
Abstract
We assessed the neuroprotective potential of α2-adrenoceptors in ischemic stroke using mice with targeted deletions of individual α2-adrenoceptor subtypes (α 2A −/−, α 2B −/−, α 2C −/−, α 2A/C −/−). The effects of the α2-adrenoceptor agonist clonidine were studied in parallel. Focal cerebral ischemia was induced with or without clonidine pretreatment by transient middle cerebral artery occlusion. Neurologic outcome and infarct volumes were evaluated on day 1. Cerebral blood flow (CBF) and mean arterial pressure were determined. α2-Adrenoceptor null mice did not display larger infarct volumes compared with wild-type (WT) mice under basal conditions (P>0.05). In line with this finding, pretreatment with clonidine did not protect from ischemic brain damage in WT mice or α 2A −/−, α 2B −/−, and α 2C −/− mice. Clonidine induced smaller infarct volumes only in α 2A/C −/− mice (P < 0.05), but this did not translate into improved neurologic function (P > 0.05). Importantly, while clonidine caused a significant decrease in arterial blood pressure in all groups, it had no blood pressure lowering effect in α 2A/C −/− mice, and this correlated with higher CBF and smaller infarct volumes in this group. In summary, we could not demonstrate a neuroprotective function of α2-adrenoceptors in focal cerebral ischemia. Careful controlling of physiological parameters relevant for stroke outcome is recommended in experimental stroke studies.
Introduction
The sympathetic nervous system is an essential regulator of cardiovascular function. Excessive activation of the sympathetic nervous system and subsequent catecholamine release (e.g., adrenaline and noradrenaline) is linked to various disease states such as hypertension, arrhythmias, or chronic heart failure. In the central nervous system, high levels of catecholamines can exert detrimental effects on neuronal tissue (Stein and Cracco, 1982; Globus et al, 1988) and have been held responsible for aggravating ischemic stroke under experimental conditions (Prass et al, 2003). The molecular effects of catecholamines are mediated by G-protein-coupled adrenoceptors. To date, nine different adrenergic receptor subtypes have been cloned, including α1A, α1B, α1D, α2A, α2B, α2C, and β1, β2, β3, (Bylund et al, 1994).
While α1-adrenergic receptors are mainly found in peripheral blood vessels where they mediate vaso constriction, β-adrenergic receptors are primarily involved in regulating cardiac function and smooth muscle tone for instance in the lungs (Philipp and Hein, 2004). Transgenic mouse models with targeted deletions in individual α2-adrenoceptor genes are useful tools to delineate the physiological and pharmacological functions of the distinct α2-adrenoceptor subtypes (Philipp et al, 2002a). Because the three α2-adrenoceptor subtypes have all been localized in the central nervous system, they are of particular interest for neuroprotection studies (Gilsbach and Hein, 2008). Indeed, α2A and α2C can serve as presynaptic negative feedback regulators and inhibit the release of catecholamines in the central nervous system on stressful stimuli (Trendelenburg et al, 2003; Gilsbach et al, 2009).
Following ligand binding, α2-adrenoceptors activate a specific subgroup of heterotrimeric G-proteins (Gi/o), leading to reduced cAMP levels due to the inhibition of adenylyl cyclase, opening of GIRK K+ channels, inhibition of neuronal Ca2+ channels, and stimulation of mitogen-activated kinase extracellular signal regulated kinases 1/2 (ERK1/2) (Cussac et al, 2001). The inhibitory function of G-protein βγ subunits on presynaptic Ca2+ channels is supposed to be the leading step in α2-adrenoceptor-mediated signal transduction observed in postganglionic sympathetic neurons (Boehm and Kubista, 2002; Stephens and Mochida, 2005; Kubista and Boehm, 2006).
Previous findings suggest that activation of α2-adrenoceptors might exert neuroprotective functions in the context of cerebral hypoxemia, especially in the developing brain (Hoffman et al, 1991a, b ) and the hippocampus (Halonen et al, 1995). However, detailed characterization of the relative contribution of the different α2-adrenoceptor subtypes is lacking and was so far hampered by the absence of subtype-specific pharmacological agonists or antagonists, respectively.
To characterize in detail the subtype-specific neuroprotective effects of α2-adrenoceptors in the mature brain, we used an in vivo model of focal cerebral ischemia/reperfusion injury in gene-targeted mice with deletions of the three α 2 -adrenoceptor subtypes and α 2A/C −/− double knockout mice.
Materials and methods
Animals
A total of 203 mice were used in this study (119 mice for transient middle cerebral artery occlusion (tMCAO) and functional outcome evaluation, 64 mice for invasive hemodynamics, and 20 mice for regional cerebral blood flow (rCBF) measurements by laser-Doppler flowmetry). Mice were maintained in a specified pathogen-free facility. All animal procedures were approved by the responsible animal care committee of the University of Würzburg and the appropriate authorities of the State of Bavaria (Regierung von Unterfranken). The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 83-123, revised 1996) and was performed in accordance with the ARRIVE guidelines (http://www.nc3rs.org/ARRIVE).
The generation of mice with targeted deletions of α 2 -adrenoceptors was reported in detail elsewhere (Gilsbach and Hein, 2008). All genotypes were backcrossed onto a C57BL/6J genetic background for > 8 to 12 generations (Philipp et al, 2002b). Eight to 12-week-old male and female α 2 -adrenoceptors−/− mice and wild-type (WT) littermates were used.
Stroke Model
All experiments complied with the recently suggested quality standards in preclinical stroke studies (Dirnagl, 2006) and the ARRIVE guidelines (http://www.nc3rs.org/ARRIVE).
We performed surgery and evaluation of all read-out parameters (SB, CK, FL, MB) while being blinded to the respective experimental groups. The tMCAO model was applied to induce focal cerebral ischemia as described elsewhere (Clark et al, 1997). Briefly, mice were anesthetized with 2% isoflurane in a 70% N2O/30% O2 mixture. A servo-controlled heating blanket was used to maintain core body temperature to 37 °C throughout the surgery. Following a midline neck incision, a standardized silicon rubber-coated 6.0 nylon monofilament (60-1720RE; Doccol, Albuquerque, NM, USA) was inserted into the right common carotid artery and advanced via the internal carotid artery to occlude the origin of the right MCA. After 60 minutes, mice were reanesthetized and the occluding filament was removed to allow reperfusion.
Determination of Infarct Size
Animals were killed 24 hours after tMCAO. The brains were harvested and cut into three 2 mm thick coronal sections using a mouse brain matrix (Harvard Apparatus, Holliston, MA, USA). The slices were stained for 20 minutes at 37 °C with 2% TTC (2,3,5-triphenyltetrazolium chloride; Sigma-Aldrich, St Louis, MO, USA) in phosphate-buffered saline to visualize the infarctions (Bederson et al, 1986a) and planimetric measurements were obtained using the ImageJ software package (ImageJ software, National Institutes of Health, Bethesda, MD, USA). Raw infarct, ipsilateral hemispheric, and contralateral hemispheric volumes were calculated as follows: Vdirect (mm3) = (area TTC section 1 (mm2) × 2.0 mm) + (area TTC section 2 (mm2) × 2.0mm) + (area TTC section 3 (mm2) × 2.0 mm), and then corrected for the brain swelling (Ginsberg et al, 2003) according to the following formula: Vcorrected (mm3) = Vinfarct × (1-(Vi–Vc/Vc), where the term (Vi–Vc) represents the volume difference between the ischemic hemisphere and the contralateral control hemisphere and (Vi–Vc)/Vc represents this difference as a percentage of the contralateral hemisphere. The resulting edema-corrected infarct volume was expressed as percentage of the control hemisphere.
Assessment of Functional Outcome
In all animals subjected to tMCAO, we studied functional outcome after 24 hours using a modified Bederson score (Bederson et al, 1986b), according to the following scoring system: 0, no deficit; 1, forelimb flexion; 2, as for 1, plus decreased resistance to lateral push; 3, unidirectional circling; 4, longitudinal spinning or seizure activity; 5, no movement. Moreover, motor function was assessed with the string test (also known as grip test), which was performed 24 hours after surgery as described previously (Braeuninger et al, 2010). For this, mice were placed midway on a rod (‘string’) between two supports and rated as follows: 0, falls off; 1, hangs onto string by one or both forepaws; 2, as for 1, and attempts to climb onto string; 3, hangs onto string by one or both forepaws plus one or both hindpaws; 4, hangs onto string by fore- and hindpaws plus tail wrapped around string; 5, escape to the supports.
Mortality was monitored until 24 hours after tMCAO, when the animals were killed.
The exclusion criteria were as follows: (1) death within 24 hours after tMCAO, (2) subarachnoid hemorrhage (as macroscopically assessed during the brain sampling), and (3) modified Bederson score = 0 (immediately after recovery from 60 minutes tMCAO).
Of the 119 animals subjected to tMCAO, 10 animals (8.4%) met at least one of these exclusion criteria and were withdrawn from the study. Dropout rates were evenly distributed between the groups.
Regional Cerebral Blood Flow Measurement
Laser-Doppler flowmetry (Moor Instruments, Axminster, UK) was used to monitor rCBF in clonidine pretreated α 2 -adrenoceptor-deficient animals and WT controls (n = 3 to 5/group) before surgery (baseline), 15 minutes after the occlusion of the MCA, 15 minutes after removal of the occluding monofilament (reperfusion), and 24 hours after surgery (Connolly et al, 1996). For this procedure, a small incision was made in the skin overlying the temporal muscle, and a 0.7-mm flexible laser-Doppler probe (model P10) was positioned perpendicular to the superior portion of the temporal bone (6 mm lateral and 2 mm posterior from the bregma). This position corresponds to the core of the ischemic territory.
Invasive Hemodynamics
For invasive hemodynamics, α 2 -adrenoceptor knockout mice and WT controls (n = 5 to 7/group) were anesthetized with 2% isoflurane in a 70% N2O/30% O2 mixture and catheterized via the right carotid artery with a high-fidelity 1.4 F Millar microtip catheter as described (Brede et al, 2002). Hemodynamic data (blood pressure and heart rate) were digitized via a MacLab system (AD Instruments, Castle Hill, NSW, Australia) connected to an Apple G4 PowerPC computer and analyzed.
Application of Study Medication
Clonidine (Boehringer Ingelheim Pharma GmbH, Ingelheim, Germany) 40 mg/kg was injected intraperitoneally 1 hour before tMCAO and 1 hour before invasive hemodynamic and rCBF analysis.
Statistics
Results are expressed as mean ± s.d. for hemodynamics, rCBF, and infarct volumes and depicted as dot plots and median for functional outcome scores. For statistical analysis, PrismGraph 4.0 software package (La Jolla, CA, USA) was used. Numbers of experiments to detect a standardized effect size on infarct volumes ≥ 0.15 were calculated via a priori power analysis with the following assumptions: α = 0.05, β = 0.2, mean, s.d. 10% of the mean. Modified Bederson score and string test score were analyzed using the nonparametric Kruskal–Wallis test with post hoc Dunn's multiple comparison test, whereas infarct volumes, rCBF, and mean arterial blood pressure values were compared using two-way analysis of variance with post hoc Bonferroni's multiple comparison test. P values < 0.05 were considered statistically significant.
Results
Invasive Hemodynamics
Mean arterial blood pressure was significantly higher (P < 0.05) in α 2A −/− and α2A/C−/− mice than in WT controls under basal conditions (WT: 74.3 ± 4.3 mmHg; α 2A −/−: 81.9 ±6.2 mmHg; α 2B −/−: 81.3 ± 6.8mmHg; α 2C −/−: 77.2 ±4.2 mmHg; α2A/C−/−: 83.3 ± 4.5 mmHg) (Figure 1). Pretreatment with the α2-adrenoceptor agonist clonidine significantly decreased mean arterial blood pressure in all groups except for the α 2A/C -deficient group (P<0.05) (WT: 63.8 ± 3.5 mmHg; α 2A −/−: 72.6 ± 9.2 mmHg; α 2B −/−: 70.4 ± 8.0 mm Hg; α 2C −/−: 62.9 ± 4.0mmHg; α 2A/C −/−: 82.4 ± 7.0mmHg) (Figure 1).
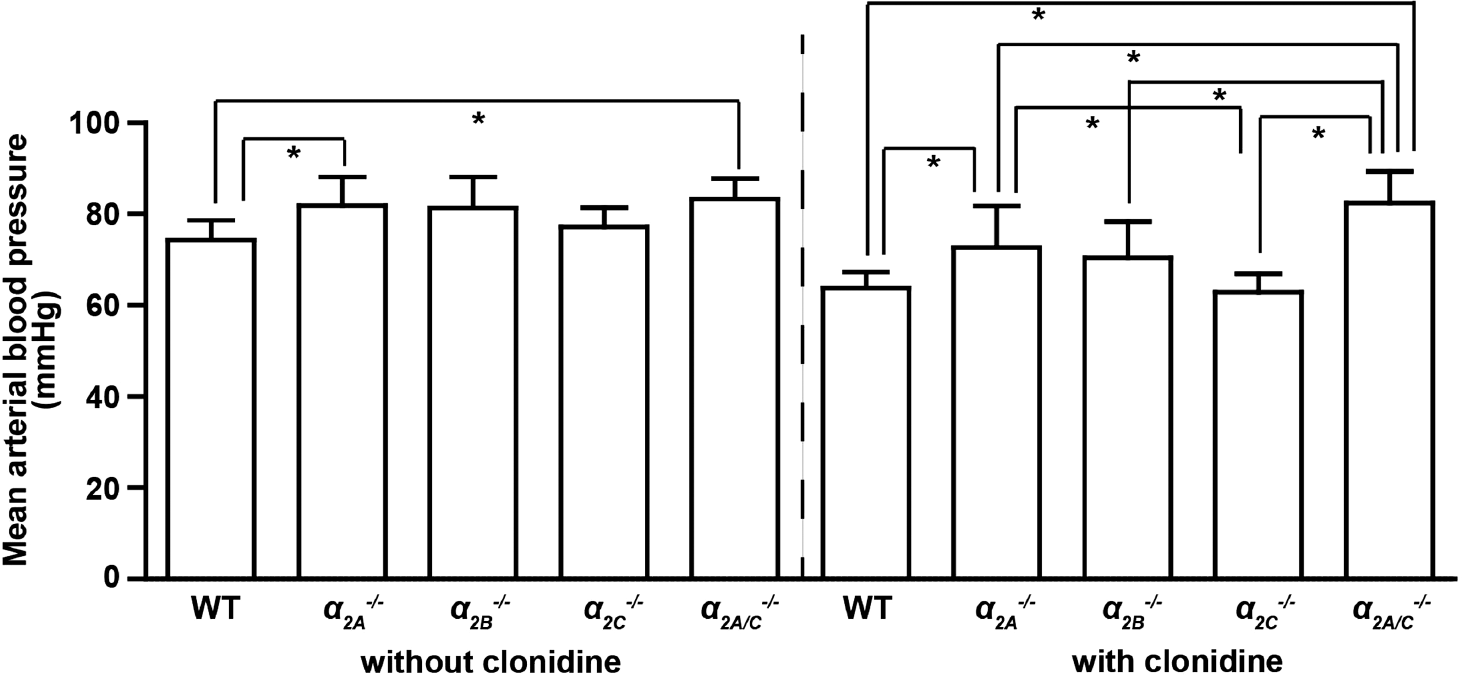
Measurement of mean arterial blood pressure. Under basal conditions (without clonidine), mean arterial blood pressure was significantly increased in α 2A −/− and α2A/C−/− animals compared with wild-type (WT) controls. After pretreatment with the α2-adrenoceptor agonist clonidine, α 2A/C −/− mice displayed significantly higher mean arterial blood pressure than WT, α 2A −/−, α 2B −/−, and α 2C −/−, mice (n = 5 to 7/group; mean ± s.d.). *P < 0.05, two-way analysis of variance (ANOVA) with post hoc Bonferroni's multiple comparison test.
Infarct Volumes
Infarct size did not differ in WT mice or gene-targeted mice under basal conditions, that is, without clonidine (WT: 50.3% ± 10.9%; α 2A −/−: 44.5% ± 14.1%; α 2B −/−: 52.7% ±13.1%; α 2C −/−: 48.6% ± 15.6%; α 2A/C −/−: 48.6% ±12.1%; P >0.05) (Figure 2). Following pretreatment with clonidine, α 2A/C −/− animals developed significantly smaller infarcts than α 2B −/−, α 2C −/−, and WT controls (WT: 53.7% ± 8.1%; α 2A −/−: 51.7% ± 13.4%; α 2B −/−: 56.6% ±12.8%; α 2C −/−,: 61.2% ± 11.8%; α 2A/C −/−: 30.2% ± 15.6%; P < 0.05 or P < 0.01) (Figure 2). No significant differences in infarct volumes were observed between clonidine-treated and untreated mice (P > 0.05).
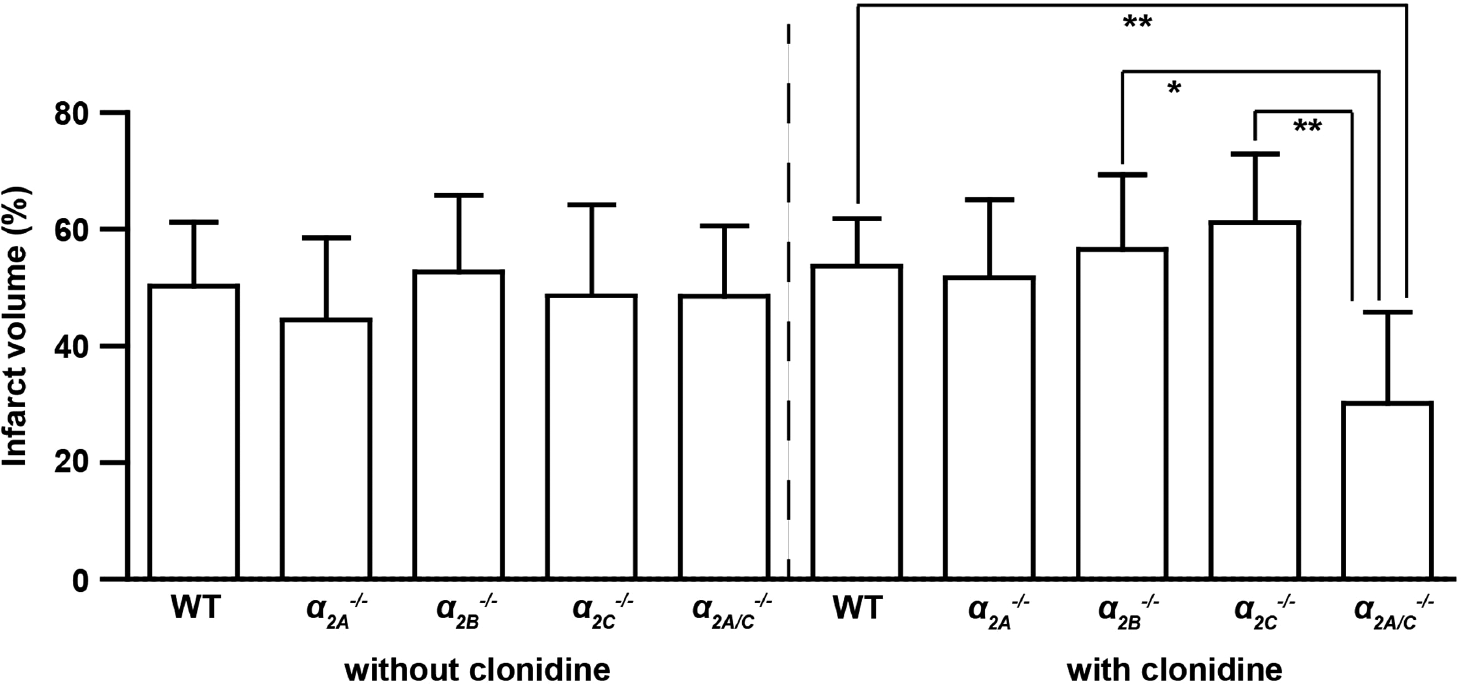
Stroke volumes 24 hours after transient middle cerebral artery occlusion (tMCAO). There were no significant differences in infarct size as assessed by infarct volumetry from 2,3,5-triphenyltetrazolium chloride (TTC)-stained brain sections between wild-type (WT) controls, α 2A −/−, α 2B −/−, α 2C −/−, and α 2A/C −/− mice without clonidine pretreatment. After pretreatment with the α2-adrenoceptor agonist clonidine, α 2A/C −/− mice developed significantly smaller infarcts than α 2B −/−, α 2C −/−, and WT mice (n = 8 to 15/group; mean ± s.d.). *P < 0.05, **P < 0.01, two-way analysis of variance (ANOVA) with post hoc Bonferroni's multiple comparison test.
Functional Outcome
Functional outcome on day 1 after tMCAO as assessed by the modified Bederson score and string test was comparable in all groups studied (P > 0.05) (Figures 3 and 4).
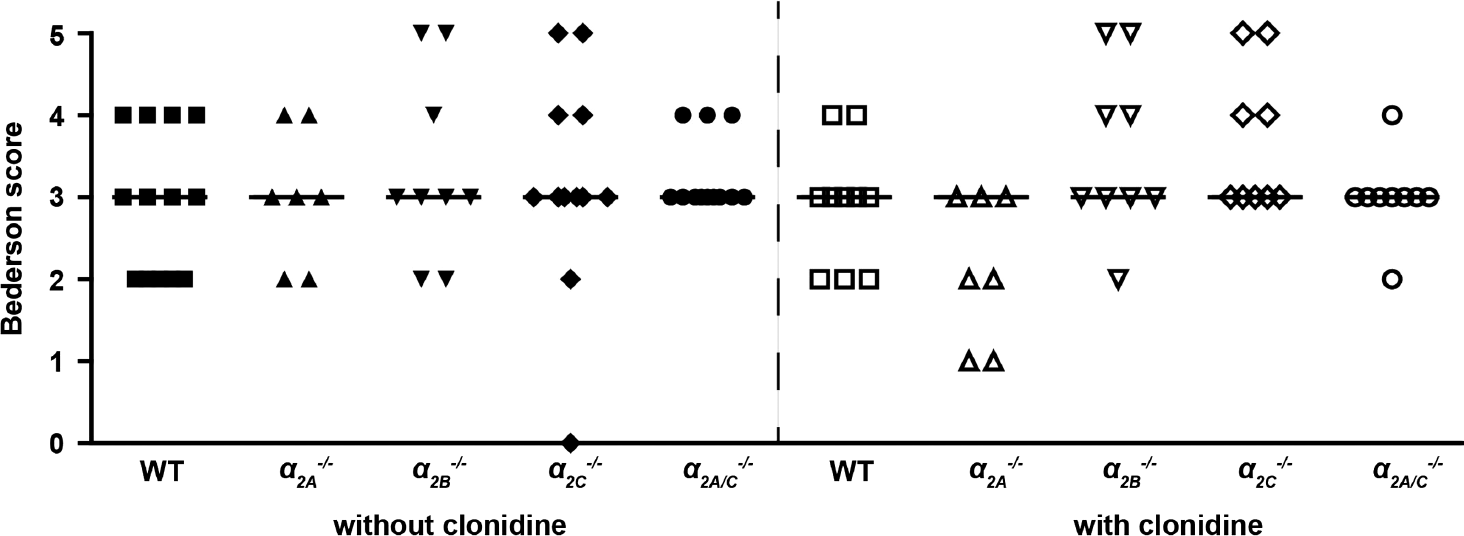
Functional outcome 24 hours after transient middle cerebral artery occlusion (tMCAO) (Bederson score). There were no significant differences between wild-type (WT) controls, α 2A −/−, α 2B −/−, α 2C −/−, and α 2A/C −/− mice without or with clonidine pretreatment (n = 7 to 15/group; horizontal bars indicate median). P > 0.05, Kruskal–Wallis test with post hoc Dunn's multiple comparison test.
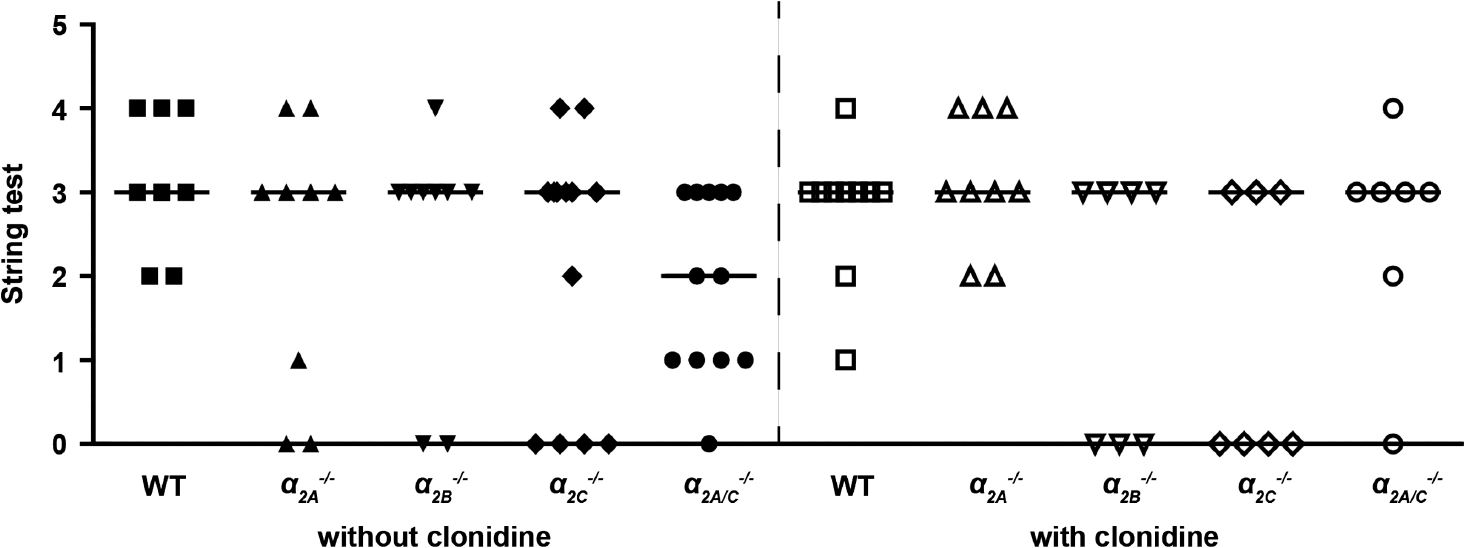
Functional outcome 24 hours after transient middle cerebral artery occlusion (tMCAO) (string test). There were no significant differences between wild-type (WT) controls, α 2A −/−, α 2B −/−, α 2C −/−, and α 2A/C −/− mice without or with clonidine pretreatment (n = 7 to 15/group; horizontal bars indicate median). P >0.05, Kruskal–Wallis test with post hoc Dunn's multiple comparison test.
Regional Cerebral Blood Flow
Regional cerebral blood flow in the ischemic hemispheres was determined by laser-Doppler flowmetry after clonidine pretreatment (Figure 5). While baseline and postocclusive rCBF was similar in all groups (P > 0.05), α 2A/C −/− mice exhibited significantly higher rCBF than the other groups at 15 minutes postreperfusion (P < 0.05). At 24 hours after tMCAO, rCBF values were again comparable (P > 0.05).
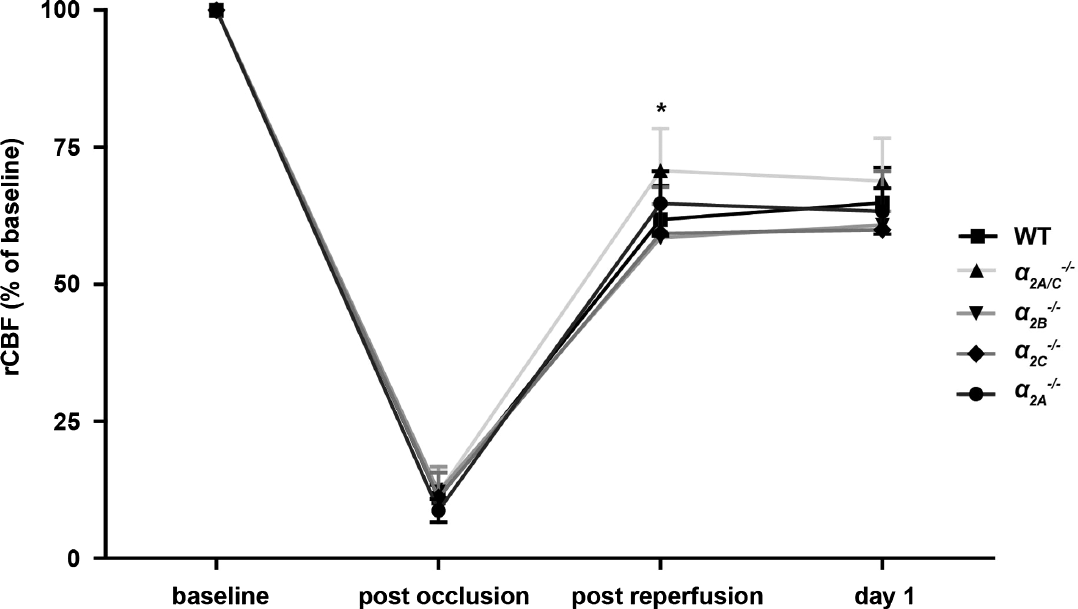
Measurement of regional cerebral blood flow (rCBF). rCBF during early reperfusion as determined by laser-Doppler flowmetry was significantly higher in clonidine-treated α 2A/C −/− mice compared with wild-type (WT) controls and α 2C −/− mice. The other time points and groups studied did not show any differences (n = 3 to 5/group; mean ± s.d.). *P <0.05, ns, not significant, two-way analysis of variance (ANOVA) with post hoc Bonferroni's multiple comparison test.
Discussion
This is the first investigation to assess the role of distinct α2-adrenoceptor subtypes in focal ischemic brain damage. We took advantage of mice with targeted deletions of specific α 2 -adrenoceptors (Gilsbach et al, 2009) and induced tMCAO. Surprisingly, both infarct volumes and functional deficits were not increased in mice lacking the different α2-adrenoceptor subtypes under basal conditions. In line with this finding, pretreatment with the α2-adrenoceptor agonist clonidine did not protect from ischemic brain damage in WT mice or α 2A −/−, α 2B −/− and α 2C −/− mice. The reduction in infarct size observed in α 2A/C −/−-mice after clonidine application could be otherwise explained by higher arterial blood pressure and increased cerebral perfusion in these animals. Our data show that α2-adrenoceptors do not confer neuroprotection in focal cerebral ischemia in contrast to what has been expected from previous studies.
Clonidine preconditioning decreased neuronal damage and improved functional outcome in a rat model of global forebrain hypoxemia (Zhang, 2004). Corresponding effects have been demonstrated for another α2-adrenoceptor agonist, dexmedetomidine, during incomplete cerebral ischemia (Hoffman et al, 1991a, b ). In addition, stimulation of α2-adrenoceptors attenuated hypoxic brain damage induced by asphyxia at or shortly before birth in rats and mice by reducing excitotoxicity and this protective effect could be ascribed specifically to the a2A subtype (Ma et al, 2004; Paris et al, 2006).
Given these data, we speculated that the α2-adrenoceptors would also mediate neuroprotection in the mature brain after focal cerebral ischemia/reperfusion injury induced by transient occlusion of the MCA. However, clonidine pretreatment in WT mice subjected to tMCAO could not reduce infarct size. In line with these findings, no increase in infarct volumes or neurologic deficits was observed in untreated mice lacking the different α2-adrenoceptor subtypes. This is in obvious contrast to the above referenced studies and the results after transient focal cerebral ischemia in rabbits (Maier et al, 1993). The exact reasons for these discrepant findings are unclear at present, but differences in the stroke models (global versus focal brain ischemia) or animal species (rat versus rabbit versus mouse) used might have a role. Moreover, since α2-adrenoceptor-mediated pathways show differential postnatal development (Schelb et al, 2001), age-dependent effects (newborn versus adult animals) might apply. Furthermore, our study differed from most of the previous investigations in that it strictly followed current quality standards in experimental stroke research (Dirnagl, 2006). Those among others included blinded evaluation of data, full disclosure of dropout rates, as well as a priori sample size calculations to minimize the risk of biased interpretation of results. Our study was designed to detect a standardized effect size on stroke volumes ≥ 0.15, with a statistical power of 0.8 translating into a β (type II error) of 1.0 to 0.8 = 0.2, respectively. A type II error, also known as a false negative, occurs when the test fails to reject a false null hypothesis, for example, α 2 -adrenoceptors−/− mice in fact develop larger strokes, but random variation between the groups obscured the difference.
Kuhmonen et al (2001) analyzed the outcome in rats subjected to tMCAO after treatment with dexmedetomidine, which like clonidine is a α2-adrenoceptor agonist and in line with our study in mice also did not detect differences in infarct sizes compared with controls. Importantly, profound effects of dexmedetomidine on mean arterial blood pressure were noted. We likewise collected physiological parameters relevant for stroke development. Clonidine pretreatment reduced mean arterial blood pressure in all experimental groups except for the α 2A/c −/− group. Accordingly, CBF was higher in α 2A/C -deficient mice after reperfusion. It is well established that final stroke size and functional outcome critically depend on CBF (Palmer et al, 1977; Shin et al, 2008). Indeed, smaller infarcts were found in clonidine-treated α 2A/C −/− mice, an observation that could have been easily misinterpreted as ‘neuroprotection’ mediated by the α 2B -adrenoceptor (the remaining α2-adrenoceptor in α 2A/C −/− mice) if blood pressure and CBF would not have been determined. Clonidine is a partial agonist with equal affinity for all three α2-adrenoceptor subtypes (Zhu et al, 1999). The fact that infarct volumes were unchanged in untreated mice lacking either subtype (A, B, C) as well as in α 2A/C −/− mice clearly argues against a major neuroprotective function of clonidine mediated via α2-adrenoceptors. However, we cannot exclude that clonidine exerts neuroprotection in stroke through other so far unknown pathways since this characteristic of clonidine might have been masked by its blood pressure lowering properties.
In summary, we found no evidence for a neuroprotective function of α2-adrenoceptors in focal cerebral ischemia. Because genetic manipulation can alter physiological parameters relevant for stroke outcome such as blood pressure or the susceptibility for pharmacological compounds, careful controlling of these parameters is recommended when performing experimental stroke studies in animals.
Footnotes
Acknowledgements
The authors gratefully acknowledge the excellent technical assistance of Gabi Köllner, Melanie Glaser, and Daniela Urlaub.
The authors declare no conflict of interest.