
Abstract
Cortical spreading depression (CSD) evokes a large cerebral blood flow (CBF) increase in normal rat brain. In contrast, in focal ischemic penumbra, CSD-like periinfarct depolarizations (PID) are mainly associated with hypoperfusion. Because PIDs electrophysiologically closely resemble CSD, we tested whether conditions present in ischemic penumbra, such as tissue hypoxia or reduced perfusion pressure, transform the CSD-induced CBF response in nonischemic rat cortex. Cerebral blood flow changes were recorded using laser Doppler flowmetry in rats subjected to hypoxia, hypotension, or both. Under normoxic normotensive conditions, CSD caused a characteristic transient CBF increase (74 ± 7%) occasionally preceded by a small hypoperfusion (−4 ± 2%). Both hypoxia (
Keywords
Introduction
Cortical spreading depression (CSD) is a wave of intense neuronal and glial depolarization, depression of synaptic activity, and massive K+ efflux increasing extracellular K+ concentration ([K+]e) to more than 50 mmol/L. Once triggered by simultaneous depolarization of a minimum critical volume of brain tissue, CSD propagates at a rate of 2 to 4 mm/min throughout the cortex irrespective of functional divisions or vascular territories (Somjen, 2001). The impact of CSD on cerebral vasculature has been subject to intense investigation. In most species under anesthesia, CSD evokes a characteristic hyperemia, nearly doubling the cerebral blood flow (CBF) at its peak. This profound CBF increase starts late during the depolarization phase and outlasts it by a few minutes (Ayata et al, 2004; Bures and Buresova, 1956; Farkas et al, 2007; Lauritzen, 1987; Mayevsky et al, 1998; Piper et al, 1991; Sonn and Mayevsky, 2000). A brief vasoconstriction occasionally precedes the vasodilation leading to an initial small hypoperfusion (Ayata et al, 2004; Osada et al, 2006; Tomita et al, 2002, 2005; Van Harreveld and Stamm, 1952; Van Harreveld and Ochs, 1957), which is augmented by nitric oxide (NO) inhibition (Duckrow, 1993; Fabricius et al, 1995), particularly when [K+]e is artificially elevated (Dreier et al, 1998). Published data also suggest that partial tissue ischemia augments the initial vasoconstriction and diminishes vasodilation, as observed during hypoxia, after bilateral carotid occlusion and in focal ischemic penumbra (Shin et al, 2006; Sonn and Mayevsky, 2000; Strong et al, 2007).
In this study, we aimed to simulate two conditions that are present in the ischemic penumbra and to quantitatively investigate the impact of reduced tissue
Materials and methods
Surgical Preparation
Rats (Sprague—Dawley, male, 265 to 500 g) were anesthetized using urethane (1.3 to 1.5 g/kg, intraperitoneal) and intubated via a tracheostomy for mechanical ventilation (70% N2O/30% O2; SAR-830, CWE, Ardmore, PA, USA). Arterial blood gases and pH were measured every 15 to 30 mins, and ventilation parameters were adjusted to maintain
Rats were placed in a stereotaxic frame (David Kopf Inst., Tujunga, CA, USA) and two burr holes were drilled under saline cooling over the right hemisphere at the following coordinates: (1) anterior 2, lateral 3 mm from lambda (parietooccipital cortex) for topical KCl application; (2) posterior 2, lateral 3 mm from bregma (frontoparietal cortex) for CSD recording. The dura overlying the cortex was removed gently, and care was taken to avoid bleeding. The steady (DC) potential and electrocorticogram were recorded with a glass micropipette filled with 150 mmol/L NaCl, 300 mm below the dural surface (Axo-probe-1A, Axon Instruments, Foster City, CA, USA). Ag/AgCl reference electrode was placed subcutaneously in the neck. Regional CBF was recorded using laser Doppler flowmetry (Periflux PF2B, Perimed, Stockholm, Sweden). The laser Doppler flowmetry probe (0.6 mm diameter) was placed in the immediate vicinity of the CSD recording electrode in contact with the pial surface away from large vessels. After surgical preparation, the cortex was allowed to rest for 30 mins under saline irrigation and covered with mineral oil to prevent drying. A cotton ball (2 mm diameter) soaked with 1 mol/L KCl was placed on the pial surface until a CSD was recorded and then removed followed by saline wash. The data were recorded continuously using a data acquisition system for off-line analysis (PowerLab, ADInstruments).
Experimental Groups
Four experimental conditions were studied as follows: normoxic normotension (
Systemic physiologic parameters in the four experimental groups
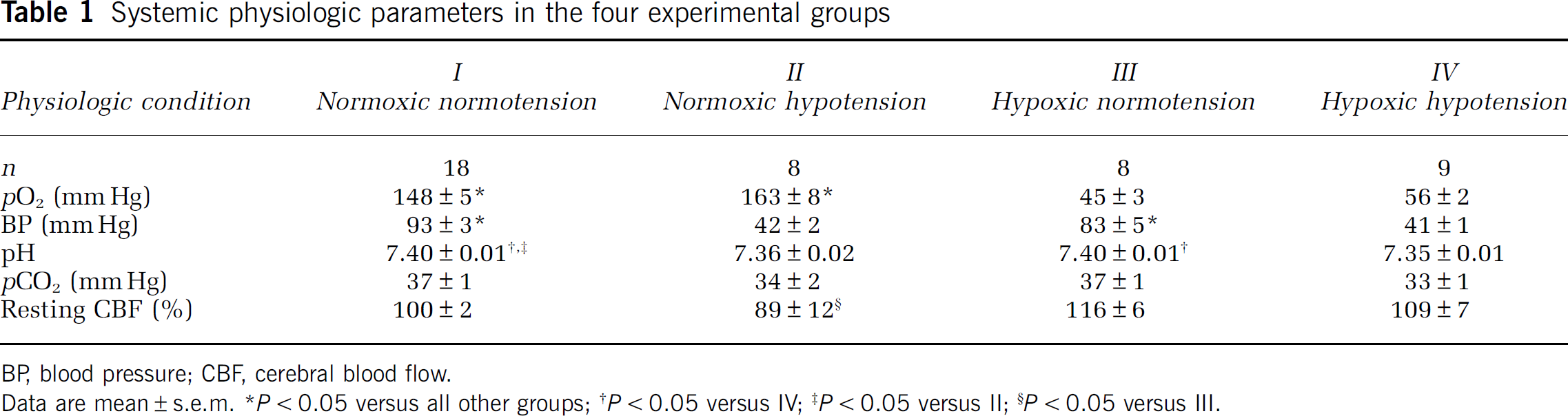
BP, blood pressure; CBF, cerebral blood flow.
Data are mean ± s.e.m.
Experimental Protocol
One to three baseline CSDs were induced in each rat at normoxic normotensive conditions, and CBF responses were recorded; at least 15 mins were allowed between each CSD induction, although occasionally two consecutive CSDs occurred after a single KCl application despite saline wash. In normoxic hypotensive, hypoxic normotensive, and hypoxic hypotensive groups, arterial
Data Analysis
Baseline CBF preceding the first CSD (i.e., initial baseline) during an experiment was taken as 100%, and residual laser Doppler signal after euthanasia was taken as biological zero CBF. The CBF response to CSD was typically triphasic (Figure 1). The following CBF deflection points were identified (labeled a—e in Figure 1A): (a) baseline immediately before CSD, (b) onset of hypoperfusion, (c) trough of hypoperfusion, (d) peak hyperemia, (e) recovery of hyperemia, (f) plateau 1 to 2 mins later. The amplitude and latency from the onset of DC shift were measured for each CBF deflection point, averaged within each of the four experimental conditions, and plotted relative to the initial baseline CBF at the beginning of the experiment under normoxic normotensive conditions (Figure 1, right panel). In addition, the duration of hypoperfusion was measured between the peak of initial increase (b) and the point when CBF recovers to the same value (see Figure 3B inset, ‘h’). Data obtained from repeated CSDs during each experimental condition in a single rat were averaged to obtain a single data point per experimental condition per rat. The amplitude of the DC shift and its duration at half amplitude were measured (see Figure 3A inset, ‘dc’) and averaged in the same way.
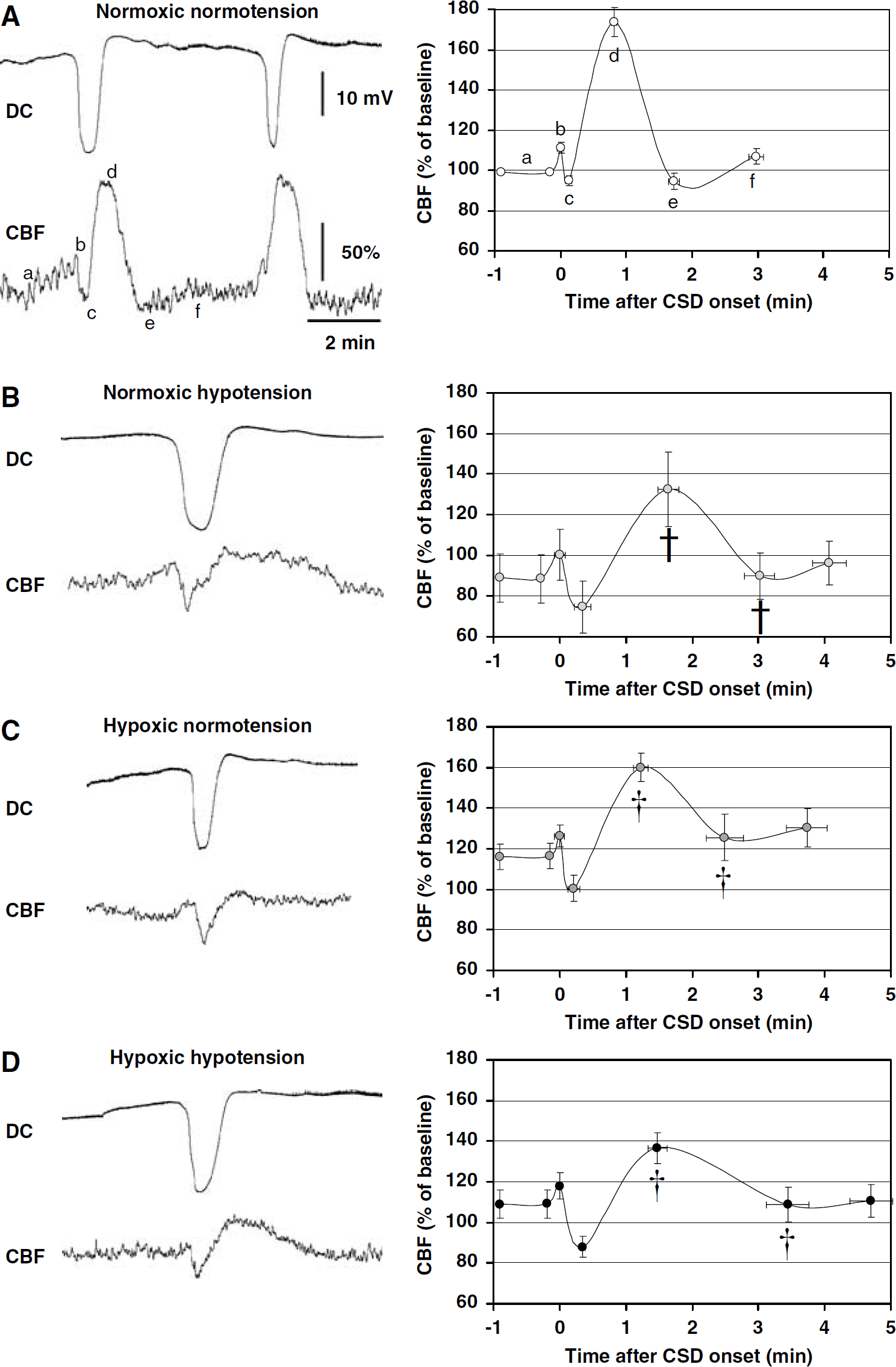
Hypoxia and hypotension transform the CBF response to CSD. Representative tracings of CBF and DC potential during CSD (left panel) and averaged CBF time course graphs (right panel) are shown from the four indicated experimental groups (
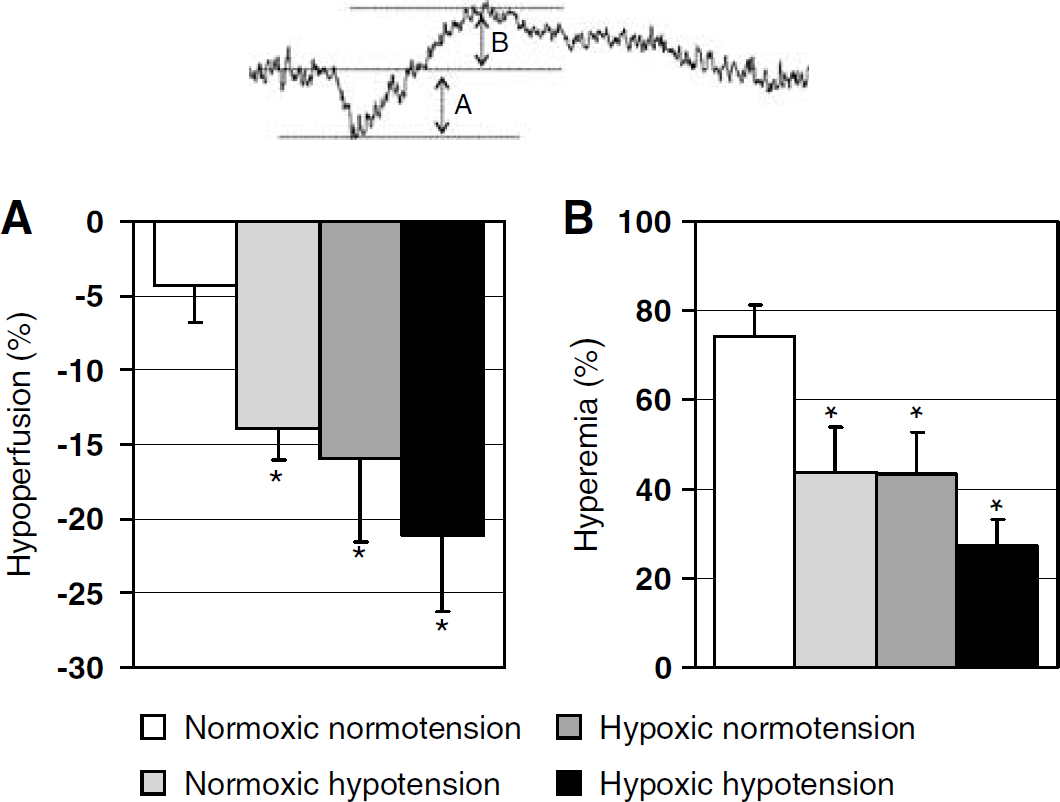
The impact of hypoxia and hypotension on the hypoperfusion and subsequent hyperemia during CSD. Both hypoxia and hypotension amplified the relative hypoperfusion (
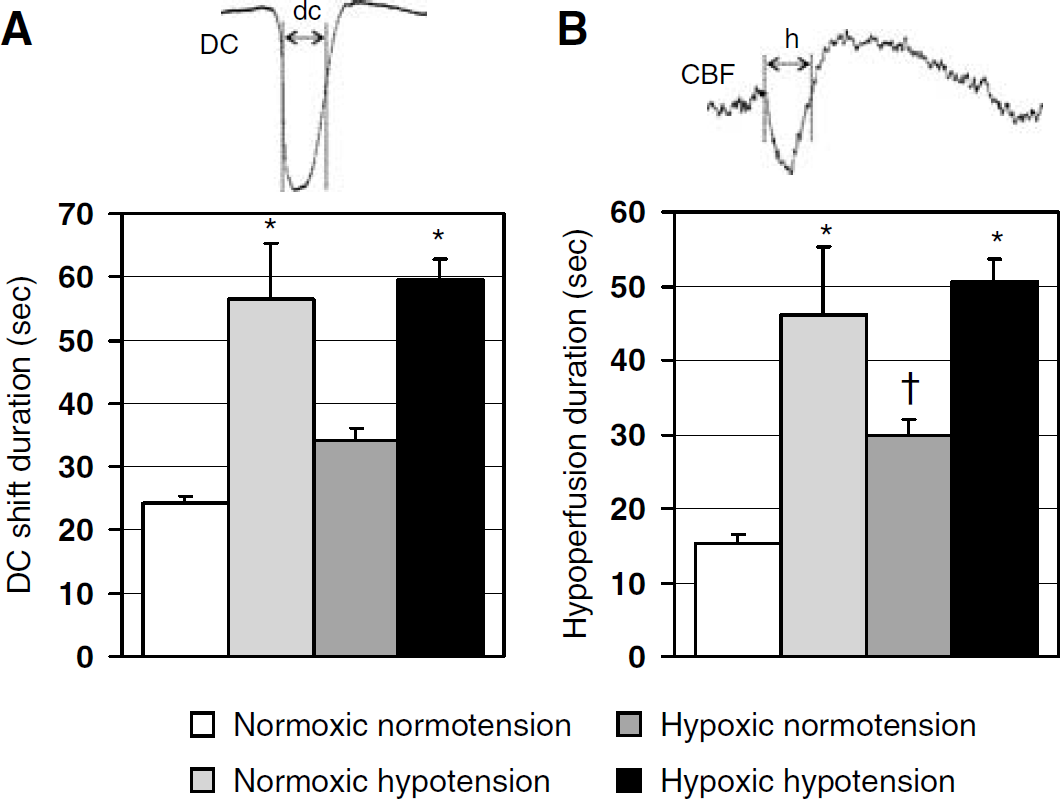
The impact of hypoxia and hypotension on DC shift and hypoperfusion durations. Hypotension significantly prolonged both the DC shift duration (
Statistical comparisons were done using one-way analysis of variance, followed by the Student—Newman—Keuls test for multiple comparisons among the four experimental groups. In addition, a forward stepwise multiple linear regression analysis was carried out using the pooled data from all rats to independently test the impacts of hypoxia and hypotension (continuous independent variables) on selected dependent variables (magnitudes of hypoperfusion and subsequent hyperemia, and durations of hypoperfusion and DC shift). The correlation between DC shift duration and the duration of hypoperfusion was tested using Pearson's product-moment correlation test on pooled data. Data were expressed as mean ± s.e.m., and
Results
Systemic physiologic parameters and baseline CBF in the four experimental groups are shown in Table 1. Hypoxia increased resting CBF in normotensive rats and, to a lesser extent, in hypotensive rats (Table 1). Normoxic hypotension reduced resting CBF only mildly (11%), suggesting that CBF autoregulation was relatively well preserved under urethane anesthesia.
Under normoxic normotensive conditions, the typical CBF response to CSD consisted of a large hyperemia (75%) that was often preceded by a small and brief hypoperfusion (Figure 1A, c). Both hypoxia (
Electrophysiological recordings showed that the DC shift duration was significantly prolonged during normoxic hypotension (135 ± 37% increase) and, to a much lesser extent, during hypoxic normotension (41 ± 9% increase;
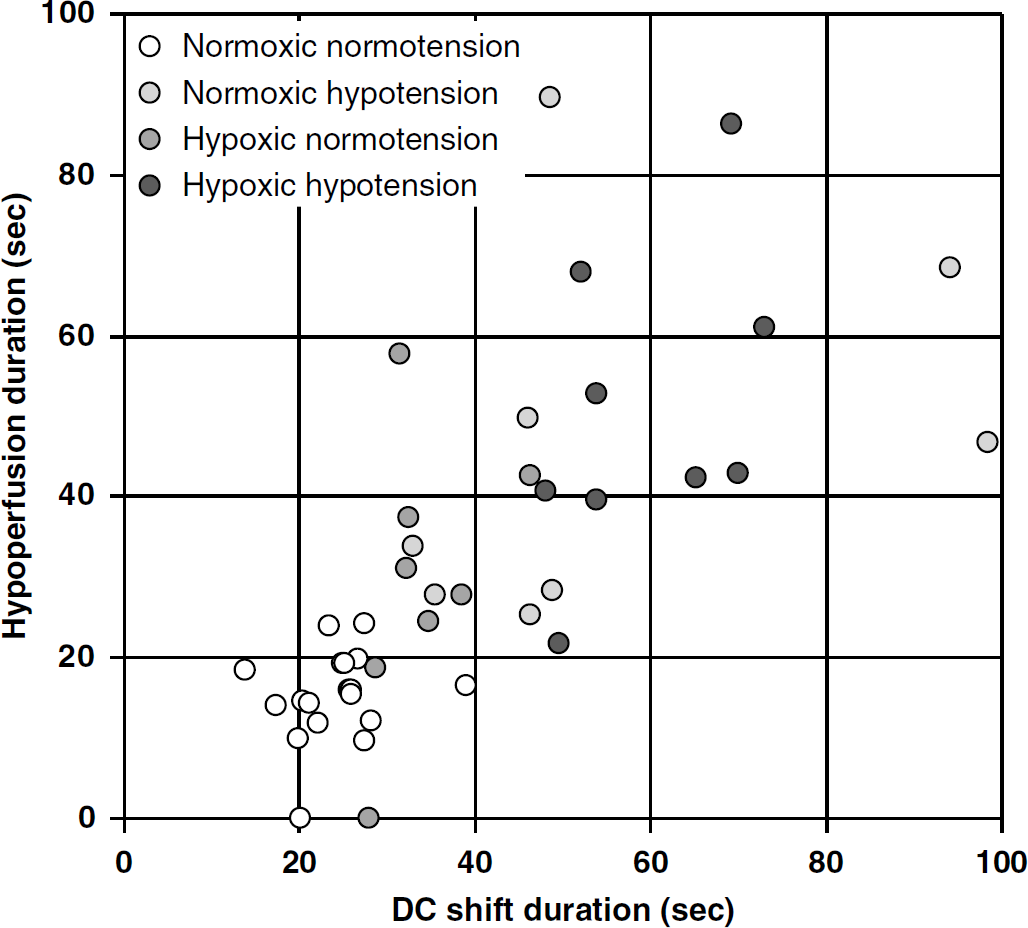
Correlation analysis between hypoperfusion and DC shift durations.
Multiple linear regression analysis of pooled data (arterial
Multiple linear regression analysis of the impact of
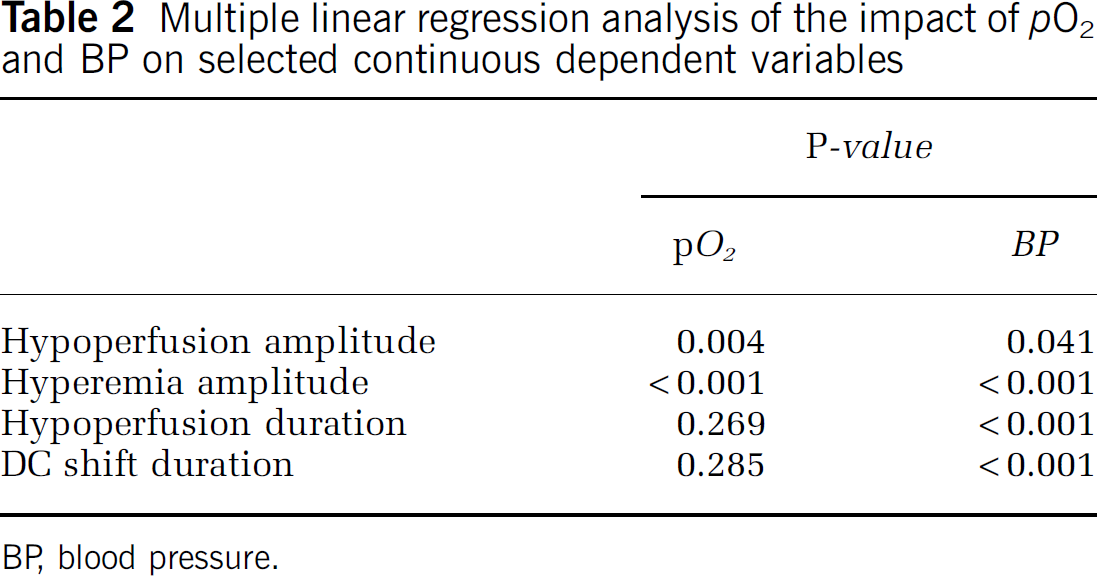
BP, blood pressure.
Discussion
The penumbra is a vulnerable region of the ischemic brain that undergoes spontaneous and repetitive periinfarct spreading depolarizations akin to CSD. In this study, we systematically investigated the impact of hypoxia or hypotension, individually or in combination, on the CBF response to CSD. Our data show that both hypoxia and reduced perfusion pressure transform the large but short-lasting CSD-induced hyperemia into a broad response consisting of hypoperfusion superimposed on the rising phase of an attenuated hyperemia. Our findings are in agreement with a previous qualitative study describing the impact of hypoxia or bilateral carotid occlusion on the CBF response to CSD (Sonn and Mayevsky, 2000) and suggest that, under moderate-to-severe hypoxic/ischemic conditions, the normal vasodilatory neurovascular coupling in rat brain is replaced by a vasoconstrictive neurovascular coupling response. Our data also support the notion that anoxic depolarization and periinfarct spreading depressions worsen the metabolism—flow mismatch in ischemic tissue not only by increasing the energy demand but also by reducing the blood supply (Shin et al, 2006). In contrast to normal CSDs that do not cause injury in the healthy cortex, depolarizations in ischemic tissue do contribute to infarct progression by metabolic and hemodynamic mechanisms, rather than being epiphenomena of focal cerebral ischemia (Busch et al, 1996).
Under normoxic normotensive conditions, the characteristic large hyperemia is often preceded by a small hypoperfusion, as previously described in anesthetized rats, rabbits, and cats (Ayata et al, 2004; Fabricius et al, 1995; Osada et al, 2006; Tomita et al, 2002, 2005; Van Harreveld and Stamm, 1952), suggesting that CSD causes vasoconstriction followed by vasodilation. More recently, selective measurements of arterial diameter in 15- to 25-day-old rats show heterogeneous vasomotor responses to CSD depending on the vessel caliber. Larger pial surface arterioles respond with a small initial constriction followed by dilation, whereas smaller intraparenchymal arterioles mainly constrict (Chuquet et al, 2007), supporting the notion that CSD exerts two opposing vasomotor effects on cerebral vessels, rather than being a universal vasodilator. Indeed, CSD does not cause hyperemia in awake rats, according to Duckrow (Duckrow, 1991; Duckrow and Beard, 1992), or in anesthetized mice (Ayata et al, 2004; Brennan et al, 2007), suggesting that the relative balance between the opposing vasomotor influences depends on the species and experimental conditions. Under physiologic conditions, the vasodilator coupling during CSD predominates in anesthetized rats. Under nonphysiologic conditions, such as systemic hypoxia and reduced cerebral perfusion pressure, the vasoconstrictive response can be augmented and become the predominant response to CSD, as previously reported in response to CSD during focal and global ischemia (Shin et al, 2006; Sonn and Mayevsky, 2000; Strong et al, 2007). The observed variability of hypoperfusion response to CSD in previous studies (Ayata et al, 2004; Bures and Buresova, 1956; Duckrow, 1993; Fabricius et al, 1995; Farkas et al, 2007; Lauritzen, 1987; Mayevsky et al, 1998; Osada et al, 2006; Piper et al, 1991; Sonn and Mayevsky, 2000; Tomita et al, 2002, 2005; Van Harreveld and Stamm, 1952; Van Harreveld and Ochs, 1957) may thus be related to differences in baseline systemic physiology (
The mechanisms by which hypoxia augments CSD-induced hypoperfusion are not known; however, the transformation during hypoxia resembled that observed upon NO synthase inhibition (Ayata et al, 2004; Dreier et al, 1998; Duckrow, 1993; Fabricius et al, 1995). Oxygen is a required substrate for NO production, and upstream regulators such as Rho-kinase can rapidly downregulate endothelial NO synthase activity during hypoxia or ischemia via allosteric mechanisms (Rikitake et al, 2005; Takemoto et al, 2002). Mild hypoxic elevation in [K+]e may contribute, as elevated [K+]e has been shown to synergize with NO synthase inhibition to invert the hyperemia into a severe spreading hypoperfusion in rats (Dreier et al, 1995, 1998). In these experiments, the pattern of oxygen-free radical chemiluminescence did not change in response to a normal CSD but showed the typical pattern associated with cellular damage when CSD caused vasoconstrictive coupling, termed inverse coupling by these authors. Hence, vasoconstrictive coupling has been proposed as an independent mechanism of neuronal damage (Dreier et al, 1998). Conclusively, under artificial cerebrospinal fluid containing hemoglobin combined with either elevated [K+]e or low glucose, they showed that CSD associated with vasoconstrictive coupling led to widespread focal pannecrosis in the absence of any preexisting energy depletion and established the term cortical spreading ischemia for this process (Dreier et al, 2000).
Hypotension alone was as effective in diminishing hyperemia despite the absence of a significant reduction in baseline resting CBF and hence tissue oxygenation. It is likely that maximal autoregulatory vasodilation was approached in the hypotensive groups, which would limit the ability of cerebral vessels to further dilate in response to CSD; low intravascular perfusion pressure may have facilitated the vasoconstrictive effect of CSD as well. In hypoxic normotensive rats in whom perfusion pressure was maintained constant, resting CBF was a reliable indicator of cerebrovascular tone and increased by only 16% (Table 1, group III), suggesting that maximal vasodilation was not reached in this group. In spite of this, the CBF response to CSD was transformed similar to the hypotensive group, suggesting that resting vascular tone may not be the most important factor determining the CBF response to CSD. It should be noted that although maximal vasodilation and reduced vasodilatory reserve might diminish the hyperemic response, they cannot fully explain the emergence of a vasoconstrictive response to CSD.
The duration of DC shift was prolonged by hypotension and hypoxia, as previously described in energy-compromised tissue (Gido et al, 1994), presumably due to the delay in restoration of ionic and water homeostasis. However, although hypoxia caused a modest prolongation of DC shift (40%), the impact of hypotension was much stronger (130%). This was paradoxical because in the presence of intact autoregulation, normoxic hypotension lowered resting CBF by only 11% (Table 1, group II), which, unlike in hypoxic rats, is not expected to cause a severe reduction in tissue
Interestingly, the prolongation of DC shift correlated with the prolongation of hypoperfusion, suggesting an association between tissue depolarization and vasoconstriction, as previously noted during periinfarct depolarizations after middle cerebral artery occlusion (Shin et al, 2006; Strong et al, 2007), and during CSDs under conditions mimicking subarachnoid hemorrhage (Dreier et al, 2002). In the latter study, on the basis of 69 experiments in rats, the durations of DC shift and hypoperfusion were highly correlated (
In summary, our data suggest that nonphysiologic conditions such as hypoxia or hypotension attenuate hyperemia and unmask a vasoconstrictive form of neurovascular coupling during CSD in rats. Whether this adverse neurovascular response takes place in the human brain after cerebral ischemia, contusion, or intracranial hemorrhage (Dohmen et al, 2007; Dreier et al, 2006; Fabricius et al, 2006) remains to be determined.
Footnotes
The authors state no conflict of interest.