
Abstract
Cortical spreading depression (CSD) is an intense depolarization wave implicated in the pathophysiology of brain injury states and migraine aura. As Cav2.1 channels modulate CSD susceptibility, we tested gabapentin, which inhibits Cav2.1 through high-affinity binding to its α2δ subunit, on CSD susceptibility in anesthetized rats. Gabapentin, 100 or 200 mg/kg, elevated the electrical threshold for CSD and diminished recurrent CSDs evoked by topical KCl, when administered 1 hour before testing. With its favorable safety and tolerability profile, gabapentin may have a role in suppression of injury depolarizations in stroke, intracranial hemorrhage, and traumatic brain injury.
Introduction
Cortical spreading depression (CSD) is a wave of neuronal and glial depolarization associated with massive K+ and glutamate efflux, and Ca2+ influx, slowly propagating in the brain tissue at a rate of 3 to 4mm/min by way of gray matter contiguity. Although the massive rise in extracellular K+ is critical for contiguous spread, glutamate adds momentum to sustain the propagation. Among pharmacological inhibitors of CSD are Ca2+ channel blockers, in particular of the Cav2.1 subtype (Kunkler and Kraig, 2004; Richter et al, 2002), possibly by reducing the release of glutamate during CSD. Gabapentin, an adjunct antiepileptic that has shown modest efficacy in migraine prophylaxis, inhibits the Cav2.1 channel by binding with high affinity and specificity to its auxiliary α2δ subunit, which both diminishes the voltage responsiveness of channels and prevents their localization to the neurotransmitter release sites (Gee et al, 1996; Hendrich et al, 2008).
As the Cav2.1 channel is a major regulator of glutamate release, and a modulator of CSD susceptibility, we tested the efficacy of gabapentin to suppress CSD. Our results show a novel acute inhibitory effect of gabapentin on CSD.
Materials and methods
A total of 32 rats (Sprague-Dawley, 200 to 450 g, male) were used to test gabapentin on CSD. Gabapentin doses and treatment protocols were chosen based on published data in other animal models of neurological diseases (Radulovic et al, 1995). Gabapentin was administered as a single 100 or 200 mg/kg intravenous dose (n = 7 and 14, respectively; Research Chemical) 60 minutes before CSD testing; intravenous route was chosen because of the saturable oral absorption kinetics, and nonlinear oral bioavailability (Radulovic et al, 1995; Stewart et al, 1993). Saline was used as vehicle control (n = 11). A subset of rats in each group was tested blindly.
Procedures
Institutional guidelines for animal care and use for research purposes were strictly followed, and study protocol was approved by institutional review board. Rats were anesthetized (isoflurane 5% induction, 1% maintenance, in 70% N2O/30% O2), paralyzed (pancuronium 0.4 mg/kg), and intubated through a tracheostomy for mechanical ventilation (SAR-830; CWE, Ardmore, PA, USA). Arterial blood gases and pH were measured every 30 minutes and ventilation adjusted to maintain arterial pCO2 between 35 and 45mmHg (Corning 178; Corning, NY, USA). Continuous measurement of blood pressure (Power-Lab; ADInstruments, Colorado Springs, MO, USA) and blood sampling were performed through a femoral artery catheter. Rectal temperature was kept at 37.0°C ± 0.1°C using a thermostatic heating pad (FHC, Bowdoinham, ME, USA). Level of anesthesia was maintained throughout the experiment to eliminate cardiovascular response to tail pinch. In all treatment groups, systemic physiological parameters were within normal range, although mild and transient blood pressure reductions were observed after 200 mg/kg gabapentin (Table 1).
Systemic and electrophysiological parameters
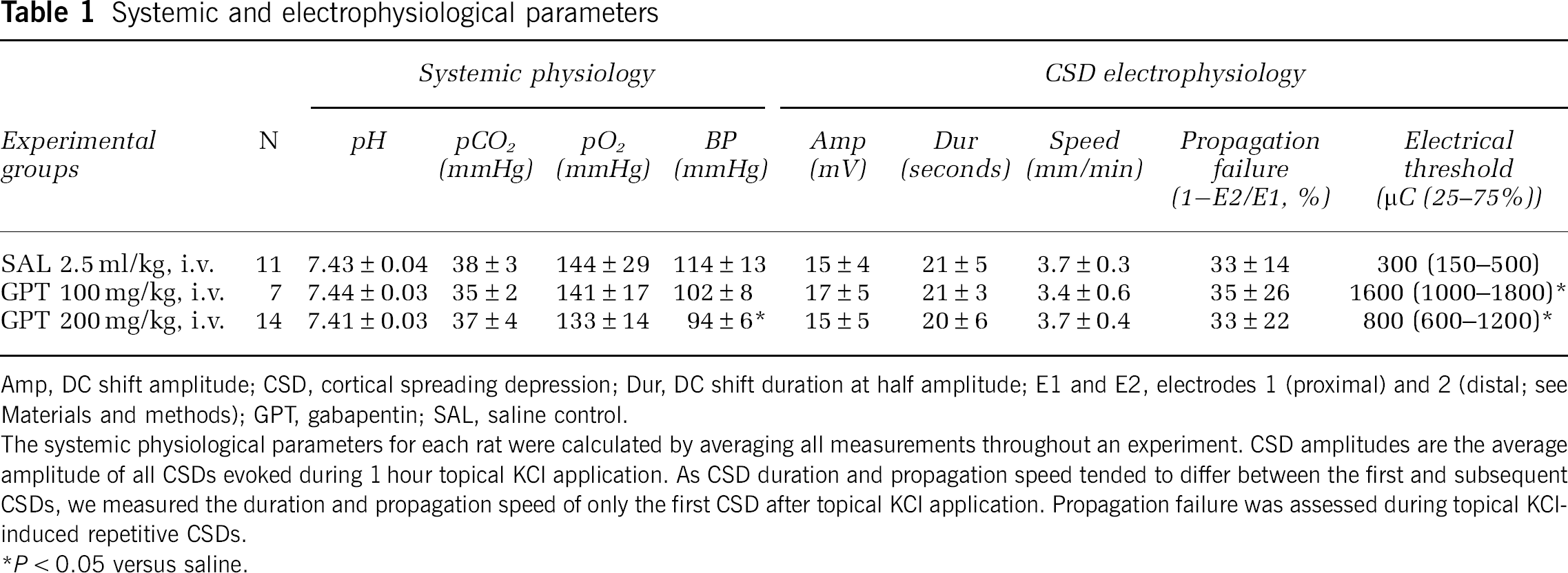
Amp, DC shift amplitude; CSD, cortical spreading depression; Dur, DC shift duration at half amplitude; E1 and E2, electrodes 1 (proximal) and 2 (distal; see Materials and methods); GPT, gabapentin; SAL, saline control.
The systemic physiological parameters for each rat were calculated by averaging all measurements throughout an experiment. CSD amplitudes are the average amplitude of all CSDs evoked during 1 hour topical KCl application. As CSD duration and propagation speed tended to differ between the first and subsequent CSDs, we measured the duration and propagation speed of only the first CSD after topical KCl application. Propagation failure was assessed during topical KCl-induced repetitive CSDs.
P < 0.05 versus saline.
Rats were placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA) and burr holes were drilled bilaterally under saline cooling at (mm from bregma): (1) posterior 7.0, lateral 2.0 (occipital, 1mm diameter for KCl application or electrical stimulation); (2) posterior 5.0, lateral 2.0 (frontoparietal, 0.5mm diameter for electrode 1); (3) posterior 3.0, lateral 2.0 (frontal, 0.5mm diameter for electrode 2). Dura overlying the occipital cortex was gently removed and care was taken to avoid bleeding. The steady (DC) potential and electro-corticogram were recorded with glass micropipettes filled with 200 mmol/L NaCl, 300 μm below pia (Axoprobe-1A; Axon Instruments, Burlingame, CA, USA). Ag/AgCl reference electrode was placed subcutaneously in the neck. After surgical preparation, cortex was allowed to recover for 15 minutes under saline irrigation. The data were continuously recorded using a data acquisition system for off-line analysis (ADInstruments).
Cortical Spreading Depression Susceptibility
We assessed CSD susceptibility using two independent methods, KCl or electrical stimulation, as described earlier (Ayata et al, 2006). For KCl-induced CSD susceptibility, we placed a cotton ball (1.5 mm diameter) soaked with 1 mol/L KCl on the pial surface and kept it moist by placing 5 μL of the same KCl solution every 15 minutes. The total number of KCl-induced CSDs detected at either recording site during 60 minutes KCl application was counted. In addition, we determined the incidence of propagation failure between the two recording sites in all treatment groups, and expressed this as the number of CSDs failed to appear at electrode 2 as percent of total CSDs recorded at electrode 1. After the end of KCl stimulation, electrical threshold for CSD was determined in the opposite hemisphere by direct cortical stimulation using a stimulator (Grass Instruments, West Warwick, RI, USA), a constant current unit (WPI, Sarasota, FL, USA), and a bipolar stimulation electrode placed on the pial surface (400 μm tip diameter, 1mm tip separation; FHC). Cathodal square pulses of increasing intensity (100 to 4000 microcoulomb, μC) were applied at 5-minute intervals by adjusting the current and duration of stimulus until a CSD was observed. At 1mA current, pulses of 100, 200, 300, and 400 milliseconds were applied, followed by 2mA current of 300, 400, and 500 milliseconds. If CSD did not occur, additional stimuli of 3 mA, 400 milliseconds, and 4 mA, 400, 500, 1000 milliseconds were applied. The logarithmic stepwise escalating stimulation protocol was chosen for its ability to distinguish group differences in CSD threshold based on our prior experience (Ayata et al, 2006). As previously observed, electrical stimulation threshold for CSD showed more variability compared with the frequency of CSDs evoked by topical KCl, which is often attributed to variations in the stimulus current-density geometry between the electrode and the cortex. To achieve sufficient statistical power with higher variability, slightly higher number of rats was studied to determine electrical threshold compared with topical KCl-induced CSDs.
In addition, we calculated CSD propagation speed by dividing the distance (millimeters) between the two recording electrodes by the CSD latency (minutes) between these sites. The CSD amplitude and duration at half-maximal amplitude were also measured.
Statistical Analysis
The systemic and electrophysiological data and the number of CSDs after topical KCl were compared using one-way analysis of variance followed by Dunnett's multiple comparisons versus control. Electrical stimulation threshold was analyzed using Kruskal–Wallis one-way analysis of variance on ranks, followed by Dunn's multiple comparisons versus control. Data were expressed as mean ± s.d. for systemic physiology, electrophysiology, and number of KCl-induced CSDs, or as median (25% to 75% range) for electrical CSD threshold.
Results
A single intravenous dose of gabapentin (100 or 200 mg/kg) 1 hour before testing elevated the cathodal stimulation threshold for CSD (Figure 1A; Table 1), and dose dependently reduced the frequency of CSDs evoked by topical KCl application by up to 30% compared with saline-injected rats (Figure 1B). Gabapentin did not significantly alter the electrophysiological properties of individual CSDs, including the propagation speed and the incidence of propagation failure between the proximal and distal recording sites (Table 1).
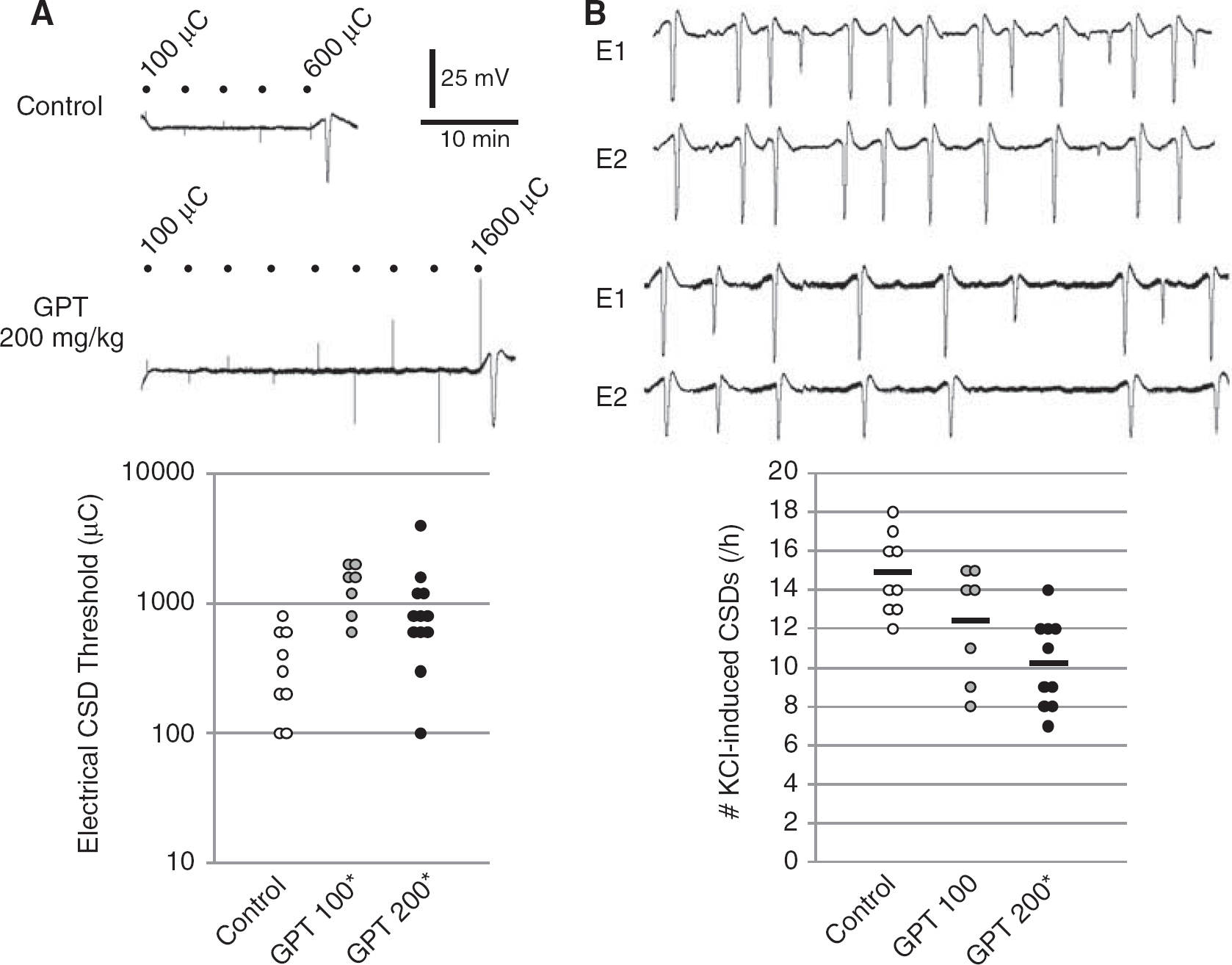
Effects of gabapentin on cortical spreading depression (CSD) susceptibility assessed by electrical stimulation (
Discussion
These data show a novel inhibitory effect of gabapentin on CSD susceptibility using two independent but complementary experimental paradigms. Importantly, CSD suppression appeared within 1 hour after a single intravenous dose, suggesting that gabapentin may suppress propagating waves of injury depolarizations akin to CSD in stroke, subarachnoid or intracerebral hemorrhage, and traumatic brain injury patients in the acute neurocritical care setting. Injury depolarizations worsen tissue outcome presumably through hemodynamic and metabolic mechanisms exacerbating the energy supply-demand mismatch (Hashemi et al, 2009; Shin et al, 2006; Strong et al, 2007). Suppression of injury depolarizations using drugs that inhibit CSD has been beneficial in animal models of stroke, and anecdotally in the clinical setting (Sakowitz et al, 2009; Shin et al, 2006). However, drugs that acutely inhibit CSD (e.g., N-methyl-D-aspartate receptor antagonists) often have neurological side effects that may limit their clinical usefulness. In that respect, gabapentin, widely used as an analgesic, adjunct antiepileptic and migraine prophylactic drug, offers an advantage with its highly favorable safety and tolerability profile. Importantly, however, intravenous administration may be required to rapidly achieve therapeutic plasma levels and CSD suppression.
Gabapentin attenuates both the stimulated presynaptic Ca2+ influx and the neurotransmitter release in synaptosomal, brain slice, and neuromuscular junction preparations (Dooley et al, 2007). Importantly, attenuation of neurotransmitter release appears to be more marked when release is evoked by intense stimulation, such as high [K+]e, rather than the more physiological stimulation paradigms. During CSD, massive elevations in [K+]e lead to large uncontrolled glutamate release, which augments CSD induction and propagation. Hence, gabapentin may suppress glutamate release during CSD more potently than during normal neurotransmission, and by this way reduce CSD susceptibility.
Gabapentin doses used in this study are clinically relevant, because they are well within the 50 to 300 mg/kg range that show efficacy in most models of epilepsy and pain in rats, and only two- to fourfold higher than the usual human doses. The plasma half-life of gabapentin is ∼100 minutes in rats compared with ∼6 hours in humans (Radulovic et al, 1995). Importantly, the inhibitory effect of gabapentin on presynaptic Ca2+ influx and neurotransmitter release becomes manifest rapidly within 30 minutes of drug application (van Hooft et al, 2002). Therapeutically, maximal antiepileptic effect is achieved within 120 minutes after a single intravenous dose (Welty et al, 1993), whereas maximal analgesic effect is attained within 30 to 60 minutes after a single intraperitoneal dose in rodents (Hunter et al, 1997).
Recent data implicated CSD suppression as a final common mechanism shared by five seemingly unrelated migraine prophylactic drugs (Ayata et al, 2006). Here, we provide evidence suggesting that gabapentin may also use this mechanism. Unlike previously tested drugs, however, gabapentin did not require chronic treatment to suppress CSD. Clinical evidence, albeit limited, supports modest efficacy for gabapentin in migraine prophylaxis. A gradual buildup of clinical efficacy has not been reported for gabapentin in migraine, the obvious constraints of trial design and reporting to assess onset of clinical efficacy notwithstanding. Nevertheless, in preliminary studies, we found that a lower dose of gabapentin (50 mg/kg, orally twice a day) for 5 weeks did not achieve CSD suppression, suggesting that chronic treatment does not augment gabapentin efficacy (data not shown).
In summary, gabapentin suppressed CSD susceptibility after a single intravenous dose. This may be a potential mechanism of action of gabapentin in migraine with aura shared by other migraine prophylactic drugs, and suggest that gabapentin may have efficacy in brain injury states where spreading depolarizations worsen tissue outcome (Williams et al, 2006).
Footnotes
The authors declare no conflict of interest.