
Abstract
The purpose of this study was to determine whether the potassium channel, TREK-1, was neuroprotective after traumatic brain injury (TBI). Since there are no selective blockers, we used TREK-1 knockout (KO) mice for our study. Wild-type (WT) and TREK-1 KO mice were anesthetized and subjected to controlled-cortical impact injury (deformation of the brain by 1.5 mm by a 3-mm diameter rod traveling at a 3 m/s). Laser Doppler perfusion (LDP) decreased by ∼80% in the injured cortex and remained at that level in both WT and TREK-1 KO mice (
Introduction
K+ channels are a diverse group of ion channels that serve multiple functions in cells. While the driving force of most cations (i.e., Na+) favors cellular depolarization and excitation, the driving force of K+ generally opposes excitability by hyperpolarizing the cell membrane. As a result, K+ channels serve an important function in controlling the excitability of a cell. Given the above, K+ channels can serve in a neuroprotective role during pathological states such as ischemia and traumatic brain injury (TBI) (Bantel et al, 2009; Chen et al, 2009; Heurteaux et al, 1993; Janahmadi et al, 2009; Liu et al, 2005; Meuth et al, 2009; Misonou, 2010; Obrenovitch, 1997). By favoring hyperpolarization, K+ channels would oppose excitation and inhibit glutamate release from nerve terminals. Aberrant release of glutamate during pathological conditions results in ‘excitotoxicity,’ a condition characterized by excessive energy consumption and pathological increases in intracellular Ca2+ (Bayliss and Barrett, 2008a ; Obrenovitch, 1997).
In the past decade, circumstantial evidence has accumulated that TREK-1 or K2P2.1 (gene name
Using TREK-1 knockout (KO) mice, Heurteaux et al (2004) reported that 70% of the KO mice died after transient forebrain ischemia (30 minutes of bilateral common carotid artery occlusion with reduction in blood pressure), whereas only 34% of the wild-type (WT) mice died. Treatment of mice with nonselective activators of TREK-1 had no effect on mice lacking TREK-1 but increased the survival rate further in WT mice. Although this study is provocative, it is possible that systemic factors could account for the outcome without a direct neuroprotective effect of TREK-1 on brain. Thus, definitive proof for a direct neuroprotective effect for TREK-1 is still in question.
The purpose of the present study was to determine whether TREK-1 was neuroprotective after TBI. For these studies, we have derived a strain of TREK-1 KO mice (Namiranian et al, 2010). Knockout and WT mice were subjected to controlled-cortical impact injury with cortical perfusion measured using laser Doppler. Contusion volume and hippocampal cell count were conducted 15 days after the injury.
Materials and methods
All protocols were approved by the Institutional Animal Care and Use Committee at the Baylor College of Medicine. TREK-1 KO mice were generated by replacing the second exon (excluding the first 13 bp), all of the second intron, all of the third exon, and the first 23 bp of the third intron of the
The surgical preparation of the mice and the protocol for TBI was similar to that previously described for our laboratory with exceptions noted below (Hannay et al, 1999; Hlatky et al, 2003; Liu et al, 2002). Mice were anesthetized with tri-bromo-ethanol (Avertin, 500 mg/kg, intraperitoneally supplemented with 160 mg/kg as needed), transorally intubated with a PE 60 cannula, and artificially ventilated using 100% O2. The respirator was adjusted to maintain end-tidal CO2 of 40 mm Hg using a microcapnograph CI240 (Columbus Instruments, Columbus, OH, USA). The microcapnograph was calibrated against direct measurement of P
The head of each mouse was fixed in a stereotaxic frame. The hair over the skull was shaved and an incision was made along the midline suture to expose the frontal and parietal plates of the skull. Using a battery-operated drill, a rectangular window was cut in both the left and the right parietal plates exposing the dura. Care was taken to avoid damaging the brain or dura by excessive heat produced by the drill or other mechanical perturbations. For TBI, we used the controlled-cortical impact injury model where a metal rod (3 mm diameter) traveling at a velocity of 3 m/s deformed the brain by 1.5 mm for 80 milliseconds. After the injury, the mice were extubated and allowed to recover from the anesthesia before being returned to the animal holding facility. Mice received an antibiotic (Baytril 100, 5 mg/kg intraperitoneally) and an analgesic (Ketoprofen, 1 mg/kg intraperitoneally) after the injury and daily for 3 consecutive days.
Cortical perfusion was monitored over the cortices ipsilateral and contralateral to the injury for 10 minutes before the injury to establish a baseline and for 60 minutes after the injury using a Perimed PIM3 laser Doppler blood perfusion (LDP) imager. Laser Doppler perfusion was recorded in six areas of interest for each mouse. The areas of interest consisted of the site of the impact, two areas adjacent to the impact site, and corresponding regions on the contralateral hemisphere (see image of skull and regions of interest in Figure 1). Laser Doppler perfusion was measured as perfusion units and normalized to the preinjury baseline.
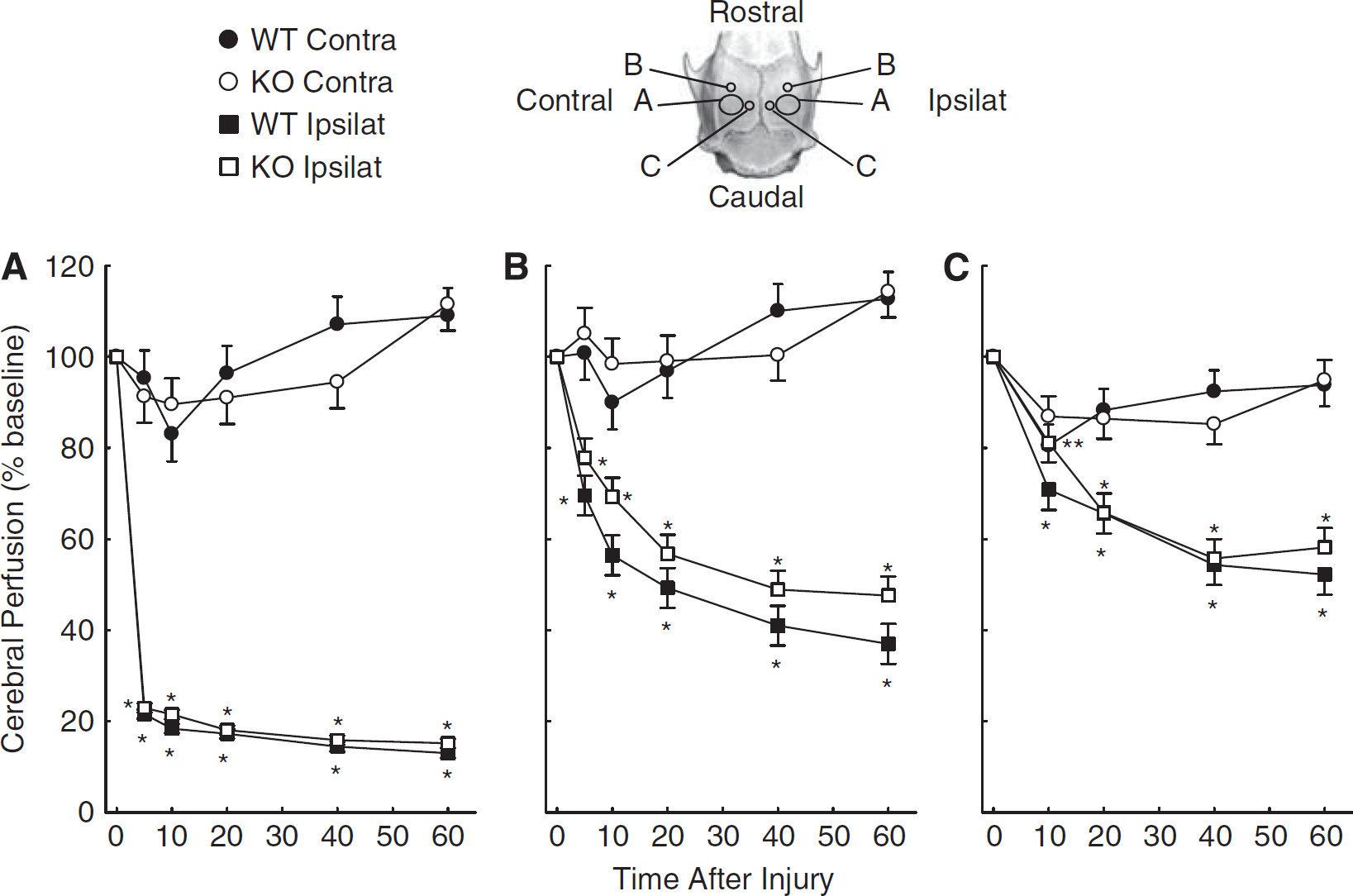
Laser Doppler perfusion (LDP) (±s.e.m.) in wild-type (WT;
Fifteen days after injury, each mouse was anesthetized with Avertin, (500 mg/kg intraperitoneally) and perfused transcardially with 0.9% saline, followed by 10% phosphate-buffered formaldehyde. The brain was removed and fixed in 4% formalin. The brain was cut coronally every 2 mm and embedded in paraffin. Representative sections (9 μm thick) were cut and stained with hematoxylin and eosin (H&E). The injury volume was calculated by measuring the cross-sectional area of injury in each H&E-stained image using image processing (Optimas Corporation, Seattle, WA, USA) and multiplying by the thickness of the tissue between the slices. Viable neurons in the CA1 and CA3 regions of the hippocampus were counted. Neurons were classified as damaged if there were signs of cytoplasmic shrinkage, basophilia, eosinophilia, or loss of nuclear detail (Hannay et al, 1999).
Experimenters were masked to the genotype during all aspects of the surgery and data collection. Data are expressed as mean±standard error of the least-squared mean (LDP) or standard error of the mean (contusion volume and cell count). Laser Doppler perfusion was analyzed using two-way repeated measures analysis of variance followed by Tukey's test where appropriate. Contusion volume and cell count were analyzed using Student's
Before the study, samples sizes were calculated using standard deviations and observed changes from published studies, mostly from our laboratory. The changes used in the sample size determination were derived from previous studies of TBI (Hannay et al, 1999; Hlatky et al, 2003; Liu et al, 2002) and focal ischemia/reperfusion in mice (Blondeau et al, 2002a ; Heurteaux et al, 2006; Meuth et al, 2009). We determined that 10 WT and 10 TREK-1 KO mice would be required using α=0.05 and β=0.8 to determine a 70% difference in contusion volume (Hannay et al, 1999; Heurteaux et al, 2006; Hlatky et al, 2003; Meuth et al, 2009), 30% differences in the densities of the CA1 and CA3 regions of the hippocampus (Blondeau et al, 2002a ; Hannay et al, 1999; Hlatky et al, 2003) and a 40% difference in LDP (Hlatky et al, 2003; Liu et al, 2002).
Relative expression of K channels were measured in brain from three pairs of adult male TREK-1 KO and WT mice to determine whether there were compensatory changes in the expression of other K channels in the TREK-1 KO mice. After flash freezing the brain in liquid nitrogen, total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA) and reversed transcribed. cDNA was quantified by real-time polymerase chain reaction using SYBR Green PCR Master Mix on an Applied Biosystems 7000 Sequence Detection System (Carlsbad, CA, USA). The efficiency of each primer set was determined to be >95%. The cycle threshold (
Results
Figure 1 shows changes in LDP in cortical areas in WT and TREK-1 KO mice. An image of the skull (directly above Figure 1B) denotes underlying cortical areas ipsilateral (Ipsilat) and contralateral (Contra) to the injury. The areas, which are denoted as A, B, and C, correspond to Figures 1A, 1B, and 1C, respectively. Site A-Ipsilat is the area directly impacted by the metal rod; sites B- and C-Ipsilat represent areas adjacent to the impact site. Figure 1A shows the area directly impacted by the metal rod and the corresponding region in the contralateral hemisphere. Laser Doppler perfusion decreased in the injured cortex by 80% (time effect
Figures 1B and 1C show LDP in areas adjacent to the impact site and corresponding areas on the contralateral cortex in WT and TREK-1 KO mice. Note that LDP decreased by ∼60% and 50% in Figures 1B and 1C, respectively, for pericontusion (ipsilateral) areas in both WT and TREK-1 KO mice (time effect
Figures 2A, 2B, and 2C show representative images of the H&E-stained coronal section obtained 15 days after injury in a TREK-1 KO mouse. Note the cavity produced by the TBI. Mean contusion volumes in WT and TREK-1 KO mice after TBI were 4.1±0.8 (
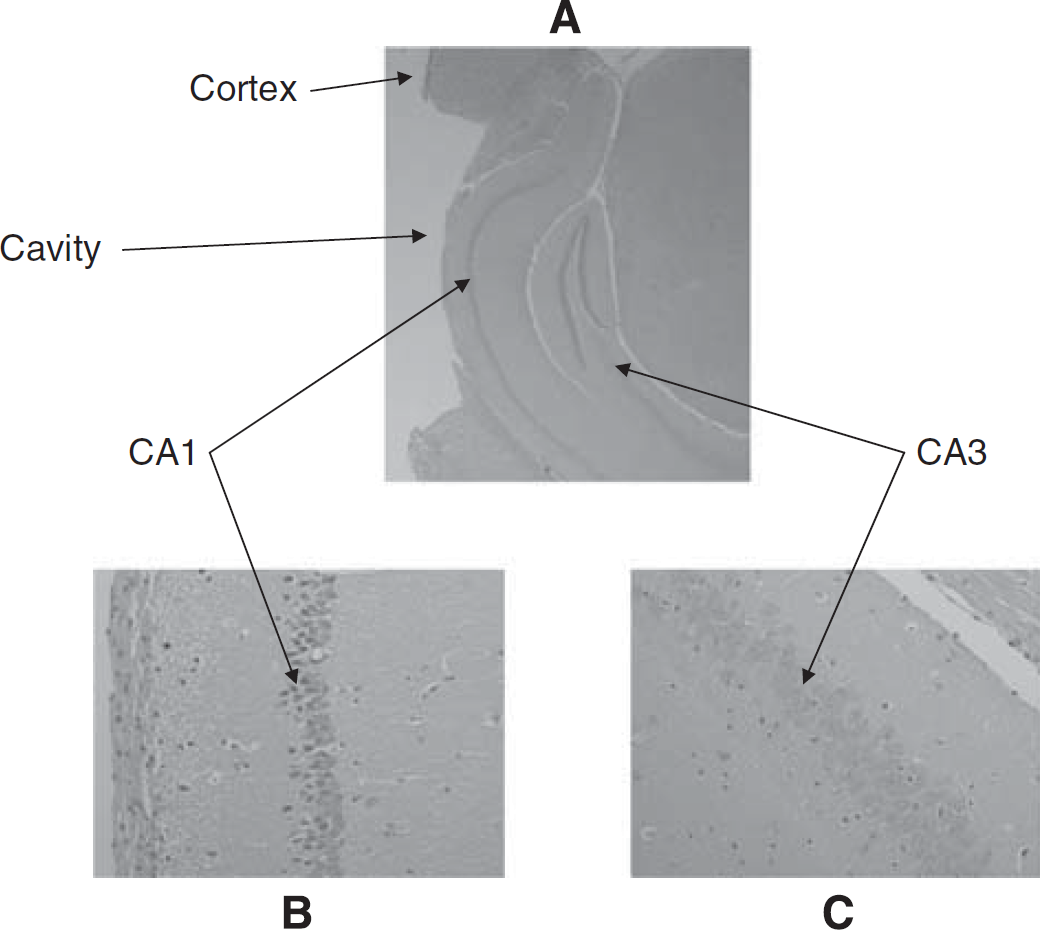
Representative coronal images of hematoxylin and eosin (H&E)-stained brain sections obtained 15 days after traumatic brain injury (TBI). Note the cavity produced by the TBI (
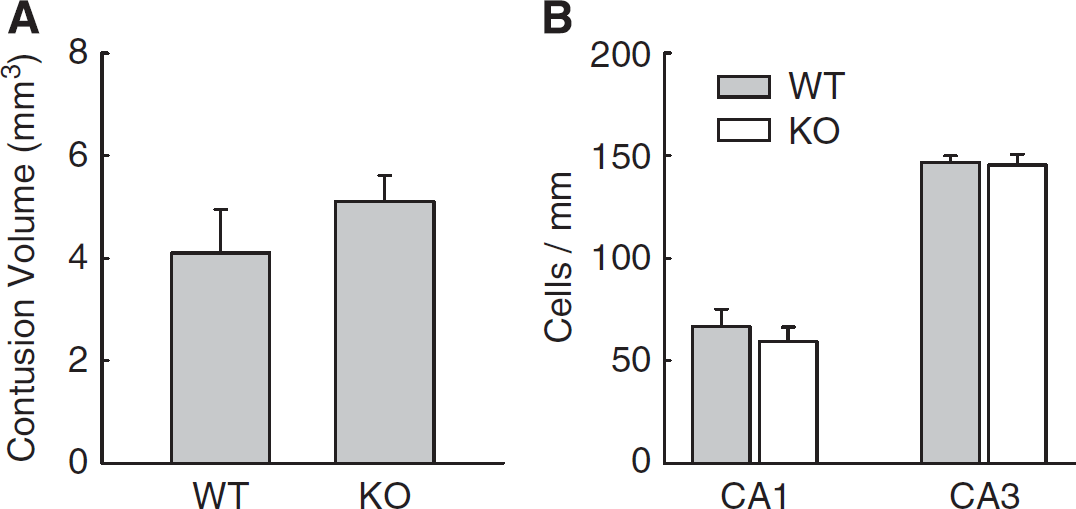
(
Figure 4 shows results from quantitative reverse transcriptase-polymerase chain reaction studies of relative expression of K2p channels, (TREK-1, TREK-2, TRAAK, TWIK-1, TWIK-2, and TASK-1) and the large conductance calcium-activated K channel (BKCa), a prominent K channel found in the brain. A ΔΔ
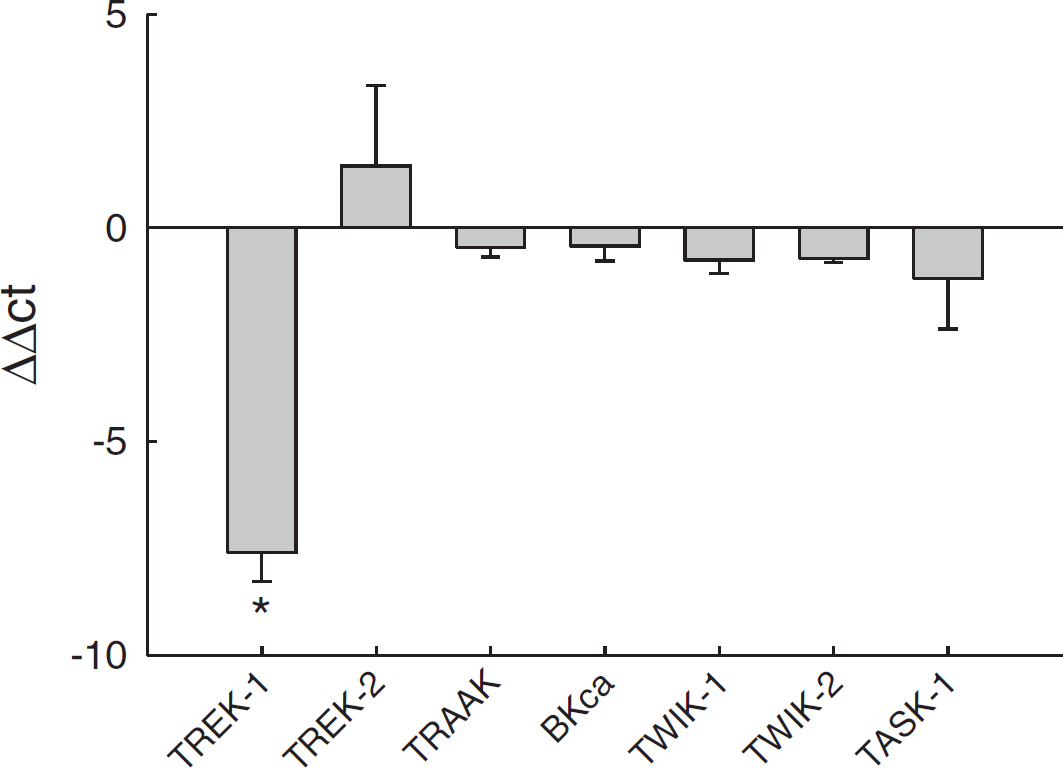
Relative expression using quantitative reverse transcriptase-polymerase chain reaction of K channels in wild-type (WT) and TREK-1 knockout (KO) mice. A ΔΔ
Discussion
In this study, we report that (1) brain injury produced by controlled-cortical impact injury was not different in mice lacking the TREK-1 K+ channel compared with WT control mice. If TREK-1 were neuroprotective after TBI, then it would be expected that TREK-1 KO mice would have a significantly greater contusion volume and fewer viable neurons in CA1 and/or CA3 neurons of the hippocampus. Since there were no differences, we conclude that TREK-1 expression does not provide protection after TBI. (2) The effects of TBI on the LDP of the contused and periimpacted cortex were not significantly different, suggesting that the presence or absence of TREK-1 does not affect changes in blood flow after TBI.
TREK-1 channels are abundantly expressed presynaptically and postsynaptically throughout the brain (Fink et al, 1996; Honore, 2007; Medhurst et al, 2001; Talley et al, 2001). The areas of expression include those areas directly affected by the TBI including hippocampus, cerebral cortex, and caudate putamen in mice (Fink et al, 1996; Medhurst et al, 2001; Talley et al, 2001). TREK-1 channels allow for the passage of K+ across the membrane at physiological ranges of membrane potentials and, thus, are considered ‘background’ or ‘leak’ channels that help to set the resting membrane potential (Honore, 2007). Since the movement of K+ through channels generally hyperpolarizes the membrane and reduces the excitability of cells, TREK-1 can potentially stabilize the membrane and oppose excitability of neurons (Bayliss and Barrett, 2008a , 2008b ; Franks and Honore, 2004; Goldstein et al, 2001; Heurteaux et al, 2004). After brain injury, a condition associated with enhanced neuronal excitability, it has been hypothesized that TREK-1 could act in a capacity to oppose and reduce the excitability. Since TREK-1 activity is resistant to hypoxia and is further activated with acidosis (Honore, 2007), conditions that accompany TBI and other forms of brain injury, TREK-1 could act to reduce energy consumption and excitation (Obrenovitch, 1997). Furthermore, it has been speculated that TREK-1 in the cerebral vasculature would assist in maintaining cerebral blood flow after the injury and further act to protect the brain (Blondeau et al, 2007). However, we did not find that the absence of TREK-1 had any effect on LDP, infarct volume, or hippocampal cell count. Given the abundance of K+ channel types including members of the K2P family, it is possible that other K+ channels take over the role of TREK-1 in the KO mice. Nevertheless, the absence of TREK-1 did not have detrimental effects on infarct volume or reductions in LDP.
In putting our results into context, we must point out that our studies do not necessarily refute previous findings. Although TBI and ischemia have similarities, they are different pathological states, which may be differently affected by the presence or absence of TREK-1. The injured brain after TBI may have sufficient ionic imbalance that K+ conductance via TREK-1 had little to no effect. Thus, the absence of TREK-1 may be detrimental after ischemia/reperfusion but not detrimental after TBI. Furthermore, we point out that previous studies involving global ischemia in TREK-1 KO mice (C57BL66J) (Heurteaux et al, 2004) were on a different background than our mice (C57BL6J and SV129). It is possible the outcome is strain dependent.
As with TREK-1, the neuroprotective properties of other K2P are controversial. Infection of organotypic hippocampal slices with virus constructs containing the coding sequence for TASK-3 showed protection from oxygen—glucose deprivation injury (Liu et al, 2005). In the same study, it was shown that neither TASK-1 nor TASK-2 afforded protection. On the other hand, mice lacking TASK-1, but not TASK-3, were reported to have larger infarct volume after transient middle cerebral artery occlusion (Ehling et al, 2010; Meuth et al, 2009). A subsequent study confirmed the protective effects of TASK-1 in permanent focal ischemia; however, the neuroprotective properties of TASK-1 could be due to a lower blood pressure in mice lacking TASK-1 (Muhammad et al, 2010). Thus, neuroprotection by some members of the K2P family, including TREK-1, remains controversial.
In summary, we find no evidence that TREK-1 is neuroprotective after TBI. Mice lacking TREK-1 showed no differences in contusion volume or hippocampal cell count when compared with WT mice. Furthermore, the presence or absence of TREK-1 is not reflected in cortical perfusion differences after TBI.
Footnotes
The authors declare no conflict of interest.