
Abstract
Chemokines and their receptors have crucial roles in the trafficking of leukocytes, and are of particular interest in the context of the unique immune responses elicited in the central nervous system (CNS). The chemokine system CC ligand 2 (CCL2) with its receptor CC receptor 2 (CCR2), as well as the receptor CXCR2 and its multiple ligands CXCL1, CXCL2 and CXCL8, have been implicated in a wide range of neuropathologies, including trauma, ischemic injury and multiple sclerosis. This review aims to overview the current understanding of chemokines as mediators of leukocyte migration into the CNS under neuroinflammatory conditions. We will specifically focus on the involvement of two chemokine networks, namely CCL2/CCR2 and CXCL8/CXCR2, in promoting macrophage and neutrophil infiltration, respectively, into the lesioned parenchyma after focal traumatic brain injury. The constitutive brain expression of these chemokines and their receptors, including their recently identified roles in the modulation of neuroprotection, neurogenesis, and neurotransmission, will be discussed. In conclusion, the value of evidence obtained from the use of
Introduction
Chemokines, or chemotactic cytokines, are classically defined by their ability to induce directional migration and activation of leukocyte subsets into inflammatory sites. Since the identification of the first human chemokines nearly two decades ago, extensive research has accumulated showing the significant contribution of these small, peptide mediators to inflammatory conditions. In the central nervous system (CNS), chemokines such as CC ligand 2 (CCL2) and its receptor CC receptor 2 (CCR2) have been implicated in neuropathologies ranging from traumatic brain injury (TBI) to autoimmune diseases.
Chemokines are classified on the basis of structural features, which in turn, give rise to their functional specificity. The two main categories recognized are CXC or α-chemokines and CC or β-chemokines; CXC chemokines have one amino-acid residue separating two conserved cysteines and are primarily chemotactic for neutrophils, whereas CC chemokines that contain two adjacent cysteines are attractants for monocytic cells and lymphocytes (Zlotnik and Yoshie, 2000). There is considerable overlap and interaction between related chemokines and their receptors, whereby one chemokine can bind to various receptors, resulting in redundancy within the signaling network.
This review aims at providing an outline of the CCL2/CCR2 and CXCL8/CXCR2 chemokine networks in the brain, their functions, and contribution to pathologic conditions. Constitutive expression of chemokines in the CNS and the proposed roles of these mediators in neurogenesis, neuroprotection, and neurotransmission will also be discussed. Finally, we will report the current progress and issues associated with drug development aimed at therapeutically targeting chemokines in the CNS and systemic diseases. Multiple database searches were conducted from 2006 to 2009 to identify relevant references, and retrieved documents were hand-searched for additional publications. References included in this review were those deemed significant to the development of a solid understanding of the chemokine networks discussed.
The macrophage chemoattractant CCL2
There are five known members of the monocyte chemoattractant protein (MCP) family, designated as CCL2, CCL8, CCL7, CCL13, and CCL12 (MCP-1–5, respectively). Each family member attracts a different subset of leukocytes after binding with different affinities to several receptors (Gouwy et al, 2004). Although CCL2, CCL7, and CCL8 are able to signal through the CCR2 receptor, CCL2 is the most potent at activating signal transduction pathways leading to monocyte transmigration (Sozzani et al, 1994).
CCL2, also known as MCP-1/JE, was the first human chemokine to be characterized (Rollins, 1996; Yoshimura et al, 1989). Its ability to activate and attract cells of the monocyte lineage including macrophages, monocytes, and microglia has been repeatedly shown by
In the brain, a positive correlation has been found between the level of
CCR2 receptor structure and intracellular signaling in the central nervous system
CCL2 binds primarily to the G-protein-coupled receptor CCR2, an interaction that is responsible for the initial phase of monocyte recruitment (Dzenko et al, 2001; Dzenko et al, 2005; Kuziel et al, 1997). Dimerized CCL2 binds the receptor, which is then internalized and removed from the cell surface. This process has been proposed to regulate extracellular CCL2 levels (Mahad et al, 2006; Tylaska et al, 2002; Zhang and Rollins, 1995). Downstream targets of CCR2 signaling include phosphatidylinositol-3 kinase, mitogen-activated protein kinases, and protein kinase C, indicating that a wide range of intracellular pathways may be involved in cellular responses elicited by CCL2 (Stamatovic et al, 2005; Wain et al, 2002).
In the CNS, CCR2 expression has been reported on various cell types, including neurons, astrocytes, microglia, neural progenitor cells, and microvascular endothelial cells (Banisadr et al, 2002, 2005; Coughlan et al, 2000; Gourmala et al, 1997; Stamatovic et al, 2005). During normal conditions, expression seems to be at consistently low levels. Astrocyte and microglial CCR2 expression seems to be quite heterogeneous and subject to significant upregulation during an inflammatory response (Andjelkovic et al, 2002; Croitoru-Lamoury et al, 2003; White et al, 2005).
Consistent with studies using
The CCL2/CCR2 network in the central nervous system
In the brain, CCL2 is predominantly produced by astrocytes and resident microglia, and to a lesser extent, by endothelial cells (Barna et al, 1994; Berman et al, 1996; Glabinski et al, 1996; Hanisch, 2002; Harkness et al, 2003). CCL2 is also released by infiltrating macrophages upon their migration into the brain parenchyma, implying the presence of autocrine regulation that perpetuates cell recruitment and activation (Clavo et al, 1996; Gourmala et al, 1997; Gunn et al, 1997; Peterson et al, 1997). Neurons are yet another source, producing detectable levels of CCL2 after brain ischemia (Che et al, 2001; Gourmala et al, 1997), transection of facial or hypoglossal nerves (Flugel et al, 2001), and lipopolysaccharide administration (Gourmala et al, 1997).
A wide range of stimuli can trigger CCL2 production and release during an inflammatory response. Treatment with lipopolysaccharide, interferon, interleukin-1 beta (IL-1β), colony-stimulating factor-1, transforming growth factor-β, and tumor necrosis factor-α (TNFα) can induce CCL2 expression in different cell types either
The CCL2/CCR2 network in brain development and neurotransmission
A distinct pattern of CCL2 and CCR2 expression has been identified at different embryonic stages in relation to the cytoarchitectural organization of the CNS, implying a role for this chemokine network during brain development (Meng et al, 1999; Rezaie et al, 2002). Treatment of rat embryonic cultures with CCL2 and CCL7 increased the differentiation of cells toward a dopaminergic phenotype (Edman et al, 2008). Complementing these data, the application of CCL2 to dopaminergic neurons
CCL2 as a modulator of blood–brain barrier permeability
The highly selective blood–brain barrier (BBB) is largely impervious to circulating leukocytes. As CCL2 production in the brain is primarily intraparenchymal, it is still unclear how this hydrophilic protein communicates with the periphery to attract blood-borne monocytes. It is conceivable that CCL2 is released directly into the bloodstream by astrocytes and brain microvascular endothelial cells, which comprise the BBB. Alternatively, CCL2 may be transported transcellularly across the BBB, possibly by interaction with specific carrier molecules, such as caveolin-1 (Ge and Pachter, 2004).
In addition to its chemotactic properties, recent evidence indicates that CCL2 has direct effects on BBB permeability (Dzenko et al, 2005; Song and Pachter, 2004). Stamatovic et al (2005) showed that exposure of astrocytes and brain microvascular endothelial cells to CCL2
The CCL2/CCR2 network in brain pathologies
Multiple Sclerosis
Multiple sclerosis (MS) is a chronic autoimmune disease characterized by extensive demyelination and inflammation, leading to a severe and progressive neurologic impairment. Inflammation has a critical role in MS, predominantly mediated by auto-reactive T cells infiltrating the brain parenchyma (Mahad and Ransohoff, 2003). The presence of CCL2 has been shown in autopsy tissue obtained from patients with both active and chronic MS, correlating with regions of hypertrophic astrocytes and macrophage infiltration, thus implicating CCL2 in this pathology (McManus et al, 1998; Simpson et al, 1998; Van Der Voorn et al, 1999). Interestingly however, CCL2 levels measured in the cerebrospinal fluid (CSF) of patients with MS was consistently attenuated compared with healthy individuals, perhaps as a result of its binding to circulating monocytes, which then downregulate CCR2 as they cross the BBB (Franciotta et al, 2001; Sindern et al, 2001).
Multiple sclerosis can be modeled in rodents by inducing experimental autoimmune encephalomyelitis (EAE), after inoculation of a myelin component, such as myelin basic protein. Elevated astrocytic
Ischemic Brain Injury
After cerebral stroke in patients, elevated CCL2 has been detected in both serum and CSF (Arakelyan et al, 2005; Losy and Zaremba, 2001). Transient occlusion of the middle cerebral artery (MCAO), an experimental model of ischemic stroke, similarly triggers CCL2 production in the rodent brain (Che et al, 2001; Gourmala et al, 1997). In transgenic
Traumatic Brain Injury
Although implicated in TBI, the precise function of CCL2 in the time course of delayed brain damage after trauma still needs to be fully elucidated. A unique role for this chemokine in TBI is substantiated by the observation that
Most studies investigating the production of CCL2 in TBI have used noncontusional models, with some involving surgical penetration of the cortex (Babcock et al, 2003; Glabinski et al, 1996) or resection of the brain tissue (Hausmann et al, 1998; Muessel et al, 2000). Lateral fluid percussion and closed head injury models should be considered superior for examining inflammation associated with mechanisms of cerebral contusion formation and thus, most accurately reproducing human TBI. A recent study using a lateral fluid percussion model showed a transient increase in CCL2 peaking at 8 to 12 h in the injured cortex (Rhodes et al, 2009). In our laboratory, CCL2 protein levels peaked between 4 and 12 h after focal closed head injury in the mouse (Semple
Although most chemokines target more than one receptor and most chemokine receptors bind to multiple ligands
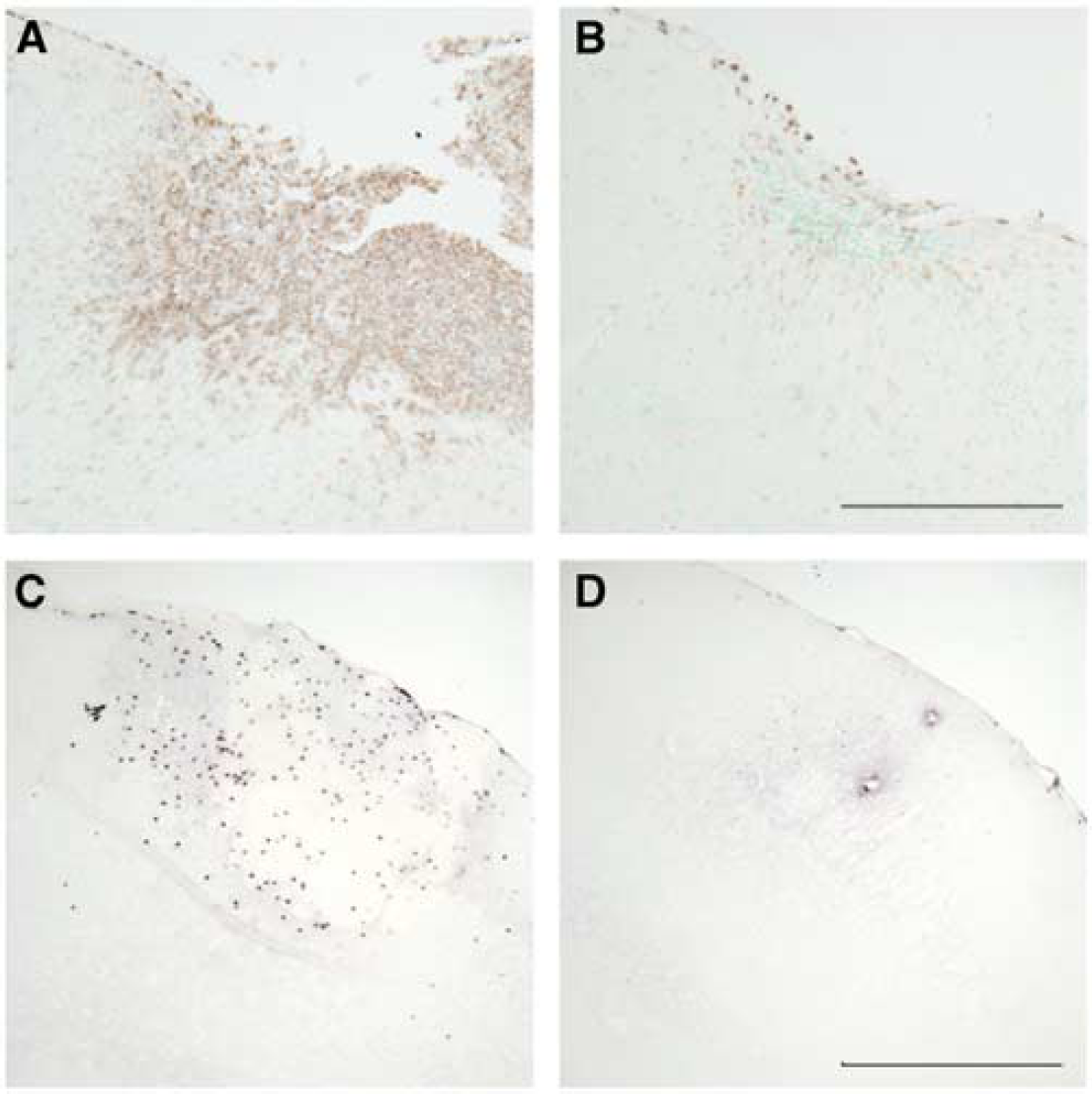
Effect of chemokine ligand or receptor deficiency on leukocyte infiltration after experimental focal traumatic brain injury. Upper microscope images illustrate the accumulation of macrophages and activated microglia in the injured cortex at 4 weeks after closed head injury, which was considerably reduced in
Neuroprotective and neurotrophic properties of CCL2-mediated signaling
Despite robust experimental evidence indicating that elevated CCL2 and subsequent recruitment of macrophages into the brain is detrimental, awareness of the fact that chemokines also possess pleiotropic and beneficial properties, beyond chemotaxis, is increasing. A potential role in tissue repair has been identified in the context of a skin wound model, with
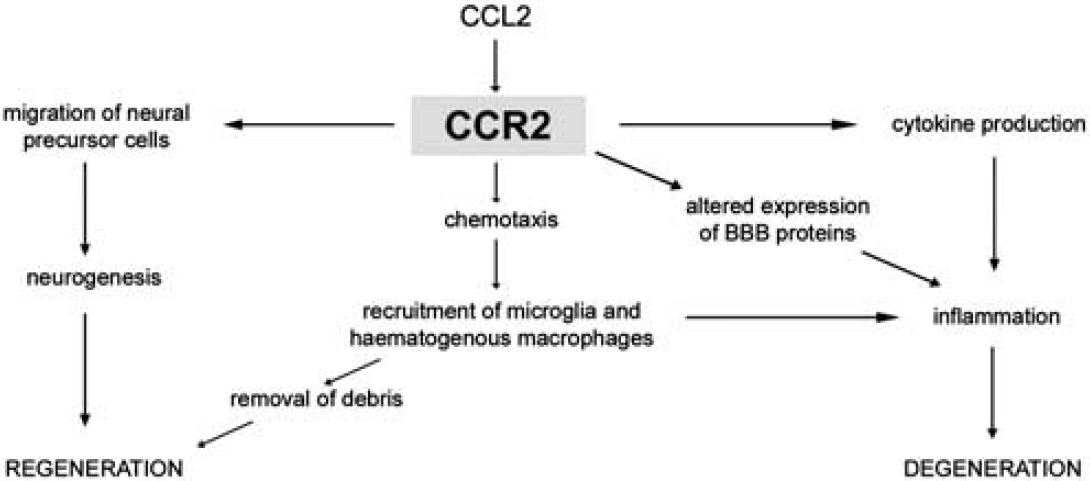
The roles of CCL2/CCR2 in brain inflammation and injury. CCL2 induces the recruitment of macrophages, production of cytokines, and direct alteration of the expression of endothelial cell tight-junction proteins to increase blood–brain barrier (BBB) permeability, which contributes to inflammation in the brain, potentially exacerbating neuronal loss. CCL2-mediated macrophage accumulation may also be beneficial, as these phagocytic cells remove myelin debris, which otherwise inhibits regeneration. Furthermore, CCL2 is chemotactic for neural precursor cells and thus, may influence repair after injury by enhancing neurogenesis.
In the brain, speculation that CCL2 may be important in neurogenesis has been gaining momentum over the past decade, as recombinant CCL2 was first shown to promote glial cell proliferation and growth
To date, the role of CCL2 in adult neurogenesis after brain injury has been investigated most thoroughly in models of stroke. Lui et al (2007) showed that MCAO induced a strong upregulation of CCL2 within the neurogenic subventricular zone. CCL2 reportedly promoted motility of adult neural progenitor cells and enhanced their differentiation into neurons, whereas the neutralization of the chemokine abolished these effects (Lui et al, 2007). Another group has also identified CCL2/CCR2-dependent migration of neuroblasts within the brain to sites of ischemic damage (Yan et al, 2007).
Current strategies for the therapeutic targeting of CCL2/CCR2
Accumulating preclinical data indicate that the CCL2/CCR2 network is a promising target to therapeutically reduce inflammatory cell infiltrates and tissue damage. Thus far, the pharmaceutical industry has primarily focused on the use of CCR2 antagonists, resulting in several compounds entering the early phases of clinical trials for application in peripheral inflammatory conditions, including rheumatoid arthritis (small molecule antagonist INCB3284, Incyte, Wilmington, DE, USA), atherosclerotic cardiovascular disease (neutralizing antibody MLN1202, Millenium Pharmaceuticals, Cambridge, MA, USA), and IgA nephropathy (CCL2-LPM, Osprey Pharmaceuticals, Saint-Laurent, QC, Canada) (Horuk, 2009).
Information on the progress of clinical trials with CCR2 antagonists is scarce, with many companies reporting either no or only slight developments in their research. This lack of progress may reflect the complexity and redundancy of the chemokine network and its interactions, thus our limited understanding of this signaling network in disease (Horuk, 2009). Application of innovative technologies may assist in effectively targeting the CCL2/CCR2 network in neuroinflammation, e.g., with the use of interference RNA to silence specific genes, RNA oligonucleotides, or dominant negative mutants of chemokine ligands to eliminate receptor function.
The question also remains as to whether CCR2 antagonists would be able to effectively cross the BBB for treatment of CNS conditions, such as TBI, MS, or stroke. In the context of TBI, we know that the BBB has increased permeability for a short period of time after injury, which allows the infiltration of serum proteins and circulating leukocytes into the CNS (Habgood et al, 2007). This time course of BBB dysfunction may provide a therapeutic window for the administration of drugs which cannot normally cross the intact BBB.
The neutrophil chemoattractants CXCL1, CXCL2, and CXCL8
Just as CCL2 is considered to be the prototypical monocyte-attracting CC chemokine, CXCL8 (also known as IL-8) is the most intensely studied CXC chemokine, first identified as a powerful mediator able to induce morphologic changes and degranulation of neutrophils (Baggiolini et al, 1989). Since then, evidence for the role of CXCL8 as a key player in neutrophil transmigration has been shown both
In rodents, two main ligands perform the same functions as human CXCL8, namely CXCL1 (also known as keratinocyte-derived chemokine, KC) and CXCL2 (macrophage inflammatory protein, MIP-2). These chemokines share 78% sequence homology, and have been recognized as the most critical CXCR2 ligands mediating the neutrophil influx characteristic of inflammatory disorders, including psoriasis, rheumatoid arthritis, atherosclerosis, and irritable bowel disease. Compounding this nomenclature confusion further, CXCL1 and CXCL2 have also been identified in humans as growth-regulated gene-alpha (GROα) and GROβ, respectively.
CXCR2 receptor structure and intracellular signaling in the central nervous system
As is the case for all known chemokine receptors, CXCR2 is a seven transmembrane G-protein-coupled receptor. Chemokines that are able to bind CXCR2 and mediate neutrophil chemotaxis contain the glutamic acid–leucine–arginine (ELR) tripeptide motif in their N-terminal domain, as opposed to non-ELR CXC chemokines (such as CXCL12), which lack this motif and generally attract lymphocytes. Upon ligand binding, CXCR2 activates G-protein-mediated phosphoinositide hydrolysis to generate diacylglycerol and inositol 1,4,5-trisphosphate, which then activate protein kinase C allowing the mobilization of calcium to initiate cellular responses (Wu et al, 1993). The ligand–receptor complex is phosphorylated and endocytosed after signaling through clathrin-dependent pathways, and once internalized, CXCR2 may be either degraded or transported back to the cell membrane for reexpression (Rose et al, 2004). Receptor endocytosis is believed to be essential for the regulation of chemotactic migration, although the evidence for this is still controversial (Rose et al, 2004; Yang et al, 1999). A recent study using receptor knockout mice has elegantly shown that functional CXCR2 is essential for the removal and thus, for the regulation of chemokines in the circulation and brain, identifying a novel scavenging role of these receptors in chemokine homeostasis (Cardona et al, 2008).
The CXCL8/CXCR2 network in the central nervous system
As with the CCL2/CCR2 network, ligands of the CXCR2 receptor appear to be expressed at low basal levels in the brain and upregulated in pathology. Reported sources of CXCL1, CXCL2, and CXCL8 include activated microglia, astrocytes, and endothelial cells; in addition, infiltrated neutrophils themselves are a major source of CXC chemokines, potentially amplifying leukocyte recruitment (Lu et al, 2005; Valles et al, 2006).
Peripherally, the receptor CXCR2 is most highly and uniformly expressed by neutrophils, although it has also been shown on eosinophils, mast cells, and on a small subset of effector T cells (Lippert et al, 2004). In the CNS, widespread neuronal CXCR2 has been detected by immunohistochemistry and
The CXCL8/CXCR2 network in brain development and neurotransmission
Recent data substantiating the involvement of CXCR2 signaling in neuronal electrical activity, neurotransmitter release, and synaptic plasticity in the CNS have contributed to the emerging concept of the ‘chemokinergic’ system as a new class of neurotransmitters (Parsadaniantz and Rostene, 2008). Giovannelli et al (1998) found that Purkinje neurons in mouse cerebellar slices respond to CXCL8 and CXCL1 treatment with a transient increase in calcium, neurotransmitter release, and impaired long-term depression. A separate study showed that CXCR2, when coexpressed on cerebellar Purkinje neurons with the Glut1 subunit of AMPA (α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate) receptors, can alter the functional profile of these ion channel receptors by increasing channel opening frequency and thus, enhancing glutaminergic activity (Lax et al, 2002). The ability of CXCL8 to modulate calcium channel excitability through CXCR2 on rat septal neurons has also been shown (Puma et al, 2001). These findings corroborate a function for CXCR2 signaling beyond neutrophil chemoattraction, which is supported by widespread constitutive CXCR2 expression throughout the adult brain.
A role for this chemokine system in brain maturation has also been suggested, as the expression of both CXCL2 and CXCR2 is widely distributed during early developmental stages in the mouse forebrain, hippocampus, thalamus, and floor plate (Luan et al, 2001). Considering the chemotactic properties attributed to this chemokine family, it is conceivable that signaling through CXCR2 may contribute to the trafficking of neuronal processes to form appropriate synapses during brain development.
Furthermore, constitutive expression of CXCR2 by human oligodendrocytes has been shown both
The CXCR2 network in brain pathologies
Multiple Sclerosis
The CXCL1/CXCR2 system seems to be involved in the pathology of MS. First, elevated CXCL8 was detected in the CSF of MS patients compared with controls, a phenomenon which is attenuated in patients receiving interferonβ-1a therapy (Lund et al, 2004). Expression of CXCR2 has been described in normal and proliferating oligodendrocytes within active MS lesions (Omari et al, 2006), as well as on activated microglia bordering the lesion (Filipovic et al, 2003). Proximal reactive astrocytes reportedly secrete the ligand CXCL1, the production of which can also be induced
Ischemic Brain Injury
A substantial number of studies have shown pronounced neutrophil infiltration in ischemic brain tissue (Emerich et al, 2002). Attenuation of neutrophil transmigration by an anti-CXCL8 antibody resulted in a 60% reduction in infarct volume after transient focal ischemia in rabbits (Matsumoto et al, 1997). The chemokine-mediated infiltration of neutrophils appears to contribute to reperfusion injury rather than to the formation of the initial infarct, as blockage of CXCR2 signaling by the noncompetitive allosteric inhibitor reparixin, was able to significantly reduce tissue damage after transient, but not permanent, MCAO in rats (Garau et al, 2005). Although reparixin binds with higher affinity to CXCR1 than to CXCR2, this compound has been shown to reduce neutrophil infiltration by 40–50%, reduce infarct volume, inhibit long-term inflammation, and improve recovery of sensorimotor function after experimental stroke (Garau et al, 2005; Villa et al, 2007). Another compound which was generated as a dual inhibitor of CXCR1 and CXCR2, DF2156A, showed similar neuroprotective effects (Garau et al, 2006).
Interestingly, several studies have detected the elevation of CXCL1 and CXCL8 in the periphery after ischemic brain injury, indicating the occurrence of systemic inflammatory events resulting from damage to the CNS. Circulating levels of CXCL8 were reportedly elevated in patients after stroke (Kostulas et al, 1998). CXCL1 was found to be increased in the plasma, liver, and lungs of mice after experimental MCAO at times preceding the peak of chemokine expression in the injured cortex and striatum (Chapman et al, 2009). These data illustrate the fact that chemokine expression in the body is both spatial and temporal, indicating that local production of chemokines within the brain parenchyma may be only one of the mechanisms contributing to the chemotactic gradients which regulate leukocyte migration.
Traumatic Brain Injury
In the clinic, we along with others have reported that CXCL8 elevated in the CSF of patients after severe TBI in both adults and children correlated with severe BBB dysfunction (Kossmann et al, 1997) and increased mortality (Whalen et al, 2000), suggesting that measurement of CSF CXCL8 may be an indicator of poor prognosis. Levels of CXCL8 were significantly higher in the CSF than in the serum, supporting intrathecal production as the prominent source. Our laboratory has previously shown that the adhesion molecule sICAM-1, the concentration of which in the CSF of TBI patients correlated with the severity of BBB breakdown (Pleines et al, 1998), is able to induce CXCL2 production in cultured mouse astrocytes and brain microvascular endothelial cells (Otto et al, 2002).
A prominent influx of neutrophils into the damaged brain parenchyma within the early hours after experimental TBI has been repeatedly shown by our group (Bye et al, 2007; Stahel et al, 2000). However, little work has been carried out to investigate the mechanisms underlying the contribution of CXCR2-mediated signaling to secondary tissue damage after TBI. We have previously shown that CXCL2 and CXCR2 are acutely increased in the ipsilateral hemisphere after closed head injury in mice, peaking between 4 and 8 h after injury (Otto et al, 2001). In a separate study, using a controlled cortical impact model in rats, Valles et al (2006) showed that production of the ligands CXCL1 and CXCL2 was increased as early as 2 h after trauma, remaining elevated for more than 24 h. Levels of the CXCR2 receptor were elevated in the ipsilateral cortex by 8 h after injury (Valles et al, 2006). Another study detected upregulated CXCR2 expression in the contused hemisphere by 4 h after lateral fluid percussion injury in mice (Rhodes et al, 2009). Recently completed work from our laboratory showing reduced accumulation of neutrophils in
Interestingly, neither CXCL2 elevation nor neutrophil infiltration was observed by our group in a rat model of diffuse traumatic axonal injury (Rancan et al, 2001), suggesting that these key features of focal brain injury are not as relevant for patterns of diffuse brain damage. Clearly, more research into the specific role of CXCR2 in the context of different forms of TBI is warranted.
Neuroprotective and neurotrophic properties of CXCR2-mediated signaling
A robust neutrophilic infiltration is often the first cellular response to acute infection or injury. Rather than contributing to tissue damage, neutrophils in infectious diseases seem to have a vital role in the containment and neutralization of bacteria to minimize tissue degradation. For example, after inoculation with
Independent of their chemotactic properties, additional neuroprotective and neurotrophic functions of the ligands CXCL8, CXCL1, and CXCL2 are emerging (Figure 3). First, it has been shown that CXCR2 signaling is involved in the resistance of astrocytes and neurons to cell death
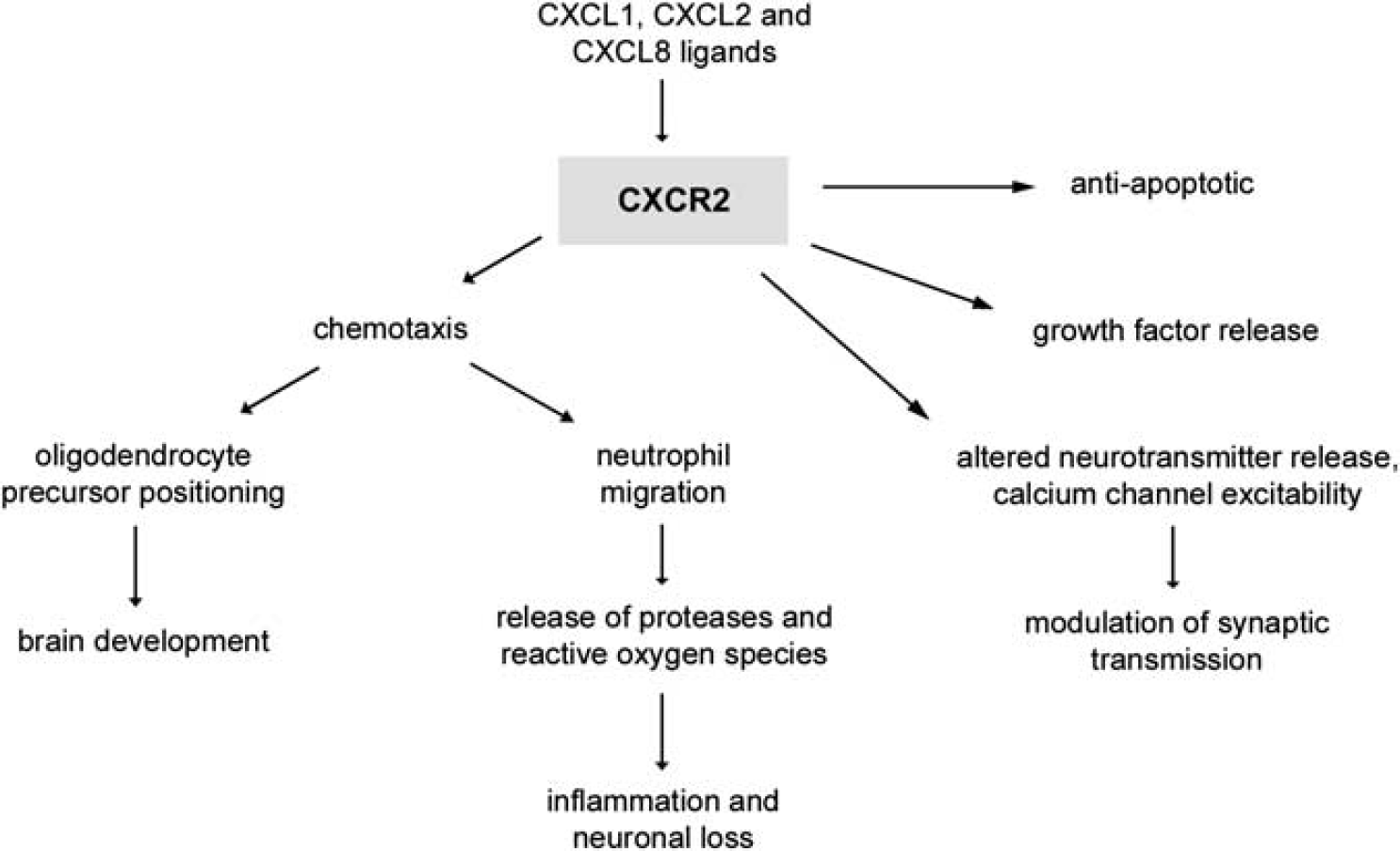
The multiple functions mediated by CXCR2 signaling in the CNS. CXCR2 is the main receptor involved in neutrophil chemotaxis, leading to cell migration into the brain during injury, infection or disease. Neutrophils perpetuate the neuroinflammatory response by the release of enzymes such as proteases, contributing to neuronal degeneration. Independent of this role, CXCR2 signaling is involved in chemotaxis of oligodendrocyte precursors during development, the release of growth factors, mediating self-defense mechanisms against Fas-initiated apoptotic cell death, and modulating synaptic transmission through altering calcium channel excitability and neurotransmitter release.
Second, neurotrophic properties have been attributed to ligands signaling through CXCR2
Both CXCL2 and CXCL8 have also been shown to be neurotrophic for cerebellar granule neurons, as treatment with these chemokines
Current strategies for the therapeutic targeting of CXCR2
Previous attempts to reduce neutrophil infiltration by the administration of neutralizing antibodies to individual chemokines have produced mixed results. This variability may depend on the model, the chemokine, and the organ being targeted. Experimentally, neutralizing antibodies against the receptor CXCR2 almost completely inhibited neutrophil infiltration in an intraperitoneal inflammation murine model, whereas blocking either CXCL1 or CXCL2 ligands alone produced only a partial attenuation (Tanimoto et al, 2007). Such studies emphasize the advantage of blocking receptors to combat the considerable redundancy within chemokine networks, as neutralization of an individual chemokine may be compensated by another chemokine binding the same receptor. Any issues caused by currently unknown differences between human CXCL8 and its murine homolog ligands may also be avoided by targeting the joint receptor, CXCR2.
The alternative approach to study the role of CXCR2 is by modeling inflammatory diseases in mice overexpressing or genetically deficient for the specific gene. It is important to remember that, although they are invaluable for defining the roles of particular molecules
Although these abnormalities provide important information on the role of CXCR2 in development and physiology, they can interfere with experimental studies investigating the role of this chemokine network in disease paradigms. Abnormalities present in knockout mice provide clues as to the possible side effects that may result from CXCR2 antagonism, indicating that patients who receive CXCR2-targeted therapeutics should be monitored carefully during clinical trials.
Conclusions
Chemokines are multifunctional mediators in the brain, both in health and pathology. The best characterized chemokine–receptor networks involving CCL2/CCR2 and CXCL8/CXCR2 have nonredundant functions not only in regulating immune cell infiltration into the CNS during neuropathology but also in physiologic processes, including neurogenesis, neuroprotection, and neurotransmission. The use of gene knockout and transgenic mice has been invaluable to increase our understanding of the different mechanisms regulated by chemokines. For conditions in which neuroinflammation is a key pathologic event such as MS, stroke, and TBI, therapeutic targeting of chemokine networks to reduce the inflammatory infiltrate and its consequences may have considerable potential benefit. Ultimately, targeting chemokines in heterogeneous conditions such as TBI with the aim of improving patient outcomes will most likely require the admission of multiple drug agents targeting several deleterious pathways, such as excitotoxicity and oxidative stress, all of which contribute to downstream neuronal degeneration and subsequent neurologic impairment.
Footnotes
The authors declare no conflict of interest.