
Abstract
Inflammation has an important function in the development of cerebral vasospasm after subarachnoid hemorrhage (SAH); however, the mediators of this inflammatory response have not been clearly identified. In this study, we have investigated the potential function of two sphingolipids, which occur naturally in plasma and serum, sphingosylphosphorylcholine (SPC) and sphingosine 1-phosphate (S1P), to act as proinflammatory mediators in cerebral artery vascular smooth muscle (VSM) cells. In rat cerebral arteries, SPC but not S1P activated p38 mitogen-activated protein kinase (MAPK). Using transcription factor arrays, two proinflammatory transcription factors activated by SPC in cerebral arteries were identified—nuclear factor-κB and CCAAT-enhancer-binding protein. Both these transcription factors were activated by SPC in a p38MAPK-dependent manner. To determine whether this contributed to vascular inflammation, an inflammatory protein array was performed, which showed that SPC increased release of the chemokine monocyte chemoattractant protein-1 (MCP-1) in cultured rat VSM cells. This increase in MCP-1 expression was confirmed in cerebral arteries. The S1P did not increase MCP-1 release. Taken together, our results suggest that SPC, but not S1P, can act as a proinflammatory mediator in cerebral arteries. This may contribute to inflammation observed after SAH and may be part of the initiating event in vasospasm.
Introduction
Subarachnoid hemorrhage (SAH) occurs after the rupture of an aneurysm on the cerebral artery wall (Sehba and Bederson, 2006). This hemorrhage leads to thrombus formation on the adventitial side of the ruptured vessel and produces local ischemia in the surrounding neural tissue (Kassell et al, 1985). In addition, in 30% to 70% of cases, SAH also results in an intense and prolonged constriction of the affected cerebral vasculature (Bederson et al, 2009). This cerebral vasospasm typically occurs after a delay of 3 to 5 days after hemorrhage reaching a maximum around 7 days. There are no efficient treatments for cerebral vasospasm and ∼50% of SAH patients will develop a subsequent infarction (Bederson et al, 2009).
The development of cerebral vasospasm is directly linked to the presence of the subarachnoid blood clot (MacDonald and Weir, 1991). However, after blood clot formation, the processes that lead to the delayed arterial constriction are unclear (Nishizawa and Laher, 2005). Several different candidates, including oxyhemoglobin, reactive oxygen species, and the vasoconstrictor endothelin-1, have been investigated, suggesting that vasospasm may be a multifactorial process (Kolias et al, 2009). Intervention studies in SAH patients who develop cerebral vasospasm have to date failed to identify a potential therapeutic target, which improves patient outcome (Nishizawa and Laher, 2005; Mocco et al, 2006). Recent studies have now suggested that inflammation is an important component in the pathological development of cerebral vasospasm (Dumont et al, 2003). Analysis of cerebrospinal fluid after SAH in patients has shown increases in inflammatory proteins such as interleukin (IL)-1β, IL-6, and C-reactive protein (Schoch et al, 2007; Hendryk et al, 2004). Although it is expected that this inflammatory response will be at least in part produced by inflammatory cell types, gene expression studies on cerebral arteries from different animal models of SAH indicate that the vascular smooth muscle (VSM) cells also undergo an inflammatory response and likely contribute directly to inflammation during the development of vasospasm. Elevation in the mRNA levels for IL-1β, IL-6, IL-8, monocyte chemoattractant protein-1 (MCP-1), and intercellular adhesion molecule-1 have been observed (Onda et al, 1999; Aihara et al, 2001) in cerebral arteries after induction of SAH. Those inflammatory markers were highest at day 7 after SAH, the peak time of cerebral vasospasm. However, although inflammation of the cerebral arteries is likely to be involved, the key proinflammatory mediators and associated signaling mechanisms, which switch on this process have yet to be identified. As vasospasm is directly related to the subarachnoid blood clot, such mediators are likely to be derived from plasma or released from blood-borne cells in serum.
Sphingolipids, produced predominantly from sphingomyelin metabolism, are now known to have important functional effects on the cardiovascular system (Alewijnse et al, 2004). Two sphingolipids, sphingosine 1-phosphate (S1P) and sphingosylphosphorylcholine (SPC), occur naturally in plasma and are elevated in serum (Yatomi, 2008; Liliom et al, 2001). The S1P concentration is ∼300 nmol/L in plasma and these levels seem to be regulated by erythrocytes that constitutively produce S1P (Yatomi, 2008). Less is known about the regulation of SPC (Nixon et al, 2008) although the plasma levels have been measured at 50 nmol/L (Liliom et al, 2001). A significant proportion of plasma-derived S1P and SPC (∼60%) are bound to lipoproteins (Okajima, 2002). An additional source of S1P and SPC comes from activated platelets. In the case of S1P, platelets do not express S1P lyase, an enzyme responsible for the breakdown of S1P, and therefore store high concentrations of S1P (Yatomi et al, 1995). When activated, the S1P is released with other platelet-derived mediators. The S1P in serum is elevated to around low μmol/L levels. The SPC is also elevated in serum (Liliom et al, 2001) but its mechanisms are unclear.
Both S1P and SPC engage a wide repertoire of intracellular signaling pathways in many different cell types (Sanchez and Hla, 2004; Nixon et al, 2008). It is now known that S1P-induced cellular effects occur predominantly through the activation of the seven-transmembrane, G-protein-coupled receptors S1P receptors, S1P1–5 (Sanchez and Hla, 2004).
After SAH, both S1P and SPC released from platelets in the blood clot would have direct contact with cerebral VSM cells in the subarachnoid space and could potentially act as mediators of vasospasm either directly or through proinflammatory effects. The aim of this study was to examine the function of S1P and SPC as potential proinflammatory mediators in cerebral arteries. Our results show that SPC, but not S1P, can induce inflammation in cerebral artery VSM cells through the activation of p38MAPK and subsequent upregulation of inflammatory transcription factors leading to the release of the chemokine MCP-1. This SPC-induced process could initiate further inflammation in other cell types and, as such, may be an important component of the inflammation associated with the development of cerebral vasospasm.
Materials and methods
Tissue Preparation and Cell Culture
Male Sprague–Dawley rats (6 weeks old, 300 to 350 g) were euthanized by inhalation of CO2 followed by cervical dislocation. All procedures were in accordance with the institutional guidelines. Cerebral arteries (middle and basilar) were immediately removed and placed into ice-cold physiological saline solution as described earlier (Coussin et al, 2002). Dissected cerebral arteries were cleaned of connective tissue and the endothelium was removed by gentle rubbing of the arterial lumen. Cleaned arteries were preincubated in serum-free cell culture medium in a humidified 5% CO2 atmosphere at 37°C overnight before
Sphingolipid Preparation
Lyophilized S1P and SPC (Sigma Aldrich, Dorset, UK) were dissolved in methanol and stored in aliquots at −20°C. Before use, methanol was evaporated and sphingolipids were dissolved in 3.6 mg/mL bovine serum albumin solution containing 10% dimethyl sulfoxide at 37°C. Concentrations of 5 μmol/L S1P and 10 μmol/L SPC were used throughout this study. These concentrations typically achieve maximal or close to maximal effects in concentration–effect curves as assessed earlier (Coussin et al, 2002, Mathieson and Nixon, 2006) (Supplementary Figure 1).
Transcription Factor Array
Rat cerebral arteries were incubated with 5 μmol/L S1P or 10 μmol/L SPC for 1 hour at 37°C. Nuclear extracts were prepared using a nuclear extraction kit (Panomics, Fremont, CA, USA). Extracts were subjected to the TranSignal function-specific protein/DNA array (Panomics) according to the manufacturer's instructions to screen for changes in the binding to 20 different transcription factor binding sites (activator protein 1, CCAAT-enhancer-binding proteins (C/EBPs), CCAAT-binding factor, cyclic adenosine monophosphate (AMP) response element-binding protein, E4F, early growth response factors, E26 transformation-specific sequence factors, GATA-3, GATA-4, hepatocyte nuclear factor (NF) 4, heat shock factor protein, myocyte enhancer factor 2, nuclear factor of activated T cells, YY1 transcription factor, NF-κB, octamer-binding transcription factor 1 (OCT-1), peroxisome proliferator-activated receptor α, Rel, Smad-binding element, Sp1). In brief, nuclear extracts were preincubated with biotin-labeled DNA-binding oligonucleotides to allow the formation of DNA/protein complexes. The complexes were separated from the free probes and hybridized to the TranSignal array. Signals were detected by horseradish peroxidase (HRP)-based chemiluminescence. Quantitation was performed as for immunoblots.
Immunoblotting
Rat cerebral arteries were incubated with 5 μmol/L S1P or 10 μmol/L SPC for the appropriate time at 37°C. As required, arteries were additionally preincubated with 30 μmol/L SB203580 (4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4pyridyl)1H-imidazole, a selective p38MAPK inhibitor (Calbiochem, Nottingham, UK) for 30 minutes. Treated tissue was homogenized with lysis buffer in a glass Braun homogenizing vessel on ice as described earlier (Coussin et al, 2002). To obtain sufficient tissue, cerebral arteries from 2 to 3 rats were pooled for each sample. Protein was measured using a Lowry assay (Bio-Rad, Hemel Hempstead, UK) to ensure equal protein loading and membranes were stained with Ponceau Red to confirm protein loading. Protein samples were fractionated by SDS-PAGE, transferred to nitrocellulose membranes, and blocked with 5% nonfat milk powder in Tris-buffered saline, pH 7.4, containing 0.1% Tween 20. Membranes were incubated overnight at 4°C with primary antibody against IκB (Santa Cruz Biotechnology, Santa Cruz, CA, USA), phospho-p38 (Cell Signaling, Danvers, MA, USA), or glyceraldehyde 3-phosphate dehydrogenase (GAPDH) followed by HRP-conjugated secondary antibody (Sigma Aldrich). Immunoreactive bands were visualized by enhanced chemiluminescence and quantitated by densitometry as described earlier (Coussin et al, 2002).
Electrophoretic Mobility Shift Assay
Rat cerebral arteries were treated with 5 μmol/L S1P or 10 μmol/L SPC for 1 hour at 37°C with or without 30 minutes preincubation in 30 μmol/L SB203580. Electrophoretic mobility shift assay (EMSA) was performed using a commercially obtained kit (Panomics) according to the manufacturer's instructions. In brief, samples were incubated with biotinylated transcription factor probe. Complexes were separated from the free probes by nondenaturing polyacrylamide gel electrophoresis. After hybridization to Hybond-N+ membrane, signals were detected by HRP-based chemiluminescence.
Inflammatory Protein Array
A7r5 cells were treated with 5 μmol/L S1P, 10 μmol/L SPC, or 25 μg/mL lipopolysaccharide (LPS) from
Enzyme-Linked Immunosorbent Assay
A7r5 cells were treated with 5 μmol/L S1P or 10 μmol/L SPC for 24 hours at 37°C. Conditioned medium was analyzed using a rat MCP-1-specific enzyme-linked immunosorbent assay kit (RayBiotech). In brief, samples were added to wells coated with anti-rat MCP-1 capture antibody for 2.5 hours. Biotinylated detection antibody was added for 1 hour followed by HRP-conjugated streptavidin. After incubation in substrate reagent for 30 minutes, stop solution was added and the samples were read immediately at 450 nm using a microplate reader. For quantitation, a standard curve was created using supplied recombinant rat MCP-1.
Immunofluorescence
Rat cerebral arteries were treated with either 10 μmol/L SPC, 5 μmol/L S1P, 1 μg/mL LPS, or vehicle in the presence of 5 μg/mL brefeldin A for 48 hours at 37°C. After stimulation, rat cerebral arteries were fixed in 3% paraformaldehyde/phosphate-buffered saline (PBS) and infused with sucrose before flash freezing as described earlier (Coussin et al, 2003). Cryosectioning was performed on a Reichert Jung cryostat E/Leica CM1900 microtome at −16°C. Sections were blocked with 3% bovine serum albumin (BSA)/PBS before incubation with primary antibody anti-MCP-1 (Millipore, Watford, UK) overnight at 4°C. Secondary antibody fluorescein isothiocyanate (FITC)-anti-rabbit IgG (Jackson Immunoresearch Europe, Suffolk, UK) was applied for 1 hour at room temperature. Subsequently, nuclei were stained with BOBO-3 (Invitrogen, Paisley, UK). Specificity of immunostaining was confirmed by the absence of fluorescence in arteries incubated with secondary antibody alone. Immunofluorescence was detected using a Bio-Rad 1024 laser scanning confocal microscope (Bio-Rad)/Olympus BX50WI equipped with a krypton-argon laser and a 40 × oil-immersion lens. The laser was fitted with either a blue (excitation 488 nm) or a yellow (excitation 568 nm) filter block.
Statistics
Data are expressed as mean ± s.e.m. Significance was tested by means of Student's
Results
Sphingosylphosphorylcholine but not Sphingosine 1-Phosphate Activates p38 Mitogen-Activated Protein Kinase in Rat Cerebral Arteries
We have shown earlier that a 15-minute incubation with SPC can increase the phosphorylation of p38MAPK in denuded rat cerebral artery (Mathieson and Nixon, 2006). This indicates an engagement of the p38MAPK signaling pathway. However, we have not previously studied the time course of SPC-induced activation in cerebral arteries. This could be important in relation to the sustained nature of cerebral vasospasm. Our data now show that phosphorylation of p38MAPK is activated within 5 minutes and reaches a peak between 15 and 30 minutes before declining toward baseline levels at 60 minutes (Figure 1A). The S1P at 5 μmol/L has no effect on p38MAPK phosphorylation over the time course of 1 hour. To ensure that the lack of effect of S1P on p38MAPK phosphorylation is due to a lower concentration (relative to SPC), we also examined the effects of 50 μmol/L S1P. At this concentration, S1P failed to induce any increase in p38MAPK activation (Supplementary Figure 2).
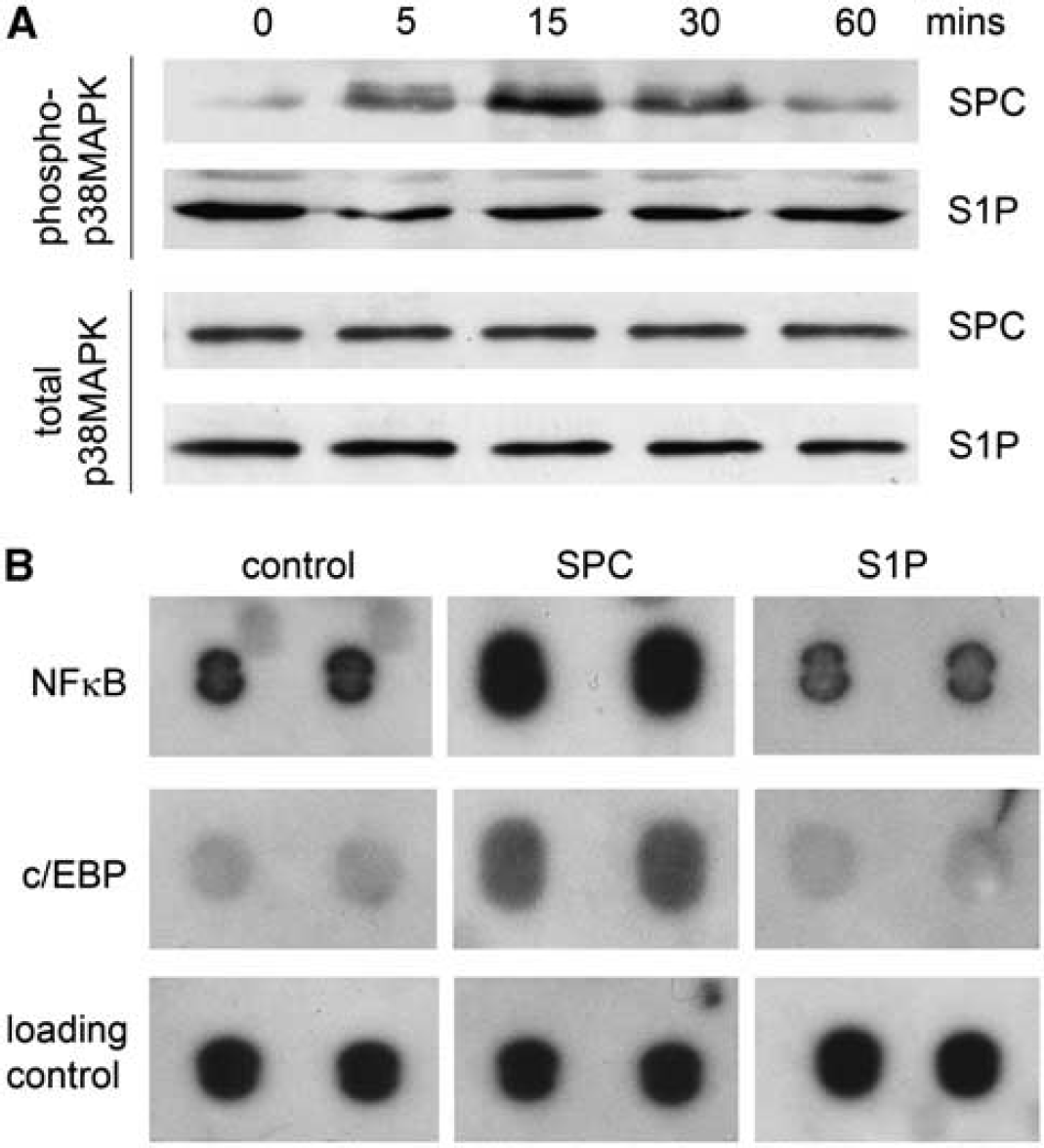
Sphingosylphosphorylcholine (SPC) activates p38 mitogen-activated protein kinase (MAPK) and the transcription factors nuclear factor (NF)-κB and CCAAT-enhancer-binding protein (C/EBP). (
Profile of Transcription Factor Activity Induced by Sphingosylphosphorylcholine and Sphingosine 1-Phosphate in Rat Cerebral Arteries
Because of the central function of p38MAPK in inflammatory intracellular signaling, we performed a transcription factor array to identify potential target proteins involved in sphingolipid signaling. As SPC and S1P have some divergence of intracellular signaling, we were particularly interested in transcription factor activity where S1P and SPC had different effects. Although this preliminary screen was limited to 20 transcription factor binding sequences, it provided information regarding the potential downstream effects of SPC and S1P. Rat cerebral arteries were treated
Activation of the Nuclear Factor-κB Signaling Pathways by Sphingosylphosphorylcholine but not Sphingosine 1-Phosphate
To confirm preliminary findings of the transcription factor arrays, we conducted further experiments on the activation of NF-κB. The activity of the transcription factor NF-κB is regulated in several ways (Hayden et al, 2006). The best-established model proposes that the inhibitor of NF-κB, IκB, sequesters NF-κB in the cytoplasm of unstimulated cells and thus maintains the transcription factor in an inactive state. On phosphorylation by IκB kinase, the phosphorylated IκB becomes ubiquitinated and subsequently degraded. This effectively releases NF-κB, which subsequently translocates to the nucleus and initiates expression of target genes. We initially examined the activation of this signaling complex by determining the degradation of IκBα, an indirect indicator for activation of the NF-κB signaling pathway. Rat cerebral arteries were incubated
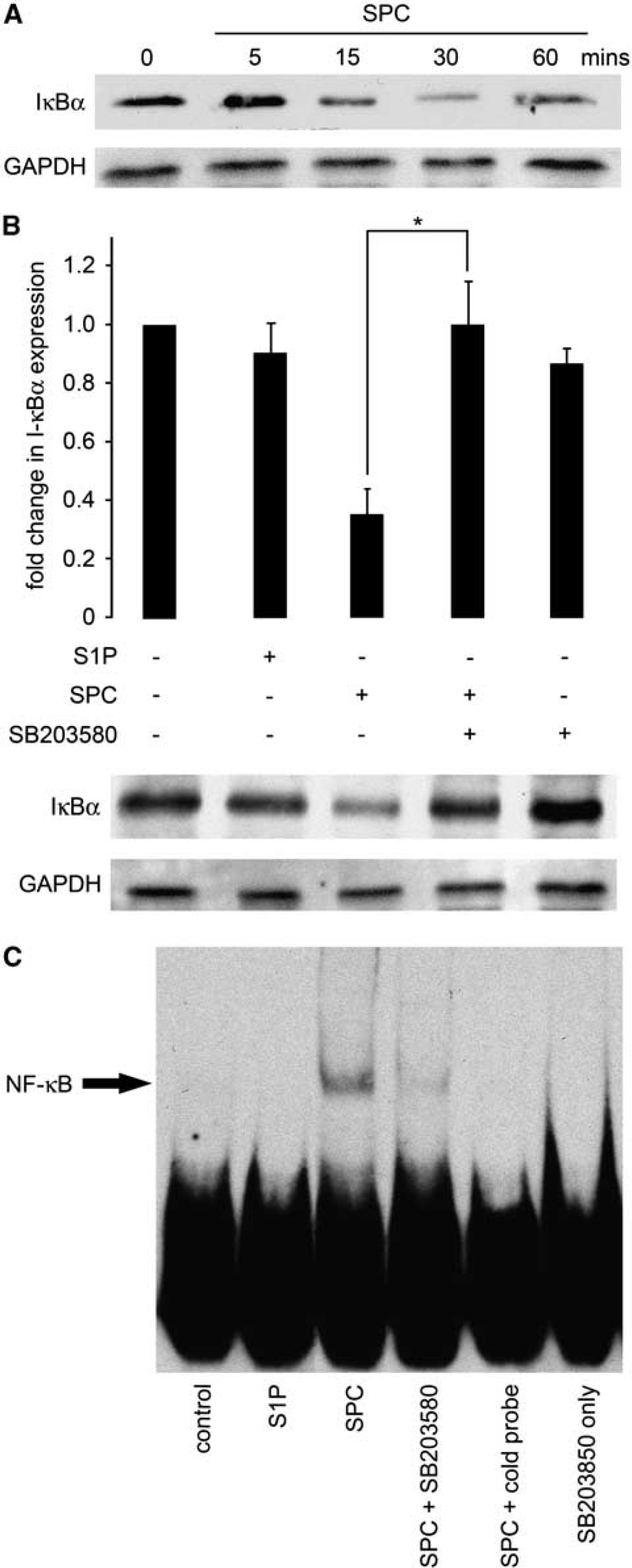
Activation of the nuclear factor (NF)-κB signaling pathway by sphingosylphosphorylcholine (SPC) is dependent on p38 mitogen-activated protein kinase (MAPK). (
Activation of the CCAAT-Enhancer-Binding Proteins Pathway by Sphingosylphosphorylcholine but not Sphingosine 1-Phosphate
The transcription factor array indicated that SPC could activate the proinflammatory transcription factor C/EBP in rat cerebral arteries. The potential binding of C/EBP to consensus oligonucleotide sequence was examined by EMSA as above. Rat cerebral arteries were treated with S1P or SPC for 1 hour at 37°C with or without pretreatment with SB203580 and nuclear extracts prepared. The SPC, but not S1P, increased C/EBP binding to DNA (Figure 3). This effect of SPC was diminished by p38MAPK inhibition.
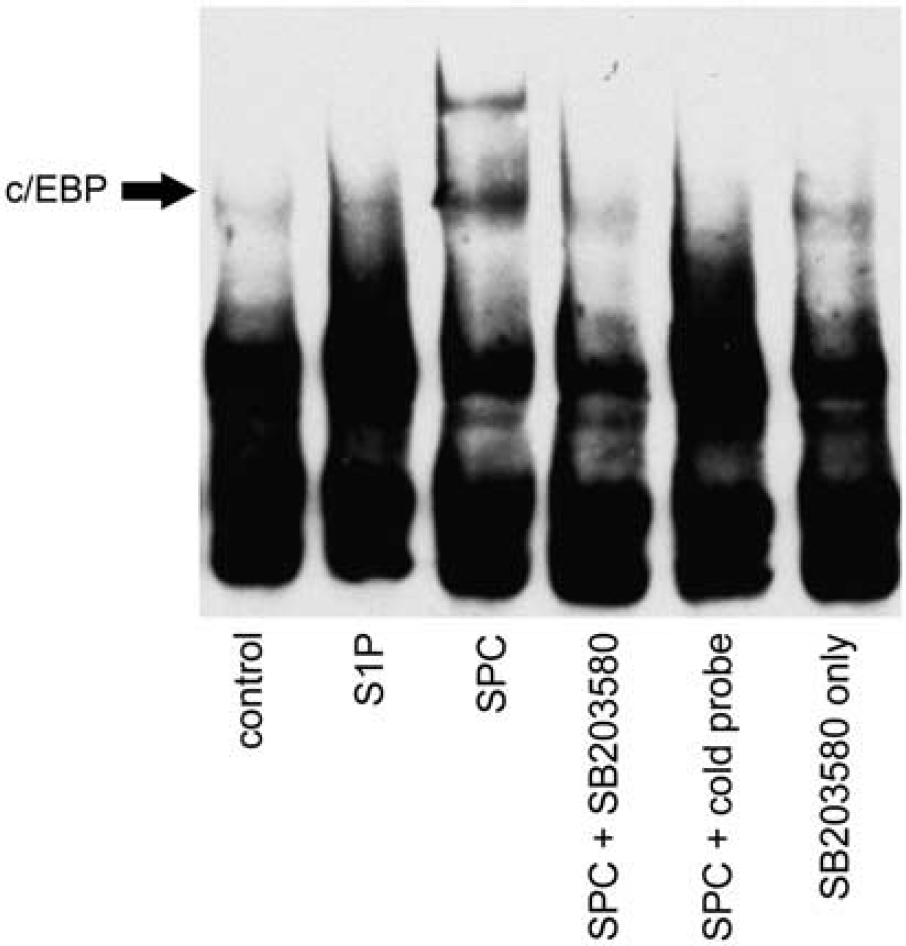
Activation of CCAAT-enhancer-binding protein (C/EBP) by sphingosylphosphorylcholine (SPC) is dependent on p38 mitogen-activated protein kinase. Nuclear fractions of cerebral arteries incubated in 10 μmol/L SPC or 5 μmol/L sphingosine 1-phosphate (S1P) as above for 1 hour after preincubation with 30 μmol/L SB203580 were subjected to electrophoretic mobility shift assay and the binding of C/EBP to DNA consensus sequences was determined. Incubation with SPC increased C/EBP binding to specific oligonucleotides. This effect of SPC was decreased by p38 inhibition. The S1P did not increase C/EBP activity. SB203580 alone also had no effect on DNA binding to this oligonucleotide. A representative electrophoretic mobility shift assay is shown (
Sphingosylphosphorylcholine-Induced Inflammatory Protein Production in Vascular Smooth Muscle Cells
As proinflammatory signaling pathways were activated in cerebral artery after SPC incubation, we investigated the downstream consequences. Cytokine protein arrays were used to identify the release of candidate inflammatory proteins. Because of the technical limitations of rat cerebral artery (tissue yields are insufficient, even after substantial pooling from multiple animals to measure release of inflammatory proteins), the rat A7r5 thoracic aorta smooth muscle cell line was used as an
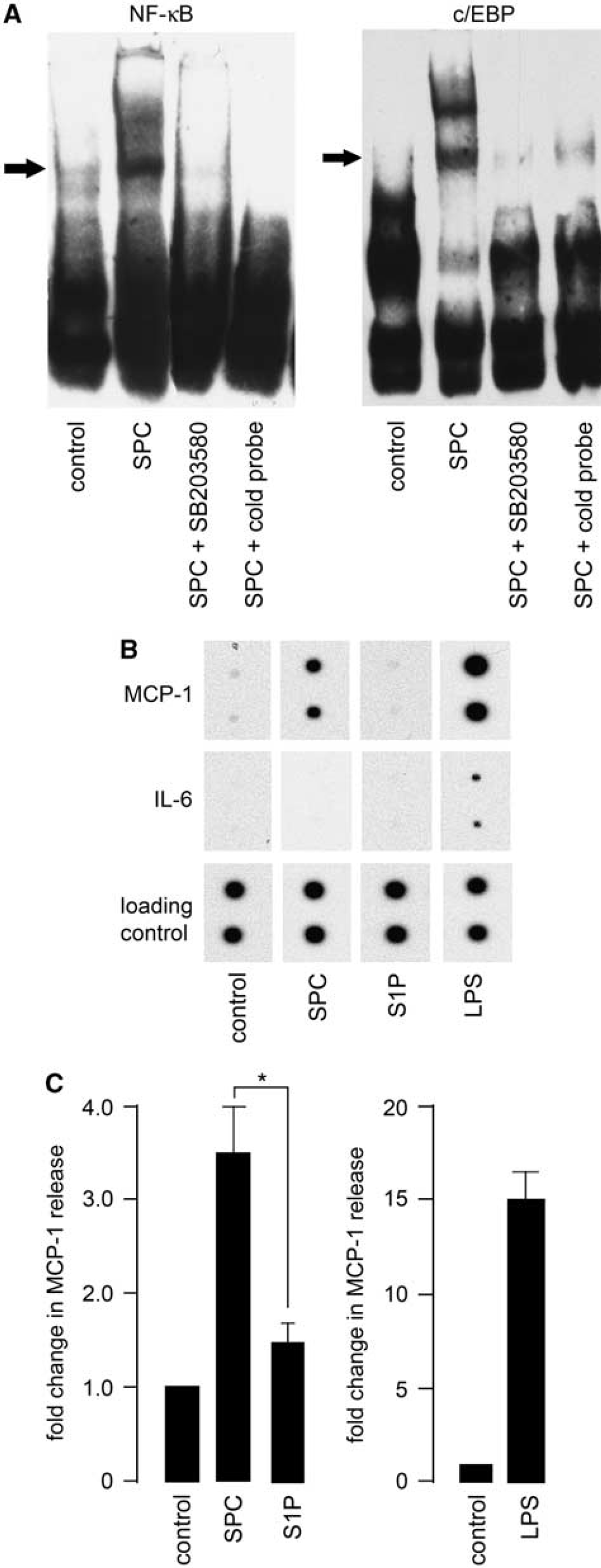
Sphingosylphosphorylcholine (SPC) induces monocyte chemoattractant protein-1 (MCP-1) release in A7r5 cells. (
To determine cytokine release, A7r5 cells were first treated with 10 μmol/L SPC or 5 μmol/L S1P for 24 or 48 hours at 37°C. Conditioned medium was analyzed using a cytokine protein array according to the manufacturer's instructions. The LPS (25 μg/mL) was used as a positive control. In LPS-treated A7r5 cells, the release of most of the cytokines detected by the array was increased as expected. The most pronounced increases were observed for IL-6, cytokine-induced neutrophil chemoattractant-3, fractalkine, granulocyte-macrophage colony-stimulating factor, MCP-1, and macrophage inflammatory protein 3α. However, incubation with SPC for either 24 or 48 hours increased the release of only one inflammatory protein, the chemokine MCP-1 (Figure 4B). The S1P did not produce an increased release in any inflammatory proteins detected by the array.
To confirm the results with the rat cytokine array, enzyme-linked immunosorbent assay was performed using A7r5 cells. Cells incubated with 10 μmol/L SPC for 24 hours had a significant increased release of MCP-1 (approximately threefold compared with control) (Figure 4C). The LPS increased MCP-1 release by ∼15-fold.
Once MCP-1 release in A7r5 cells was established, we tested whether
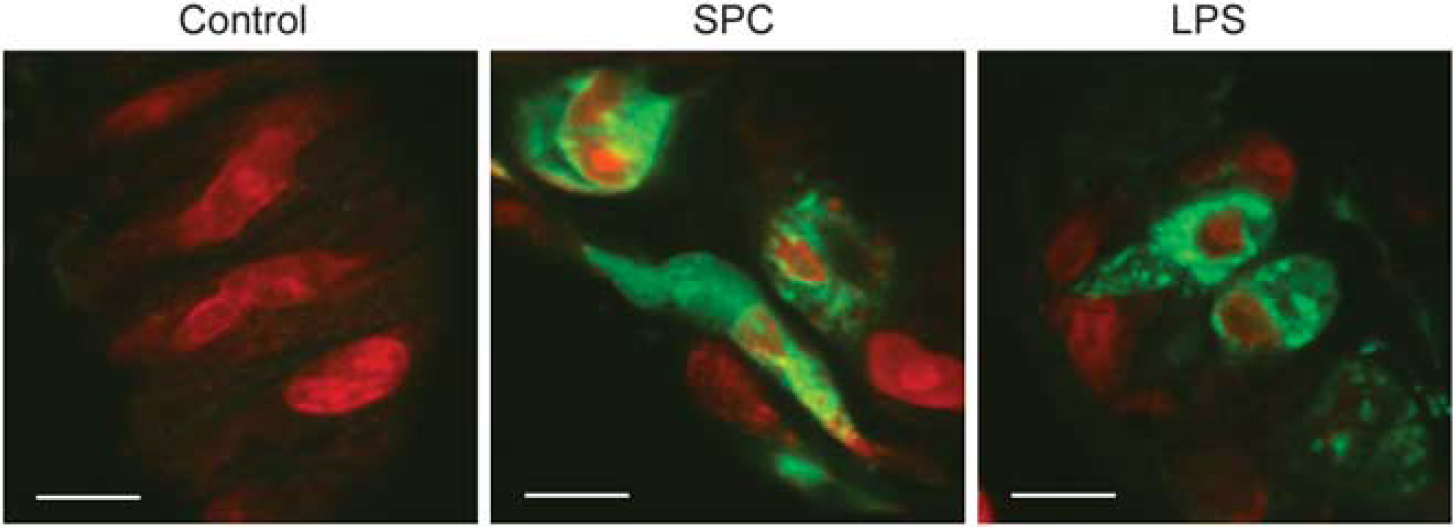
Sphingosylphosphorylcholine (SPC) induces monocyte chemoattractant protein-1 (MCP-1) production in rat cerebral artery vascular smooth muscle cells. Rat cerebral arteries were treated
Discussion
A potential function for sphingolipids in the development of vasospasm has previously been suggested through contractile effects on cerebral arteries. We (Coussin et al, 2002; Mathieson and Nixon, 2006), and others (Shirao et al, 2002), have shown that S1P and SPC can induce a constriction of cerebral arteries
The findings presented in this study are only of potential relevance to vasospasm if SPC levels are increased during SAH. A recent clinical study has shown that in patients who have developed SAH, the concentration of SPC in the cerebral spinal fluid is significantly increased ∼17-fold after 7 days (Kurokawa et al, 2009). This provides direct evidence that SPC is elevated in SAH and could therefore contribute to vasospasm. The mechanisms of this increase are not known. It is likely that increased levels of SPC are due to release from activated platelets (Liliom et al, 2001). However, in SAH it has also been shown that erythrocytes are an important component in the development of vasospasm (Macdonald et al, 1991). Although it has been reported that S1P accumulates in erythrocytes (Yatomi, 2008), SPC metabolism in erythrocytes has not been assessed. As both S1P and SPC are increased in serum, it remains a possibility. Sphingolipids are therefore probably increased in the subarachnoid space by several different pathways within the area of a cerebral aneurysm. In this study, we have used maximal, or just below maximal, concentrations of S1P and SPC (as assessed by concentration–effect curves previously in rat cerebral arteries) (Coussin et al, 2002; Mathieson and Nixon, 2006). These are in the range typically used by many other studies in different cell types. Although the concentrations of SPC measured from cerebral spinal fluid
Although inflammation may have a primary function in cerebral vasospasm, the proinflammatory mediators have not yet been clearly identified. A recent study has shown that cerebral ischemia itself can contribute to a localized inflammation (Maddahi and Edvinsson, 2010), which may have a function in SAH pathology. In this study, the ability of SPC to upregulate MCP-1 in a reasonably selective manner (at least in the rat A7r5 VSM cell line) suggests a potentially specific function for this sphingolipid in the inflammation associated with vasospasm after blood clot formation. Although we cannot state that this selectivity occurs in the rat cerebral artery, microarray experiments similar to that performed by Vikman et al (2007) to determine inflammatory gene expression could provide further evidence of this. Interestingly, MCP-1 is upregulated in the cerebral spinal fluid of patients with SAH (Gaetani et al, 1998) and may be of particular relevance to clinical vasospasm. A recent study has suggested that MCP-1 levels are directly correlated with an outcome of angiographically demonstrated vasospasm and could therefore be a biomarker for predicting onset (Kim et al, 2008). In an experimental model of SAH, MCP-1 expression was increased in cerebral artery VSM cells (Lu et al, 2009), demonstrating that the VSM cells could directly contribute to the observed increase in MCP-1 levels. Our data now provide a potential novel mediator, naturally occurring in plasma and serum, which can increase MCP-1 expression in rat cerebral arteries. Our results also delineate an intracellular mechanism for the SPC-induced increase in MCP-1 expression. We now show that in cerebral arteries, SPC, through the activation of p38MAPK, can increase the activity of two proinflammatory transcription factors, NF-κB and C/EBP. This leads to enhanced specific DNA binding. Both these transcription factors are closely associated with an increase in inflammatory cytokines and chemokines in VSM cells (Sekine et al, 2002; Dwarakanath et al, 2004). Although NF-κB and C/EBP belong to the families of transcription factors, identifying the specific isoforms of each transcription factor involved in SPC-mediated MCP-1 expression is beyond the scope of this study. The MCP-1 gene promoter region contains promoter sites for both C/EBP and NF-κB (Sekine et al, 2002; Ueda et al, 1997).
Other inflammatory proteins, in addition to MCP-1, are upregulated during SAH and likely also contribute to the inflammation associated with vasospasm. The SPC-induced effects are therefore likely to be part of a larger inflammatory response involving multiple cell types and intracellular pathways. With regard to VSM cells, it is also possible that S1P (which did not lead to increased expression of any inflammatory proteins in this study) may have other actions, which have not yet been characterized. It is also acknowledged that the main inflammatory cells in SAH are likely to be leukocytes, which will drive the increase in inflammatory mediators. However, SPC could have an important function in initiating this response by stimulating the infiltration of monocytes into the vessels wall through expression of MCP-1. Therefore, although the SPC-induced MCP-1 expression is unlikely to contribute to the majority of inflammation, it may be sufficient to have an important and primary function shortly after SAH. Until the site of action of SPC in cells is uncovered and SPC pharmacology further developed, it will be difficult to confirm the relative importance of this sphingolipid. Although SPC can act as a low-affinity agonist at S1P receptors, the divergence of SPC and S1P signaling observed in our studies (current study and Mathieson and Nixon, 2006) indicate that this is not the case in cerebral arteries. Preliminary studies with the limited S1P receptor antagonists available had no effect on SPC-induced MAPK activation (data not shown). A direct intracellular action of SPC also cannot be ruled out.
In conclusion, our study has shown that SPC can act as a proinflammatory mediator in cerebral artery VSM cells by upregulating expression of the chemokine, MCP-1. The mechanism of this upregulation occurs through the activation of proinflammatory transcription factors. This provides evidence that SPC is a novel link between blood clot formation and the initiation of inflammation. Further
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.