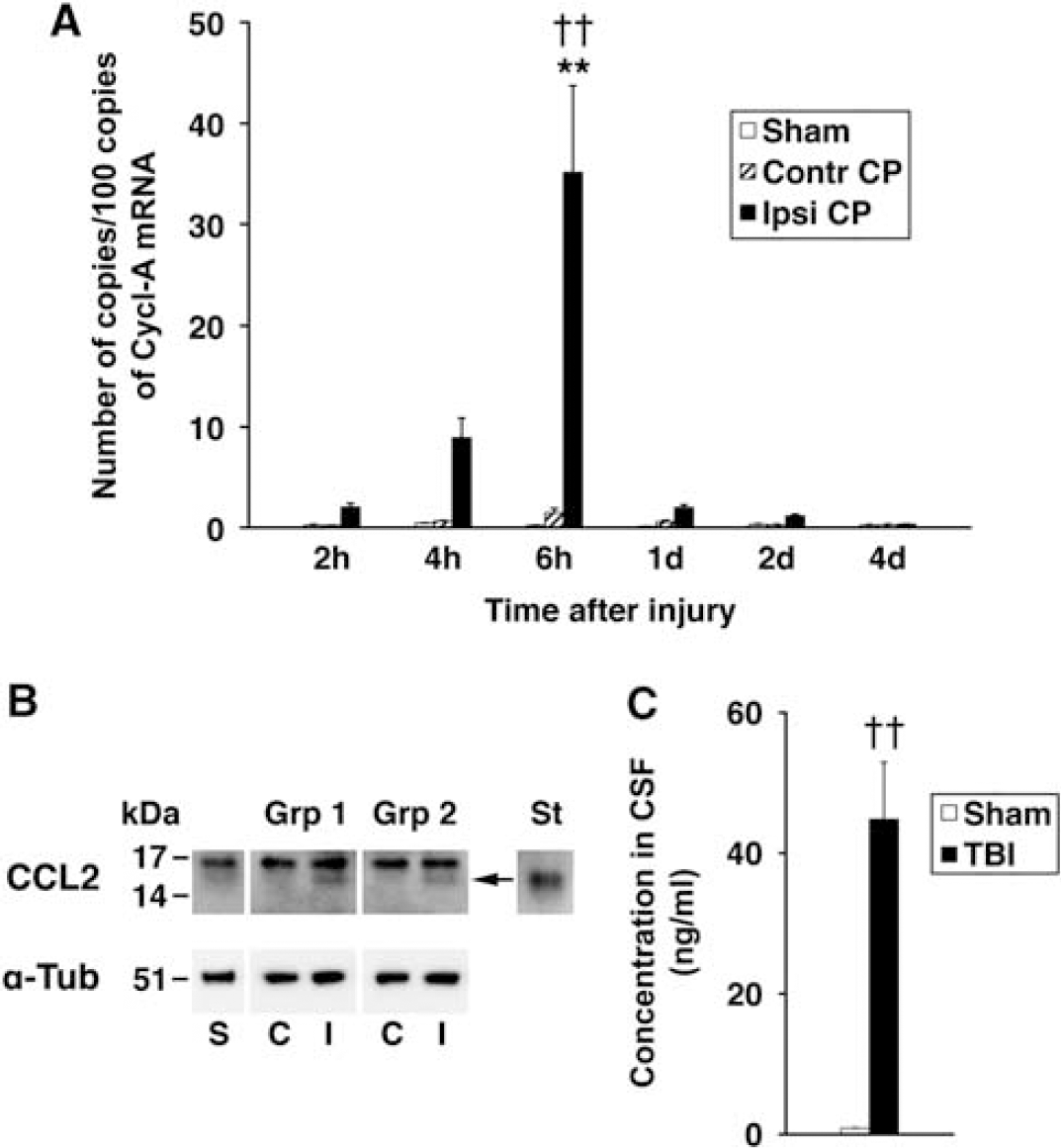
Posttraumatic production of CCL2 by the lateral ventricle choroid plexus (CP). (
Results
Posttraumatic Increase in Choroidal CCL2 Synthesis
Posttraumatic changes in CCL2 expression in the lateral ventricle CP were initially analyzed using real-time reverse transcriptase (RT) polymerase chain reaction. This analysis showed a gradual increase in CCL2 mRNA, which peaked at 6 hours after injury (Figure 1A). This increase in choroidal synthesis of CCL2 was transient and at 1 day after TBI, CCL2 mRNA in the ipsilateral CP dropped abruptly to a level, which was not significantly different from that found in the contralateral CP or in the CP from sham-injured rats. No changes in CCL2 mRNA in the contralateral CP or the CP from sham-injured animals were observed.
We next used Western blotting to determine whether the posttraumatic increase in choroidal CCL2 synthesis observed at the message level is also associated with increased choroidal production of CCL2 protein. The choroidal tissue was harvested at 6 hours post-TBI, a time point at which a peak in posttraumatic increase in CCL2 mRNA was observed. Consistent with RT-PCR analysis, an increase in CCL2 protein expression was observed in the ipsilateral CP in two independent groups of animals (Figure 1B). This increase in choroidal CCL2 synthesis was followed by a 50-fold elevation of CCL2 concentration in the CSF collected from the cisterna magna at 6 hours after injury when compared with sham-injured animals (Figure 1C).
Injury-Dependent Changes in CCL2-Immunopositive Staining of the Choroidal Tissue
The immunohistochemical analysis of choroidal tissue harvested at 6 hours after sham injury showed a distinct pattern of CCL2-positive staining of choroidal epithelium associated with the apical domain of epithelial cells (Figure 2A). Similar pattern of immunostaining for CCL2 was observed in both the contralateral (Figures 2B–2G) and ipsilateral (data not shown) CPs harvested at 6 hours post-TBI. Double immunostaining of choroidal tissue with anti-CCL2 antibody and an antibody to β-catenin, a component of the adherens junction complex, demonstrated a clear separation of the CCL2-immunoreactive product from this basolateral marker (Figures 2C-2E). The apical localization of the CCL2-immunoreactive product was further confirmed by double immunostaining of the choroidal tissue with anti-CCL2 antibody and an antibody to AQP1, a water channel expressed apically in choroidal epithelial cells (Praetorius and Nielsen, 2006; Figure 2G). Intense cytoplasmic CCL2-positive staining of epithelial cells in the ipsilateral, but not contralateral, CP was associated with the Golgi complex (Figures 2H and 2I). The localization of CCL2 to the Golgi complex was consistent with increased synthesis of this secreted protein occurring after TBI. Unlike apical CCL2 staining that was rather evenly distributed throughout the CP, the Golgi-associated CCL2 immunostaining was found in clusters of epithelial cells scattered through the choroidal tissue. Such clusters of cells were observed as early as 4 hours after TBI. The number of epithelial cells with the Golgi-associated CCL2 immunostaining peaked at 6 hours post-TBI, which was in line with the results from the real-time RT-PCR analysis and Western blotting. The distribution of CCL2-immunopositive staining in the contralateral or ipsilateral CP was confined to the epithelial cells. The CCL2-immunoreactive product was not associated with other types of cells normally present in the choroidal tissue, such as endothelial and epiplexus cells or stromal macrophages.
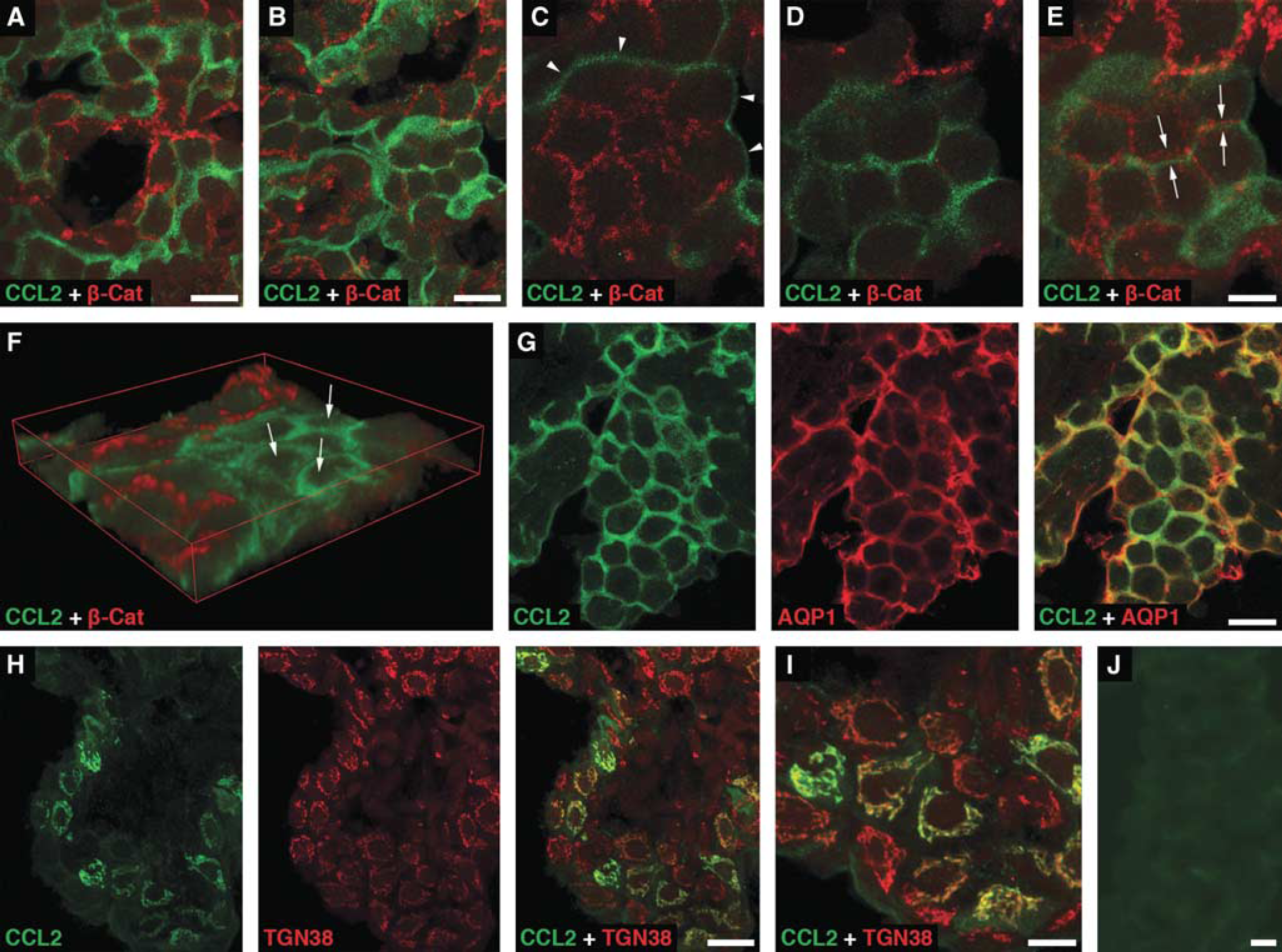
Immunohistochemical localization of CCL2 in the lateral ventricle choroid plexus (CP). Confocal microscopy images of choroidal tissue harvested at 6 hours after sham injury (
Polarity of CCL2 Secretion by Choroidal Epithelium
To test whether IL-1β induces CCL2 synthesis in the choroidal epithelium and to determine the direction of CCL2 release (across the apical versus basolateral membrane), we used an
Under control conditions, the low-level production of CCL2 was observed. This constitutive secretion of CCL2 was polarized, with the chemokine being largely released across the apical (CSF-facing) membrane of choroidal epithelium. In dose-response studies, the epithelial monolayers were exposed to 2, 10, 100, and 1,000 pg/mL of IL-1β for 6 hours (Figure 3A). CCL2 secretion was increased 25 and 120 times over its constitutive production when the monolayers were exposed to IL-1β at concentrations as low as 2 and 10 pg/mL, respectively. The exposure of epithelial cells to IL-1β at 100 pg/mL resulted in a 290-fold increase in CCL2 synthesis relative to control. The choroidal production of CCL2 appeared to reach its maximum capacity when the monolayers were incubated with IL-1β at a concentration of 1 ng/mL. With all IL-1β concentrations tested, except 1 ng/mL, the larger amounts of CCL2 were secreted across the apical versus basolateral membrane of choroidal epithelium. In time-course studies, the cells were incubated with 10 pg/mL of IL-1β for 1, 3, 6, and 24 hours (Figure 3B). At this concentration of IL-1β, CCL2 secretion was predominantly apical throughout the 24-hour observation period. The CCL2 levels in the apical and basolateral chambers increased rapidly between 1 and 6 hours of exposure to IL-1β; however, later on, the CCL2 levels in the apical chamber increased at much slower rate, whereas no further increase in CCL2 concentration was observed in the basolateral compartment.
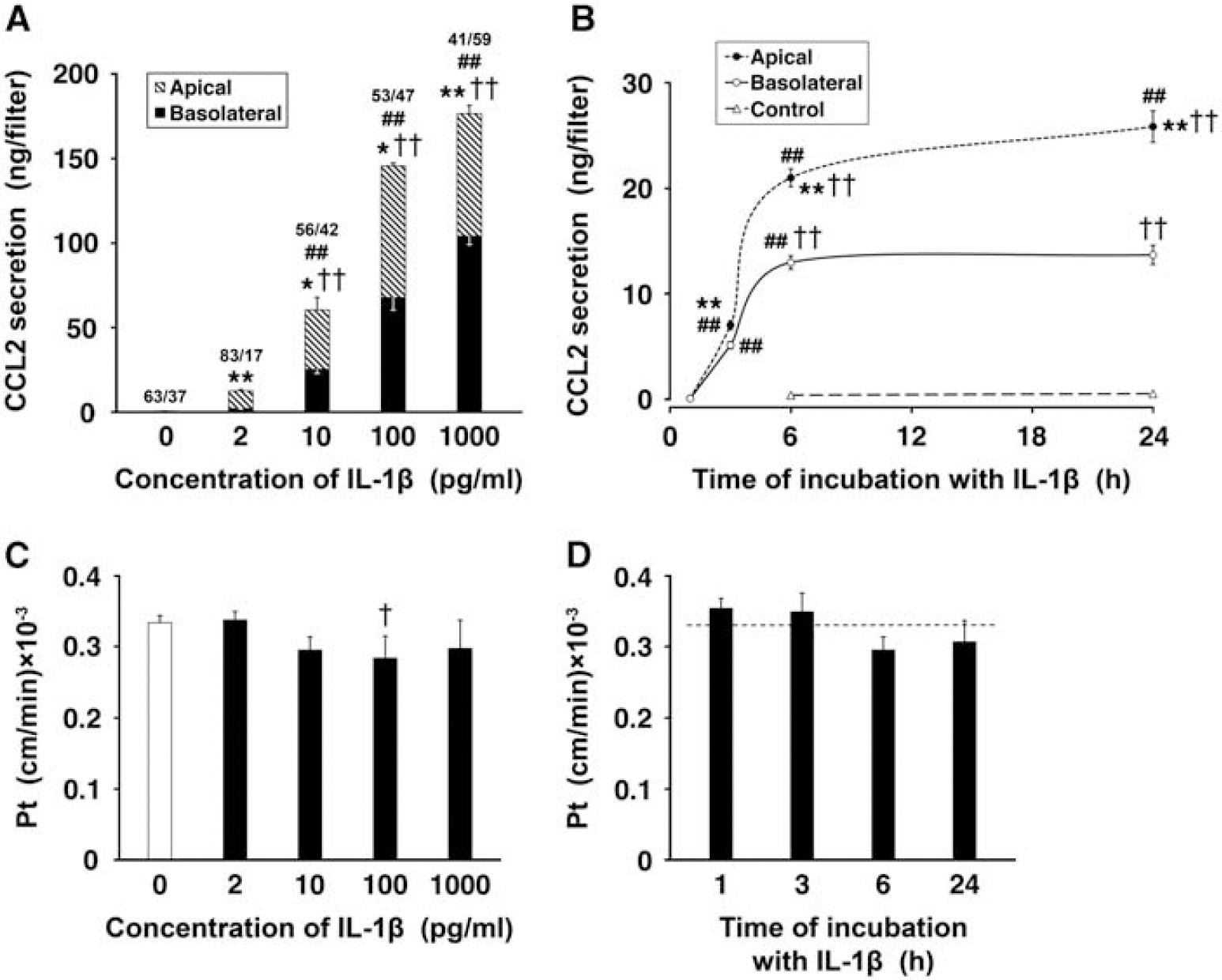
Secretion of CCL2 by primary cultures of choroidal epithelial cells. The cells were seeded on Transwell Clear inserts as described in Materials and methods. The experiments were performed 5 days after the cells reached confluence (
The possible paracellular diffusion and/or transcellular transfer of CCL2, which might have had an effect on our estimates of polarity of chemokine secretion by epithelial monolayers, was also evaluated. To this end, recombinant rat CCL2 was added to either the apical or basolateral chamber, and the media were sampled from both compartments 6 hours later. The levels of CCL2 measured in the respective opposite chambers at the end of the incubation period were higher than those resulting from constitutive secretion, which was assessed in parallel in separate monolayers. However, the amounts of CCL2 that actually permeated the monolayers represented only 0.6% to 1.6% of the amount of the chemokine added to each chamber. Similar low permeability to recombinant human CCL2 was observed for both control monolayers and the epithelial cells pretreated with IL-1β. This suggested that IL-1β did not enhance a leak and/or transcellular transfer of CCL2 across the epithelial monolayer. The exposure of epithelial cells to IL-1β or CCL2 also did not cause any apparent changes in the integrity of the monolayers, as shown by the lack of increase in permeability to sucrose (Figures 3C and 3D, and data not shown).
Monocyte Trafficking Across the Blood—Cerebrospinal Fluid Barrier
Posttraumatic increase in choroidal synthesis of CCL2 was followed by monocyte recruitment to the ipsilateral CP; however, there was a delay between an increase in the production of this chemokine and the accumulation of monocytes in choroidal tissue. Monocytes infiltrating the ipsilateral CP were found at 1 day after injury (Figures 4B and 4C), whereas at 6 hours post-TBI, these inflammatory cells were only sporadically observed to accumulate in the ipsilateral CP. Monocytes did not infiltrate the whole ipsilateral CP, but only accumulated in certain areas of the choroidal tissue. Monocytes did not accumulate in the contralateral CP (Figure 4A), which was consistent with the lack of changes in CCL2 synthesis in the contralateral CP (see Figures 1A and 1B).
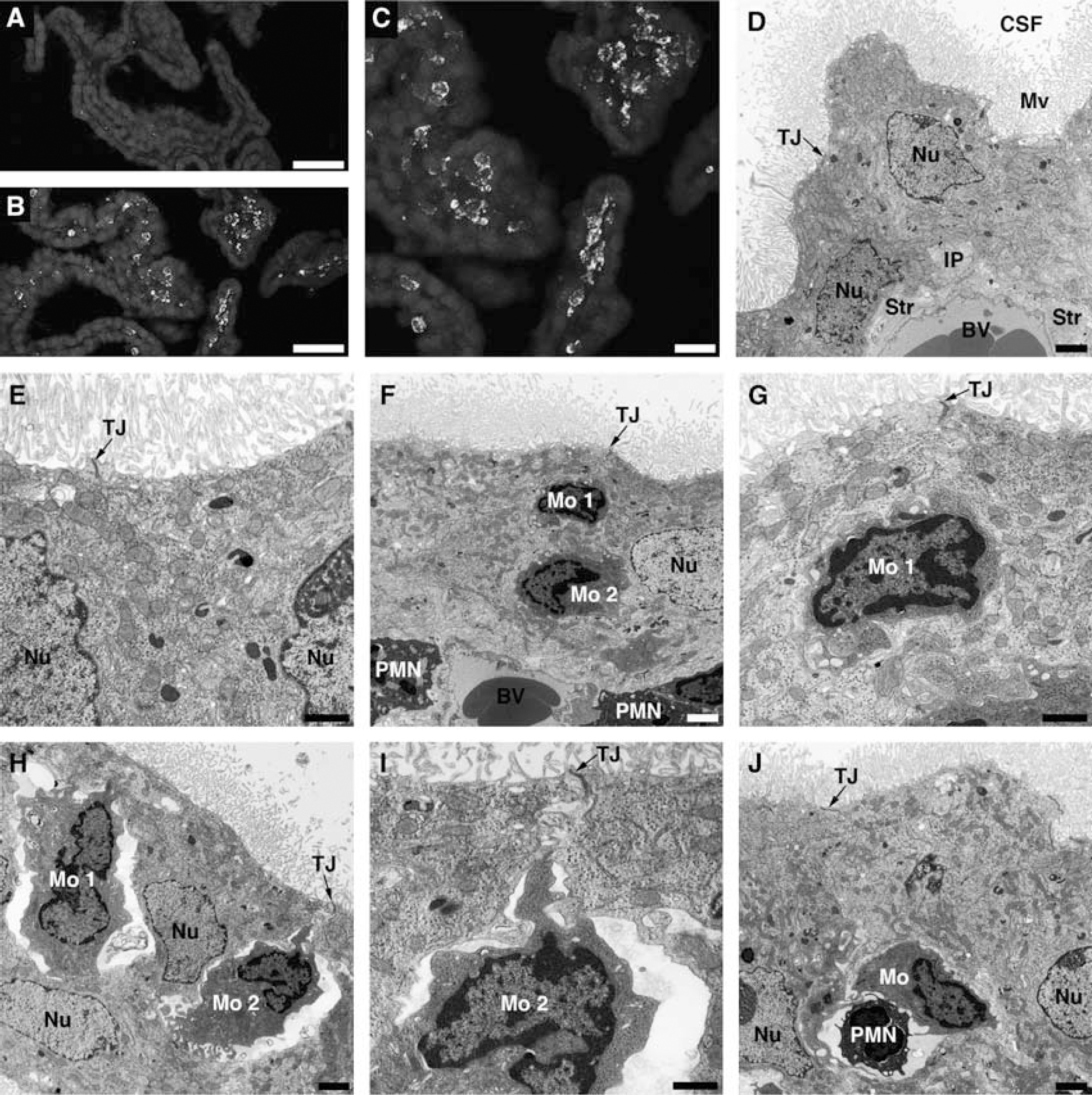
Posttraumatic accumulation of monocytes in the lateral ventricle choroid plexus (CP). (
To demonstrate monocyte trafficking across the BCSFB, transmission electron microscopy was used. Ten rats were analyzed at 1 day post-TBI, and in 8 out of these 10 animals (80%), the infiltration of the ipsilateral CP by monocytes was observed. The contralateral CP had normal morphology (Figures 4D and 4E), similar to the morphology of choroidal tissue from sham-injured rats (data not shown). The choroidal epithelial cells had well-defined microvilli and the lateral membrane was folded into elaborate interdigitating processes near the base of the cells. The space between adjacent epithelial cells was narrow and the tight junctions between the cells were well discernible. In the ipsilateral CP, monocytes were found to reach the intercellular space between the epithelial cells and were apparently moving toward the apical domain of choroidal epithelium (Figures 4F–4I). The space between invading monocytes and the choroidal epithelial cells was occasionally found to be narrow (Figures 4F and 4G), but predominantly an enlarged, rather than tight, space between migrating monocytes and the epithelial cells was seen in the ipsilateral CP (Figures 4H and 4I). We were also able to find monocytes invading the ipsilateral CP in tandem with neutrophils (Figure 4J). Usually, one to four such events were observed in the areas of the ipsilateral CP where the accumulation of inflammatory cells occurred. The movement of monocytes between the epithelial cells toward the apical domain of choroidal epithelium did not appear to affect the integrity of tight junctions.
Discussion
Monocytes invading the brain parenchyma in response to ischemia or TBI have detrimental effect on neuronal survival and functional recovery after injury (Chen et al, 2003; Dimitrijevic et al, 2007; Semple et al, 2010a). The influx of these inflammatory cells is driven by monocyte chemoattractants, such as CCL2, whose synthesis is rapidly increased in the injured cortex (Semple et al, 2010a; Szmydynger-Chodobska et al, 2010). In the present study, we demonstrated that neurotrauma also results in a rapid increase in production of CCL2 by the lateral ventricle CP located ipsilaterally to injury. This increase in choroidal CCL2 synthesis was not associated with posttraumatic accumulation of monocytes in the choroidal tissue, but resulted from production of CCL2 by the choroidal epithelium. Indeed, although monocytes can produce CCL2 in response to proinflammatory mediators (Colotta et al, 1992), these inflammatory cells were rarely found in the ipsilateral CP at 6 hours post-TBI, a time point at which a maximum increase in choroidal CCL2 synthesis was observed. An increase in choroidal production of CCL2 was associated with a significant elevation of CCL2 concentration in the CSF, which was comparable to the levels of this chemokine found in the CSF of patients with severe TBI (Semple et al, 2010a).
These observations raise the question about the importance of the CP as a source of CCL2 in the injured brain. Using the primary cultures of choroidal epithelial cells, we found that the rate of apical secretion of CCL2 in response to IL-1β is relatively stable during the first 6 hours of incubation with the cytokine (see Figure 3B) and amounts to 15 ng/h per cm2 of surface area of epithelial monolayer. In these experiments, the epithelial monolayers were exposed to 10 pg/mL of IL-1β, the average concentration of IL-1β found in ventricular CSF in patients with severe TBI at 6 hours after injury (Shiozaki et al, 2005). To extrapolate these
The immunohistochemical analysis of choroidal tissue demonstrated that CCL2 is produced by the epithelial cells. This chemokine did not appear to be synthesized by other types of cells normally present in the choroidal tissue, such as endothelial and epiplexus cells or stromal macrophages, in both sham-injured and traumatized rats. These results are in line with the previous studies, in which
Interestingly, both in sham-injured rats and in animals subjected to TBI, a distinct CCL2-positive staining of the apical surface of choroidal epithelium was observed. This finding was consistent with the results from
The experiments involving the epithelial monolayers showed that CCL2 is secreted across both the apical and basolateral membranes of choroidal epithelium. This finding is in line with the previous studies, in which bidirectional secretion of chemokines by intestinal epithelia has been demonstrated and found to be necessary for leukocyte migration across this epithelial barrier (McCormick et al, 1995, 1998). In our
The lack of changes in paracellular permeability of epithelial monolayers after exposure to CCL2 contrasts with increased permeability of the blood-brain barrier observed in response to this chemokine under both
The above-discussed features of choroidal epithelium, such as the ability to synthesize CCL2 in response to injury and bidirectional secretion of this chemokine across the apical and basolateral membranes of choroidal epithelial cells, strongly suggest that the BCSFB has a role in posttraumatic invasion of monocytes. This idea is further supported by our electron microscopic analysis of choroidal tissue. Similar to the movement of neutrophils (Szmydynger-Chodobska et al, 2009), the migration of monocytes across the BCSFB appeared to involve the paracellular pathway. The movement of monocytes along the paracellular pathway was frequently associated with the widening of space between invading inflammatory cells and the adjacent epithelial cells (see Figures 4H and 4I), a phenomenon not observed for migrating neutrophils (Szmydynger-Chodobska et al, 2009). Interestingly, monocytes were sometimes found to invade the ipsilateral CP in tandem with neutrophils (see Figure 4J). The synergistic interactions between monocyte and neutrophil chemoattractants (Gouwy et al, 2004) could have a role in the movement of these two types of inflammatory cells together across the BCSFB.
Presently, it is not well defined how monocytes, or other inflammatory cells, invade the brain parenchyma after crossing the BCSFB. Also, little is known about the possible expression of cell adhesion molecules at ependymal and pial/glial linings bordering the CSF space. Monocytes have the ability to produce a variety of matrix metalloproteinases (Newby, 2008), and the activation of these metalloproteinases have been shown to be a critical factor in the recruitment of inflammatory cells to the brain tissue (Toft-Hansen et al, 2006). Carried by the bulk flow of CSF, monocytes may passage from the lateral cerebral ventricle to the cistern of velum interpositum located above the third cerebral ventricle, and may also enter the subarachnoid CSF space near the injury site. Our previous studies (Chodobski et al, 2003) suggest that these parts of the brain CSF space have an important role in influx of peripheral inflammatory cells into the brain parenchyma. The investigations of experimental autoimmune encephalomyelitis in rodents, an animal model of multiple sclerosis, have also shown that peripheral inflammatory cells, such as T cells, may enter the neural tissue from the CSF by moving along the perivascular, Virchow-Robin, space (Bartholomaus et al, 2009). Our preliminary observations (unpublished data) suggest that after injury, monocytes may use the same route to invade the brain parenchyma from the CSF.
Footnotes
Acknowledgements
The authors thank Ms Julie Sarri for her technical assistance, Ms Virginia Hovanesian for her help in acquiring and processing confocal microscopy images, and Ms Carol Ayala for her help with electron microscopy.
The authors declare no conflict of interest.