
Abstract
Angiotensin II receptor blockers (ARBs) have a potent ability to inhibit oxidative stress and advanced glycation, in addition to their protective effects originated from blood pressure lowering and angiotensin II type 1 receptor (AT1)-blockade. To obtain a pharmacological tool to dissect the mechanisms of ARBs’ protective benefits in experimental stroke, we synthesized a novel ARB-derivative, R-147176, which is 6,700 times less potent than olmesartan in AT1-binding inhibition and therefore has a minimal antihypertensive effect, but retains marked inhibitory effects on oxidative stress and advanced glycation. We evaluated the effect of R-147176 (10–30 mg/kg per day), administered orally or intravenously, on brain infarct volume in transient thread occlusion and photothrombotic models in rats. The antioxidative and antiinflammatory properties were also investigated. R-147176 significantly reduced infarct volume, without influence on blood pressure, in both models. R-147176 significantly reduced the numbers of ED-1-positive cells and of TUNEL-positive cells, and protein carbonyl formation in the damaged brain. This ARB derivative, despite its significantly lower AT1 affinity and virtually no antihypertensive effect, ameliorated ischemic cerebral damage through antioxidative and antiinflammatory properties. These findings suggest potential usefulness of R-147176 as a pharmacological tool to investigate the ARBs’ protective effect in experimental stroke and open new therapeutic avenues.
Introduction
Antihypertensive inhibitors of the renin-angiotensin system, such as angiotensin II receptor blockers (ARBs), are known to protect against ischemic cerebral injury, at least in part, independently of blood pressure (Ito et al, 2002; Groth et al, 2003; Engelhorn et al, 2004; Lou et al, 2004; Forder et al, 2005; Brdon et al, 2007). The challenge is therefore to gain further insights into the blood pressure-dependent and -independent mechanisms of neuroprotection of ARBs, to be able to optimize the design of the clinical trials.
We demonstrated earlier that, in addition to protective effects by blood pressure lowering and angiotensin II type 1 receptor (AT1)-blockade, ARBs have a unique ability to inhibit oxidative stress and advanced glycation (Miyata et al, 2002; Nangaku et al, 2003; Izuhara et al, 2005). The relation between the decreased blood pressure and the reduction in local oxidative stress and advanced glycation, in the drug-induced tissue benefits remained moot.
To obtain a pharmacological tool to dissect the mechanisms of ARBs’ protective benefits in an experimental stroke, we developed TM2002, a novel derivative of edaravone, a molecule known for its cerebroprotective effect. This compound is characterized by a low AT1 affinity but a powerful inhibition of oxidative stress and advanced glycation (Izuhara et al, 2008a). It protected diabetic kidneys and brain ischemic areas despite minimal effects on blood pressure (Takizawa et al, 2007). The relation of these advantages with the actual structure of edaravone remained to be settled.
To further dissociate blood pressure from oxidative stress and to exclude any edaravone related effect, we synthesized a novel, nontoxic ARB derivative, R-147176, characterized by a weak affinity for the AT1 and AT2 receptors (and thus a minimal antihypertensive effect), but a striking inhibition of oxidative stress and advanced glycation (Izuhara et al, 2008b). This compound is not toxic. It is 6,700 times less effective than olmesartan in AT1-binding inhibition, but strongly inhibits advanced glycation and oxidative stress in several different types of rat renal injury models (Izuhara et al, 2008b).
In this study, we tested its cerebroprotective benefits in two different rat models of ischemic cerebral injury, i.e., a model of transient ischemia induced by thread occlusion, mimicking clinical cardioembolic stroke, and a model of permanent ischemia induced by photothrombotic occlusion, mimicking clinical atherothrombotic stroke. We show that R-147176 ameliorates ischemic cerebral damage in both rat models, independently of blood pressure changes, through its antioxidative and antiinflammatory effects.
Materials and methods
R-147176
R-147176 has been selected among 139 newly synthesized olmesartan-derivative imidazole compounds on the basis of low AT1 affinity, potent inhibitory activities for advanced glycation end products (AGEs) and for oxidative stress, and appropriate pharmacokinetic parameters (Izuhara et al, 2008b).
On the basis of Ki values, R-147176 was 6,700 times less potent than olmesartan in terms of AT1-binding inhibition (Izuhara et al, 2008b). R-147176 also showed low binding affinity for the human AT2 receptor (Izuhara et al, 2008b).
Animal Studies
All animal experiments were performed in accordance with the guidelines of the Committee on Ethical Animal Care and Use of Tokai University.
Thread occlusion rat model: Experimental model. Male Sprague-Dawley rats (Charles River, Japan), weighing 331±15 g, underwent a transient focal brain ischemia through intraluminal thread occlusion of the middle cerebral artery (MCA) as described earlier (Longa et al, 1989; Takizawa et al, 2007). Briefly, under isoflurane anesthesia (induction 4%, maintenance 2.0% in 30% oxygen and 70% nitrous oxide), the left common and external carotid arteries were isolated and ligated. A 4 to 0 nylon thread introduced into the internal carotid artery from the carotid bifurcation and advanced to the origin of the MCA was withdrawn 2 h later. Brain and rectal temperatures were maintained at around 37°C to 38°C with a warming pad. Mean arterial blood pressure (MABP), arterial blood gases, and plasma glucose were monitored through the tail artery. Cortical cerebral blood flow (CBF), measured with a laser-Doppler flowmeter (FLO-C1, Neurosci Inc., Japan), was expressed as a percentage of the baseline value.
Animals with transient focal ischemia were divided into two groups: Group 1 (experimental group: n=10 for infarct volume and immunohistochemical evaluation; n=13 for oxyblot and pentosidine analysis) received 20 mg/kg of R-147176 dissolved in 0.5% carboxymethylcellulose (CMC) by oral gavage 10 mins after MCA occlusion and again on each of the subsequent 7 days. Group 2 (control group: n=8 for infarct volume and immunohistochemical evaluation; n=12 for oxyblot and pentosidine analysis) received only 0.5% CMC orally on the schedule of Group 1. All animals were killed with an overdose of pentobarbital at 7 days after the start of occlusion.
Infarct volume measurement. The method has been fully described earlier (Takizawa et al, 2007). Tissue fixation was achieved at the time of killing by transabdominal-arterial perfusion fixation with 100 mL of phosphate-buffered saline, followed by 100 mL of 4% paraformaldehyde in 0.01 mol/L phosphate buffer. After removal from the skull, the brain was stored in 4% paraformaldehyde in 0.01 mol/L phosphate buffer (pH 7.4) for 24 h. Coronal brain sections were obtained at 2.0-mm intervals, stained with hematoxylin and eosin, and photographed. Infarct areas were measured in the cerebral cortex and caudoputamen with the help of NIH Image (The National Institute of Health, USA). The infarct volume (mm3) was calculated by multiplying each area with the distance between sections. Infarct size was measured by an examiner (TU) blinded to the animal's experimental status.
Immunohistochemistry. Coronal brain sections fixed with 4% paraformaldehyde and incubated in a 5 mmol/L solution of hydrogen peroxide for 10 mins were exposed to 5% normal goat serum for 10 mins.
ED-1 was used as a marker of microglia/macrophages (Schroeter et al, 1997). The treated sections were incubated with mouse antirat ED-1 (Chemicon, Temecula, CA, USA) at 4°C for 3 h in a humidified chamber, and subsequently incubated at room temperature for 1 h with biotinylated antirat IgG (Vectastain Elite ABC peroxidase kit, Burlingame, CA, USA), followed for 30 mins by ABC reagent. The bound antibody was visualized with 3,3′-diaminobenzidine and hydrogen peroxide.
TUNEL staining was performed by indirect immunohistochemistry with a kit (TACS2 TdT-Blue Label in situ Apoptosis Detection: Trevigen, Gaithersburg, MD, USA).
One observer (TU), blinded to the experimental protocol, counted the number of ED-1 and TUNEL-positive cells in each of three predetermined areas (0.62 mm2) per high-power field (× 400). The average in each cell count of three predetermined areas was evaluated.
Protein oxidation detection and pentosidine measurement. Levels of oxidatively modified proteins were quantified, as described earlier(Takizawa et al, 2007), with an OxyBlot protein oxidation detection kit (Integen, NY, USA) both in infarcted and noninfarcted hemispheres.
Brain tissue content of pentosidine, a representative AGE, was measured in both infarcted and noninfarcted hemispheres. The tissue was minced, rinsed with chloroform–ethanol mixture, dried under vacuum, and hydrolyzed in 500 μL of 6 N HCl for 24 h at 110°C under nitrogen. Pentosidine was analyzed by means of reverse-phase HPLC as described earlier (Nangaku et al, 2003).
Photothrombotic occlusion rat model: Experimental model. Male Sprague-Dawley rats (SLC, Japan), weighing 278±11 g, underwent brain artery photo thrombotic occlusion as described earlier (Takizawa et al, 2007). Briefly, under halothane anesthesia (4% induction/ 2% maintenance), the left MCA was occluded by a photochemical reaction (Umemura et al, 1993). The body temperature was maintained at 37.5°C±0.5°C with a warming pad. The head of the optic fiber was placed on a 3-mm diameter bone window in the skull base. Photoillumination (wavelength 540 nm, 600,000 lux) using a xenon lamp (L4887, Hamamatsu Photonics, Japan) was performed through the optic fiber for 10 mins after an intravenous injection of Rose Bengal (20 mg/kg) through the tail vein.
Neurologic deficit at 24 h after the occlusion was evaluated according to the neurologic scores (Bederson et al, 1986).
The dose-dependent effect of the oral administration of R-147176 on infarct volume was measured in 30 rats divided into three experimental groups (n=10 in each group). Group 1 (low-dose group) and group 2 (high-dose group) received once a day, by gavage, 10 and 30 mg/kg of R-147176 dissolved with 0.5% CMC, respectively, during 7 days before and 10 mins after MCA occlusion. Group 3 (control group) received, by gavage, only 0.5% CMC on the schedule of groups 1 and 2.
The effect of the intravenous administration of R-147176 on infarct volume was measured in 16 animals divided into two groups (n=8 in each group): group 1 (experimental group) received 10 mg/kg of R-147176 dissolved in saline with DMSO by intravenous bolus injection, 10 mins and 2 h after the occlusion. Group 2 (control group) received bolus injections of only saline with DMSO on the schedule of group 1. All animals subjected to photothrombotic occlusion of MCA were killed with an overdose of pentobarbital 24 h after the start of occlusion.
Infarct volume measurement. Infarct volume was measured in coronal, 2-mm thick brain slices incubated in 2% 2,3,5-triphenyltetrazolium chloride in phosphate-buffered saline for 30 mins at room temperature, photographed subsequently with a digital camera, and eventually stored in 4% paraformaldehyde. Infarct areas and volume were assessed on the photographs by the method used in the thread occlusion models.
Statistical analysis
All values were expressed as mean±s.d. One-way ANOVA followed by Fisher's protected least significant difference was used to analyze the statistical significance of differences in physiologic parameters, neurologic deficits, infarct volume, the numbers of ED-1 and TUNEL-positive cells, oxyblot, and pentosidine content among the groups.
Results
Thread Occlusion Rat Model
The oral administration of either vehicle or R-147176 failed to produce any statistically significant change in MABP, measured at 0 mins, 10 mins, and 2 h after the MCA occlusion, and 10 mins and 1 week after reperfusion (Figure 1A). Brain and rectal temperatures, arterial blood gases, blood glucose, CBF, and hematocrit did not differ statistically significantly between the two groups (Table 1).
Physiological parameters in rats subjected to thread occlusion model

R-147176 significantly (P<0.05) reduced the infarct volume from 188.5±73.2 mm3 in the control group to 115.2±47.3 mm3 in the experimental group (Figure 1B). Neocortical and striatal infarct volumes also decreased significantly from 99.0±43.9 and 89.6±33.2 mm3, respectively, in the control group to 55.8±36.4 and 59.4±37.3 mm3 (P<0.05), respectively, in the experimental group.
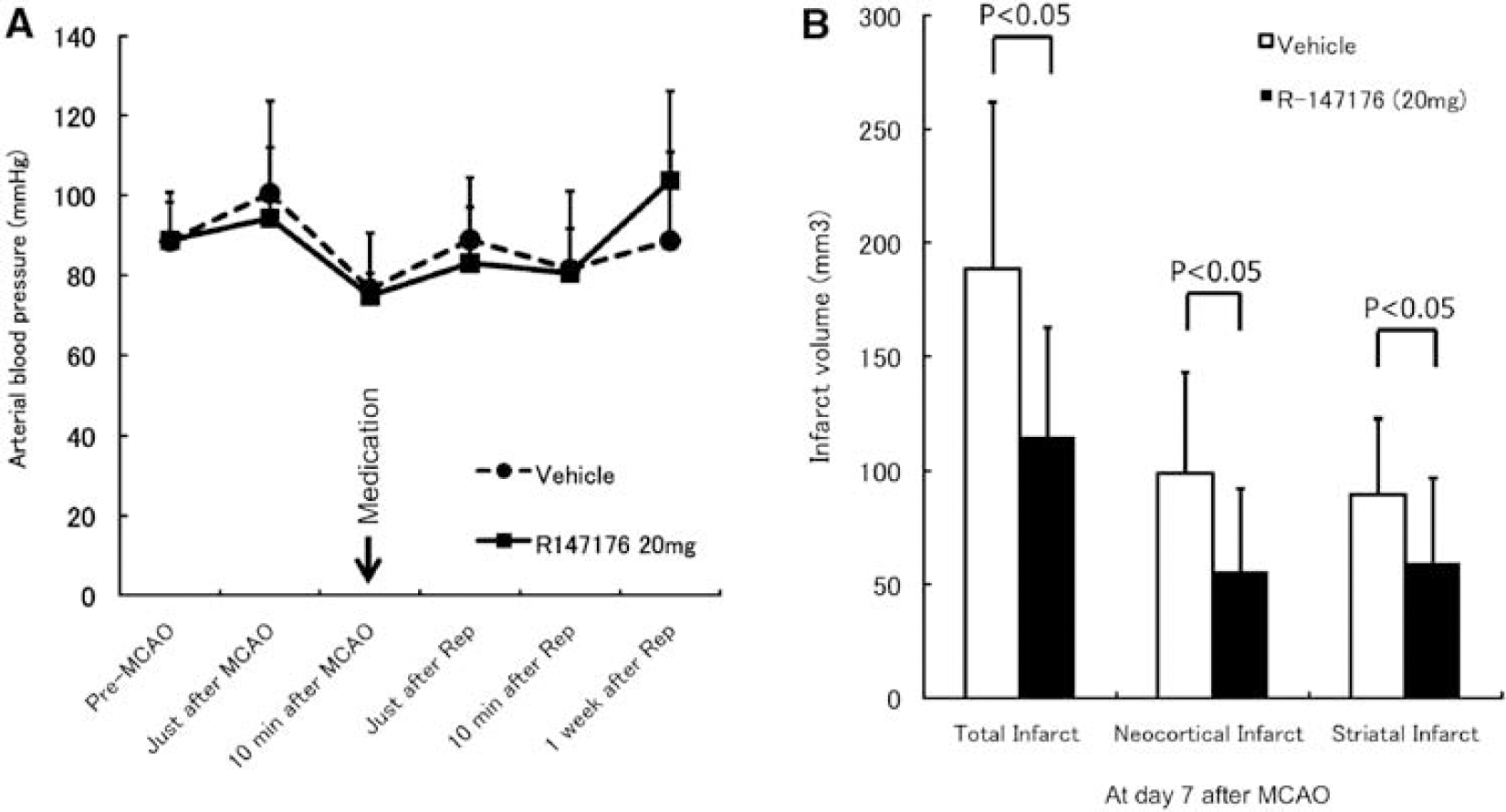
Effect of R-147176 on blood pressure and infarct volume in rats with transient focal ischemia. (
The cellular response in the ischemic brain tissue was evaluated by immunohistochemical staining for ED-1, a marker of microglia/macrophages, and by TUNEL assay, a method to detect apoptotic cells. R-147176 significantly (P<0.05) reduced the numbers of ED-1-positive cells in the penumbra, but not in the cortical infarct (Figure 2A). It also significantly reduced TUNEL-positive cells in the infarcted cortex (P<0.01) and penumbra (P<0.01) (Figure 2B), a result taken to indicate that R-147176 rescued neurons from apoptotic cell death.
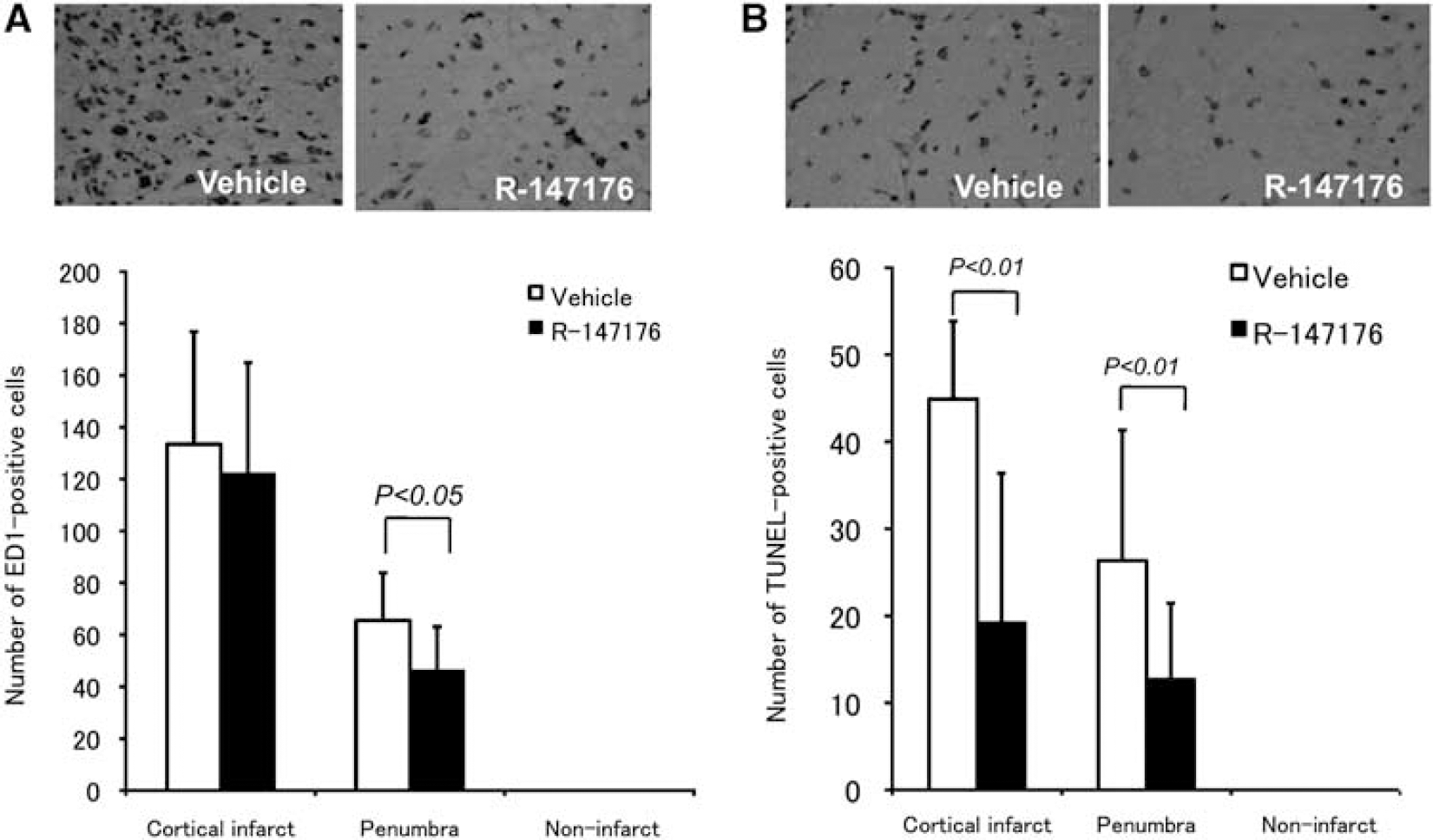
R-147176 reduced inflammatory response and apoptosis in rats with transient focal ischemia. (
Protein carbonyl formation, in brain tissues of rats subjected to a 2 h occlusion followed by a 7-day reperfusion, was also reduced by R-147176. The optical density values of bands corresponding to protein carbonyls fell markedly (P<0.01) in infarcted brain in the experimental group as compared with the control group (Figure 3A).
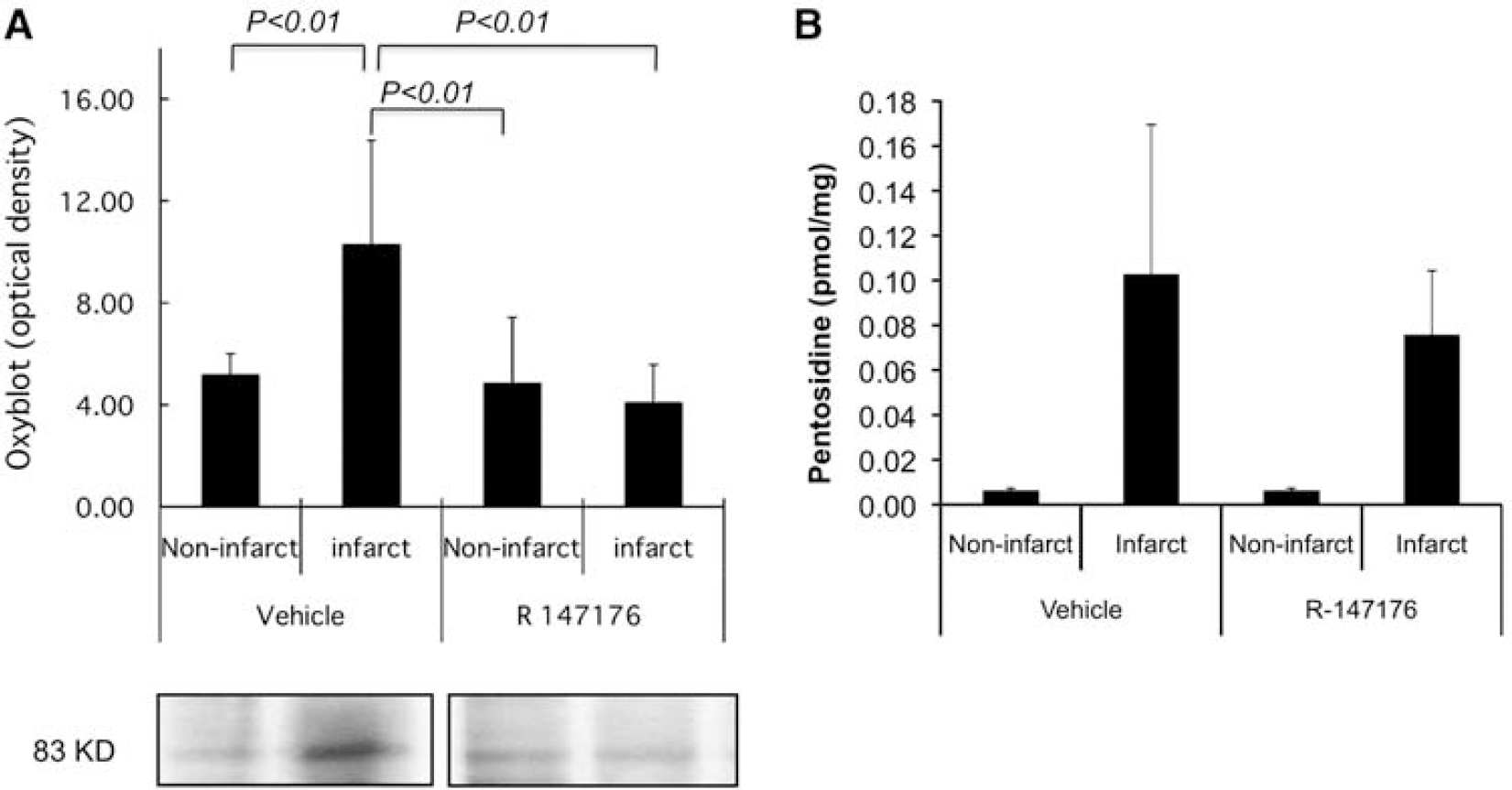
R-147176 reduced protein carbonyl formation and pentosidine in brain tissues in rats with transient focal ischemia. (
Advanced glycation-related brain damage, assessed by brain pentosidine content was lower in the experimental group (0.08±0.03 pmol/mg protein) than in the control group (0.10±0.07), but these changes failed to reach statistical significance (P=0.36, Figure 3B).
Photothrombotic Occlusion Rat Model
Oral administration of R-147176 did not influence MABP measured 1 h after the MCA occlusion in the control and experimental groups (Figure 4A). Neurologic deficit score did not differ significantly between control, low, and high R-147176 groups (2.7±0.5, 2.4±0.5, and 2.3±0.5, respectively). At both dosages (10 and 30 mg/kg), R-147176 significantly (P<0.01) reduced total and cortical infarct volume from 320.6±30.1 and 236.4±55.0 mm3, respectively, in the control group to 252.3±55.0 and 169.8±52.0 mm3 and to 254.8±48.0 and 172.0±40.5 mm3, respectively, in the experimental groups (Figure 4B).
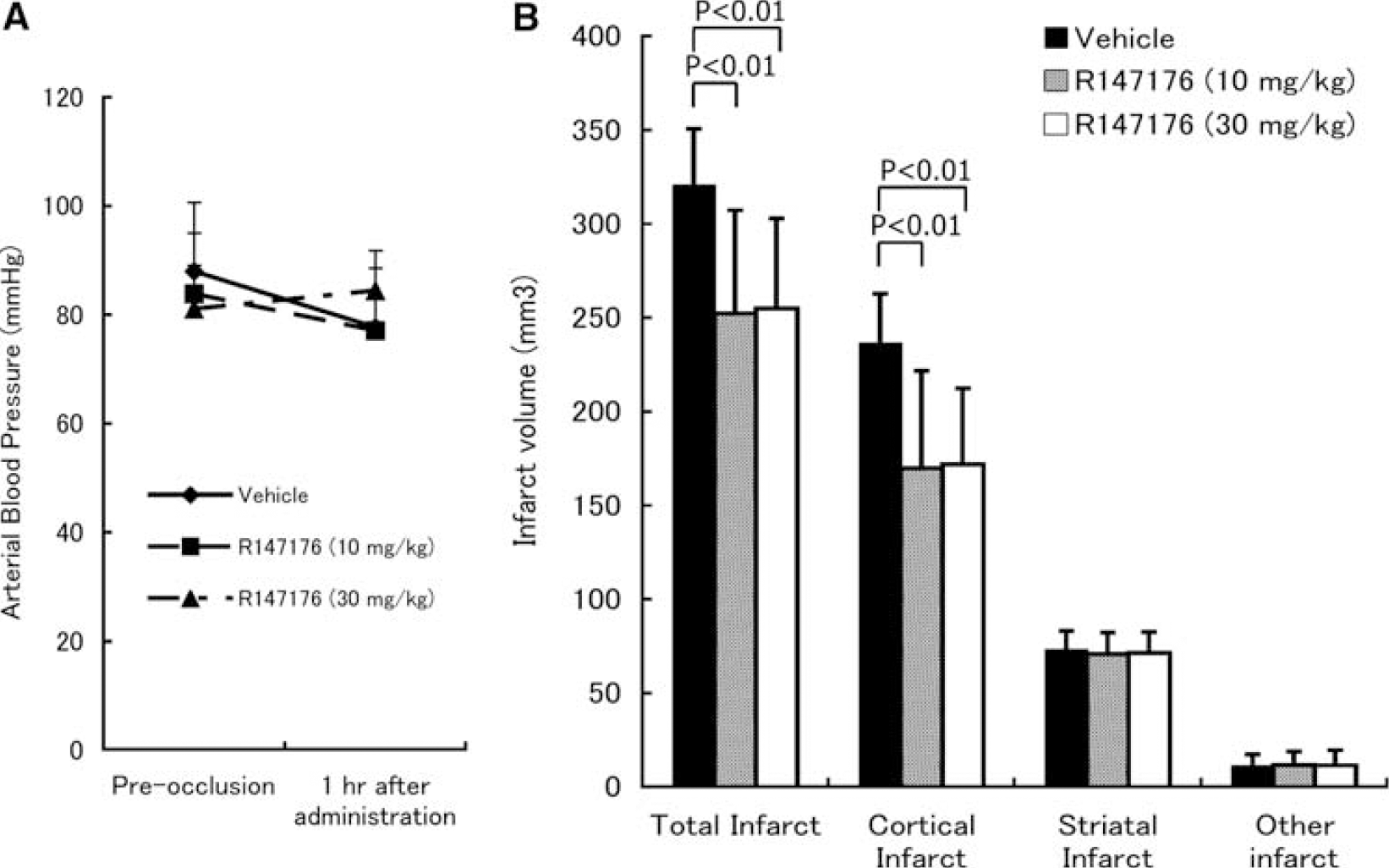
Effect of oral administration of R-147176 on blood pressure and infarct volume in rats with photothrombotic focal ischemia. (
Intravenous administration of R-147176 failed to modify MABP in the control and experimental groups (data not shown). The neurologic deficit score was ameliorated (P<0.05), from 2.4±0.5 in the control group to 1.9±0.4 in the experimental group (Figure 5A).
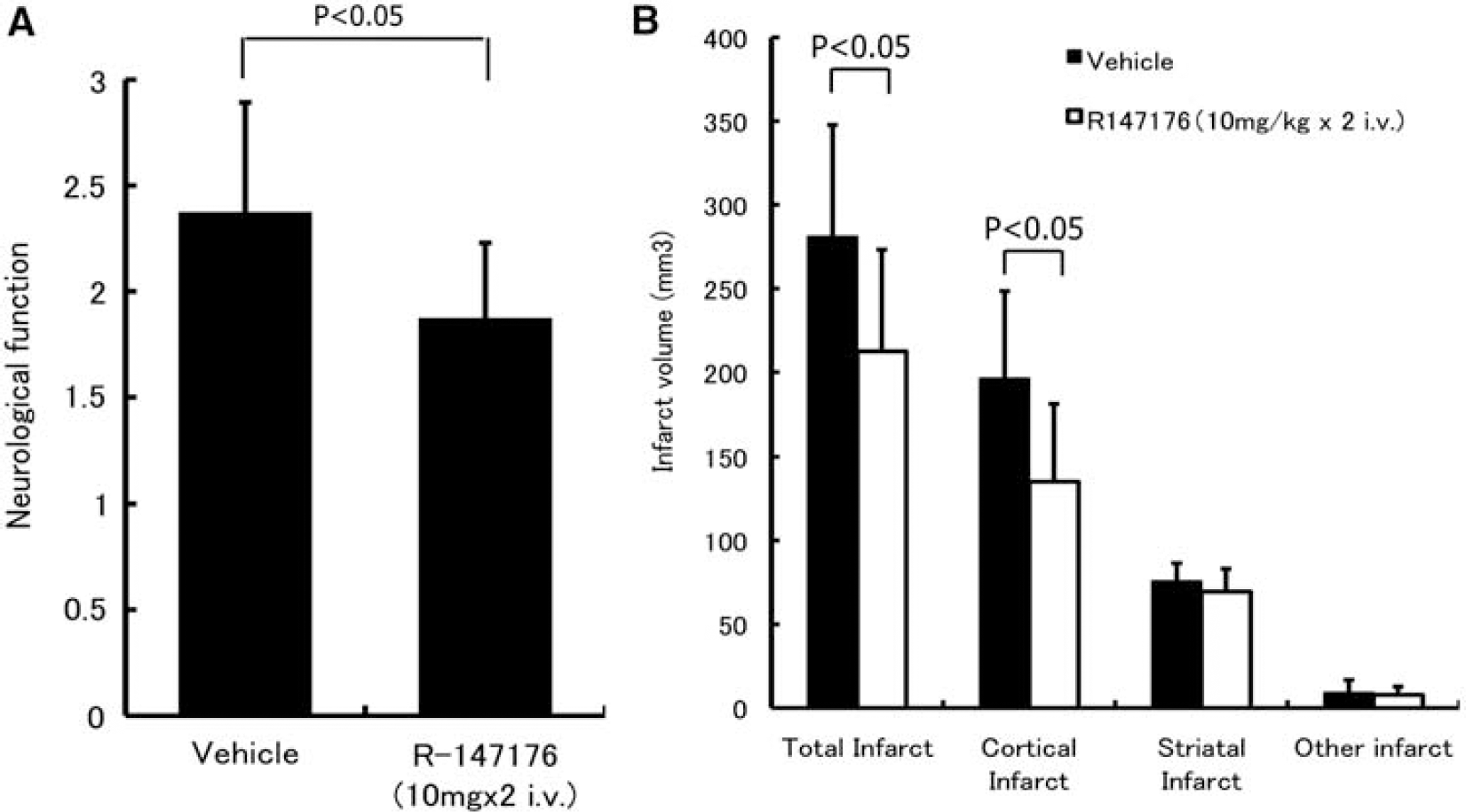
Effect of intravenous administration of R-147176 on neurologic grading score and infarct volume in rats with photothrombotic focal ischemia. (
Total infarct volume was significantly (P<0.05) reduced from 281.1±66.6 mm3 in the control group, to 212.4±60.8 mm3 (P<0.05) in the experimental group (Figure 5B). Cortical infarct volume also decreased significantly (P<0.05) from 196.2±52.2 mm3 in the control group to 135.2±45.9 mm3 in the experimental group (Figure 5B).
Discussion
The present data confirm and further extend the conclusions drawn from our earlier experiments on ischemic rat brains administered an edaravone derivative, TM2002 (Takizawa et al, 2007): the powerful inhibition of oxidative stress by R-147176 protects the brain against temporary or pemanent ischemia as shown by a reduced infarct volume. These results were obtained without significant modifications of blood pressure, a finding anticipated as a result of R-147176's low affinity for AT1 receptor, confirming their independence from changes in blood pressure. Furthermore, their similarity with those achieved with the edaravone derivative, TM2002, show that they are independent of the latter's structure.
If the level of angiotensin II receptor affinity played a critical role, a parallel effect on angiotensin II receptor affinity on the one hand and cerebral protection on the other hand would be expected. AT1 blockade by ARBs prevents excessive blood vessel contraction and results in an enhanced relaxant effect of the unopposed cerebrovascular AT2 receptors (Wackenfors et al, 2006). Earlier studies showed that ARBs improve recovery from ischemic stroke by preventing blood flow decrease in the marginal zone of ischemia because of increased capacity of cerebral arteries to dilate when CBF decreases (Nishimura et al, 2000; Ito et al, 2002; Engelhorn et al, 2004). Studies of AT2 receptor knockout mice support a positive role of central AT2 receptor stimulation in ischemic brain lesions (Iwai et al 2004). However, R-147176 has a little inhibitory effect on AT1 and AT2 receptors binding (Izuhara et al, 2008b), and no effect on CBF during ischemia and reperfusion in this study. Furthermore, R-147176, by contrast to an equal concentration of losartan, does not affect (1) angiotensin II-induced Ca2+ influx in human aortic vascular smooth muscle cells (50 μmol/L each), assessed by laser scanning confocal microscope, and (2) systolic blood pressure in an angiotensin II-infused hypertensive rat model (20 mg/kg per day each) (Supplementary Information). Therefore, an impressive protective influence in the two different ischemic cerebral injury models suggest that brain benefits of ARBs are not necessarily associated with its protective effects by blood pressure lowering and AT1-blockade, although the trivial effects of R-147176 on AT1 as well as AT2 and AT4 receptors binding (Faure et al, 2006) cannot be denied.
The central role played by the inhibition of oxidative stress in the cerebroprotective effects of R-147176 is supported by the demonstration of decreased brain tissue levels of protein carbonyls. TM2002 administration also reduced protein carbonyl formation and the number of AGE-positive cells (Takizawa et al, 2007), strongly implying therefore that oxidative stress and advanced glycation are key to the devastating effects of cerebral ischemia. Such a compound with a high AT1 affinity and minimal intrinsic properties against oxidative stress would also represent an ideal complementary tool for mechanistic comparative studies.
To further elucidate the mechanisms involved in cerebral ischemic damage, we assessed the local inflammatory response to ischemia. Strikingly, oxidative stress was accompanied by cellular inflammation, which was attenuated by R-147176: the number of ED-1 cells, taken as markers of macrophages, fell significantly in the penumbra and that of TUNEL-positive cells, taken as evidence of apoptosis, was more than halved in the infarcted cortex and in the penumbra.
Antiinflammatory effects of ARB, unrelated to blood pressure decrease, have been demonstrated in vivo in an experimental stroke model (Lou et al, 2004). ARB decreased the expression of NF-κ B, TNF-α, IL-β, and eliminated the macrophage infiltration in cerebral microvessels (Zhou et al, 2005), through the moduration of chemokine monocyte chemoattractant protein 1/ C-C chemokine receptor 2 system (Dai et al, 2007) in spontaneously hypertensive rats. Recently, Chen et al (2008) suggested that candesartan (an ARB) suppressed chronic renal inflammation in spontaneously hypertensive rats, independently of AT1 blockade. Improved renal redox homeostasis was ascribed to a direct antioxidant effect and it was suggested that renal inflammation was mitigated by a redox sensitive NF-κ B suppression. These results wholly support our conclusion that oxidative stress is implicated in a number of chronic lesions and further suggest that NF-κ B is an interface between oxidative stress and cellular inflammation. Of note in this context, interaction of AGEs with cellular targets leads to oxidative stress through activation of NF-κ B (Yan et al, 1994; Lander et al, 1997).
Extended clinical trials are required before any advocation of R-147176 use in cerebral disorders. R-147176's advantage over ARBs lies in its absence of hypotensive effect in patients with a normal or even low blood pressure: it should prevent any aggravation of acute ischemia in the penumbral region (Yatsu and Zinvin, 1985; Fischberg et al, 2000; Castillo et al, 2004; Semplicini et al, 2006; Wityk and Lewin, 2006). This drug is safe in the acute and subacute toxicity studies (Izuhara et al, 2008b). No acute toxicity was observed in mice at 2 weeks after a single dose in the range from 500 to 2000 mg/kg and in rats at 1 week after a single dose in the range from 300 to 2,000 mg/kg. Subacute toxicity was also assessed in rats administered 200 mg/kg of R-147176 daily for 2 weeks. They exhibited no abnormal serum or urine biochemistry. In respect to the pharmacokinetics of R-147176, assessed in rats administered an oral dose of 50 mg/kg, the calculated plasma Tmax, Cmax, and T1/2 were 2 h, 41 μg/mL, and 2 h, respectively. However, more toxicological data including chronic toxicity will be essential for R-147176's clinical trials.
The strength of our experimental data relies also on the use of different ischemic models and routes of administration as suggested by the stroke therapy academic industrial round table (STAIR, Savitz and Fisher, 2007) in the wake of the recent failure of NXY-059 in the first stroke acute-ischemic NXY trial.
Our present findings suggest potential usefulness of R-147176 as a pharmacological tool to dissect the mechanisms of the protective effect of ARBs in an experimental stroke and also open new avenues for the treatment of stroke.
Footnotes
Acknowledgements
The skillful technical assistance of Saori Kohara, Hiroko Yuzawa, Saori Sekino, Yoshiko Itoh, Johbu Itoh, and Yoko Takahari (Tokai University) is gratefully acknowledged.
The authors declare no conflict of interest.