Excitotoxicity is the major cause of many neurologic disorders including stroke. Potassium currents modulate neuronal excitability and therefore influence the pathological process. A-type potassium current (IA) is one of the major voltage-dependent potassium currents, yet its roles in excitotoxic cell death are not well understood. We report that, following ischemic insults, the IA increases significantly in large aspiny (LA) neurons but not medium spiny (MS) neurons in the striatum, which correlates with the higher resistance of LA neurons to ischemia. Activation of protein kinase Cα increases IA in LA neurons after ischemia. Cultured neurons from transgenic mice lacking both Kv1.4 and Kv4.2 subunits exhibit an increased vulnerability to ischemic insults. Increase of IA by recombinant expression of Kv1.4 or Kv4.2 is sufficient in improving the survival of MS neurons against ischemic insults both in vitro and in vivo. These results, taken together, provide compelling evidence for a protective role of IA against ischemia.
Excitotoxic cell death is the major cause of many neurodegenerating disorders including stroke (Choi and Rothman, 1990; Rothman and Olney, 1986). Potassium currents have crucial roles in regulating neuronal excitability and therefore influence the ischemic outcome. Preclinical studies have shown that application of pharmacological agents activating ATP-sensitive potassium channels (Heurteaux et al, 1993) or large-conductance Ca2+-activated potassium channels (Gribkoff et al, 2001) has protective effects on neurons after global or focal ischemia. A-type potassium current (IA) and delayed rectifier potassium current (Ikd) are the major voltage-dependent potassium currents (Kv) in neurons. Studies have shown that the enhancement of Ikd is associated with apoptosis (Redman et al, 2007; Yu et al, 1997). Application of TEA (tetraethylammonium), a nonspecific Ikd channel blocker, has protective effects on neurons after cerebral ischemia (Wei et al, 2003). The roles of IA after ischemia are less clear. It has been shown that IA is progressively increased in ischemia-resistant dentate granule cells after transient forebrain ischemia (Zou et al, 2005). The underlying mechanisms and functional significance of alterations of IA after ischemia remains to be elucidated. Kv channels are modulated by protein kinases, causing changes in gating kinetics, current density, and channel trafficking (Jonas and Kaczmarek, 1996). Moreover, the expression, distribution, and activities of protein kinases have been altered after ischemia (Tanaka, 2001). The effects of protein kinases on Kv channels after ischemia deserve further studies.
One of the intriguing features following transient cerebral ischemia is the selective cell death. In the hippocampus, 90% of the CA1 pyramidal neurons die after ischemia while CA3 neurons and dentate granule cells remain intact (Pulsinelli et al, 1982). In the striatum, the medium spiny (MS) neurons are highly vulnerable to ischemia whereas the interneurons, such as large aspiny (LA) neurons, are resistant to the same insult (Chesselet et al, 1990; Pulsinelli et al, 1982). Since IA is crucial in determining the neuronal excitability and thus potentially affects the ischemic outcome, its functional phenotypes might differ between ischemia-vulnerable and ischemia-resistant neurons. The present study investigated the IA currents in ischemia-vulnerable MS neurons and ischemia-resistant LA neurons after transient forebrain ischemia, examined the potential causal relationship of IA and neuronal protection after ischemia.
Materials and methods
Male adult (100 to 180 g) and timed pregnant (E18) Wistar rats (Charles River Laboratories, Wilmington, MA, USA) were used in the present study. Kv1.4 and Kv4.2 double knockout (KO) mice were used in some experiments. Kv1.4+/- mice were generated as described (London et al, 1998). Breeding heterozygous littermates generated Kv1.4−/- and genetically matched wild-type (WT) controls. Kv4.2−/- mice were originally obtained from Dr Jeanne Nerbonne, Washington University Medical School. Kv4.2−/- mice were bred by homozygous mating. Kv1.4−/-/Kv4.2−/- double KO mice were generated by crossbreeding the two single KO strains. The Kv1.4−/-, Kv4.2−/- mice and double KO mice exhibited normal growth, development and fertility. All experimental protocols were approved by the Institutional Animal Care and Use Committee of Indiana University School of Medicine in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Transient Forebrain Ischemia and Brain Slice Preparation
Transient forebrain ischemia was induced using the four-vessel occlusion method (Pulsinelli and Brierley, 1979) with modifications (Ren et al, 1997). The animals were anesthetized with a mixture of 1% to 2% halothane, 33% O2, and 66% N2. The common carotid arteries were isolated after which a silicon-tube loop was placed loosely around each common carotid artery to allow subsequent occlusion of these vessels. The vertebral arteries were electrocauterized. A temperature probe (0.025“ OD; Physitemp, Clifton, NJ, USA) was inserted beneath the skull in the extradural space, and the brain temperature was maintained at 37°C. Glass microelectrodes filled with 2 mol/L NaCl were used to record ischemic depolarization. The microelectrode was advanced 3.0 mm below dura into the neostriatum. The recordings were performed with an amplifier (Neuroprobe, Model 1600; A-M Systems, Carlsborg, WA, USA). The duration of ischemic depolarization was determined by measuring the period from the beginning of the extracellular direct current potential reaching −20 mV to the point where the potential started to repolarize after recirculation. Transient forebrain ischemia was produced by occluding both common carotid arteries to induce ischemic depolarization for ∼22 minutes. Cerebral blood flow resumed immediately upon release of the carotid artery clasps.
Brain slices were prepared from animals before ischemia and at different interval after reperfusion using procedures similar to those previously described (Pang et al, 2002). Briefly, the animals were anesthetized with ketamine-HCl (80 mg/kg, intraperitoneally) and decapitated. The brains were quickly removed and immersed in ice-cold artificial cerebrospinal fluid, which was composed of the following (in mM): 130 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.4, 295 to 305 mOsm/L. Transverse striatal slices of 300 μm thickness were cut using a vibratome (VT 1000; Leica, Nussloch, Germany) and incubated in artificial cerebrospinal fluid for ≥ 1 hour at room temperature (∼24°C) before being transferred to the recording chamber.
Cell Cultures
For neuronal culture, the striatum was dissected from E18 embryo in an ice-cold Hank's balanced salt solution without Ca2+ and Mg2+ (Invitrogen, Carlsbad, CA, USA) and incubated with 0.125% trypsin (Invitrogen) for 30 minutes at 37°C. Tissues were then triturated with fire-polished Pasteur pipettes (Fisher Scientific, Pittsburgh, PA, USA) and centrifuge at 1,000 for 10 minutes. Cells were resuspended in Neurobasal medium containing 2% B27 (Invitrogen) and plated on 24-well plates or 12 mm glass cover slips (Fisher Scientific) coated with 0.01% (w/v) poly-l-lysine (Sigma, St Louis, MO, USA) at a density of 2 × 105 cells per well or per cover slip. Neurons were put into a standard incubator (Taibai Espec, Osaka, Japan) maintained at 37°C in 95% air, 5% CO2. In some experiments, cortical neurons were cultured from 1-day-old Kv1.4−/-/Kv4.2−/- double KO and WT pups.
HEK293 cells were cultured in Dulbecco's Modified Eagle Media supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen).
Transfection of Recombinant Kv Channels
The FuGENE6 transfection reagent (Roche Molecular Biochemicals, Indianapolis, IN, USA) was used for Kv channel transfection. Cultured neurons at 2 days in vitro (DIV) were transfected with pEGFP-N1, or a mixture of pEGFP-N1 and Kv constructs (1:1). To transfect one well of neurons in a 24-well plate, 1.5 μL FuGENE6 was diluted with 100 μL B27-free neurobasal media. After 5 minutes incubation at room temperature, 1 μg of plasmid DNA was added. The mixture was then added to the neuronal culture following 15 minutes incubation at room temperature.
Intrastriatal Delivery of Adeno-Associated Virus and Quantification of Neuronal Survival After Ischemia
To prepare AAV1-Kv4.2, the rat Kv4.2 cDMA was excised from rKv4.2-pcDNA3.1 and was used to replace green fluorescent protein (GFP) of pssAAV-GFP (Lowery et al, 2009) using a NheI/XbaI digestion to create pAAV-Kv4.2. Viral stocks of AAV-Kv4.2 (serotype 1) were prepared using the triple-transfection method and purified on a cesium chloride gradient by ultracentrifugation (Howard et al, 2008; Lowery et al, 2009).
Rats were anesthetized with 2% halothane and placed in a stereotaxic apparatus (KOPF, Tujunga, CA, USA). Intrastriatal (0.7 mm anterior to bregma, 3.0 mm lateral from the midline, and at a depth of 4.0 mm) infusion of adeno-associated virus (AAV) vectors (AAV1-GFP and AAV1-Kv4.2, 1.0 × 1011 viral genomes/each AAV, 4 μL each) were performed at the rate of 0.5 μL/min over 10 minutes using a 30-gauge beveled needle. After infusion, the needle was left in place for 15 minutes. In both control and ischemic groups, GFP only or GFP + Kv4.2 was injected into the left or right striatum of the same animal to reduce the variability of sampling and ischemic insult. Thirteen days after AAV infusion, rats were subjected to 22 minutes transient forebrain ischemia followed by 24 hours reperfusion and fixed with 4% paraformaldehyde. Brains were cut into 40 μm serial coronal sections. A total of six sections (collected every fifth section) were obtained from each animal using injection needle track as a reference (three sections roastal and three sections causal to the track). GFP-positive neurons were distinguished from glial cells by their smooth somata and extensive dendritic branches. The GFP-positive neurons in the striatum of each section were counted under fluorescence microscope (X200, Olympus BX50; Olympus Optical, Tokyo, Japan) using double-blind methods. The numbers of GFP-positive neurons in the sections of each animal were summed and grouped into four groups: GFP control, GFP ischemia, GFP + Kv4.2 control, and GFP + Kv4.2 ischemia. The neuronal survival was presented as the percentage of GFP-positive neurons in ischemic animals against that in control ones in AAV1-GFP alone and AAV1-GFP plus AAV1-Kv4.2 groups respectively.
Oxygen/Glucose Deprivation and Cell Death Assay
Cultured neurons at DIV 10 were subjected to oxygen/glucose deprivation (OGD). Cultures were placed in an anaerobic chamber (ThermoForma, Marietta, OH, USA) and washed with deoxygenated, glucose-free balance salt solution containing (in mM): 116 NaCl, 5.4 KCl, 0.8 MgSO4, 1.0 NaH2PO4, 26.2 NaHCO3, 1.8 CaCl2, bubbled with 95% N2 and 5% CO2 for 30 minutes. Then, cultures were aerated with an anaerobic gas mixture (5% CO2, 10% H2, and 85% N2) at a constant pressure of 0.15 bars to remove residual oxygen. The anaerobic chamber was humidified and maintained at 37°C. Oxygen/glucose deprivation was terminated by replacing balance salt solution with normal culture medium and retained to the culture incubator. Control cultures were performed by exposing to balance salt solution containing 20 mmol/L d-glucose and maintained in the culture incubator.
The neuronal death after OGD was assessed using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. After incubation with 100 μL MTT (5 mg/mL; Sigma) at 37°C for 5 hours, the media was removed, and 1 mL dimethyl sulfoxide (DMSO) was added into each well to dissolve crystals, of which 100 μL was added into 96-well plate to measure the absorbance at 550 nm using a plate reader (Expert 96UV, AsysHitech, Eugendorf, Austria).
The effects of transfection of recombinant Kv on neuronal viability were evaluated using methods for analysis of neuronal survival similar to those previously described (Mattson et al, 1995). Briefly, the number of GFP-positive intact neurons in the identical field (× 10 objective) was counted under fluorescent microscope at 24 hours before and 24 hours after 4 hours OGD. Most neurons that died during the time interval were absent. Remaining neurons with intact neurites and a soma with smooth round appearance were considered viable. Neurons with degenerating features (such as the vacuolated or swollen soma, beady dendrite, and fragmentation) were identified as nonviable. The value was compared between the cultures transfected with GFP plus recombinant Kv and those transfected with GFP alone.
Electrophysiological Recording
Recording electrodes were prepared from borosilicate glass (Warner Instruments, Hamden, CT, USA) using a horizontal electrode puller (P-97; Sutter Instruments, Novato, CA, USA). Electrodes were filled with an intracellular solution containing (in mmol/L): 145 KCl, 1 MgCl2, 10 EGTA, 0.2 CaCl2, 10 HEPES, 2 Na2ATP, and 2% neurobiotin (Vector Laboratories, Burlingame, CA, USA), pH 7.4, 290 to 295 mOsm/L. For recording from brain slice, oxygenated artificial cerebrospinal fluid was used as extracellular solution. For recording on cultured neurons or HEK293 cells, the extracellular solution contained (in mM): 144 NaCl, 6 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, pH 7.3, 300 to 305 mOsm/L. The flow rate of extracellular solution was adjusted to 2 to 3 mL/min. Recordings were carried out at room temperature. Cells were visualized with an infrared-differential interference contrast or fluorescent microscope (BX50WI; Olympus Optical) and a CCD camera. Whole-cell patch-clamp recordings were performed with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA, USA). After tight-seal (> 1 GΩ) formation, the electrode capacitance was compensated. Immediately after establishment of whole-cell configuration, the resting membrane potential was obtained by direct reading from the amplifier. The membrane capacitance, series resistance, and input resistance of the recorded neurons were measured by applying a 5-mV (10 milliseconds) hyperpolarizing voltage pulse from a holding potential of −60 mV The series resistance was 8 to 12 MΩ. Cells with a series resistance > 10% of the input resistance were discarded. The membrane capacitance reading was used as the value for whole-cell capacitance. During the experiment, the membrane capacitance and series resistance were periodically monitored. Cells with a series resistance change > 20% during the experiment were excluded from the analysis. Signals were filtered at 2 kHz and digitized at a sampling rate of 5 kHz using a data-acquisition program (Axograph 4.6; Axon Instruments).
At a holding potential of −60 mV, the voltage-dependent outward potassium currents were evoked by voltage steps (from −80 mV to +70 mV in 10 mV increments, 400 milliseconds) following a 300-millisecond hyperpolarizing pulse of −120 mV in the presence of tetrodotoxin (1 μM) and CdCl2 (300 μM) to block voltage-activated Na+ and Ca2+ currents, as well as Ca2+-activated potassium currents. Taking advantage of the rapid inactivation at depolarized membrane potential, IA was isolated by subtracting the currents evoked after depolarized prepulses (+ 10 mV, 100 mV) from those evoked without depolarized prepulses. In some experiments, 4-AP (4-aminopyridine; 10 mmol/L) and TEA (20 mmol/L) were applied to examine the pharmacological characteristics of IA.
The current density of IA for each neuron was obtained by dividing the membrane capacitance from current amplitude. The current amplitude of IA was measured at the peak of each current (∼4 milliseconds after the onset of the command pulses). The steady-state activation curves were established similarly to those previously reported (Deng et al, 2004). Briefly, the conductance (G) was calculated using the following equation: G = I/(Vm–Vk), where I was the current amplitude, Vm was the command potential, and Vk was the reversal potential of potassium (Vk = −98 mV). The conductance was then normalized with respect to the maximum value and plotted as a function of the membrane potential during the test pulse. The resulting activation curves were fitted with a normalized Boltzmann distribution: G/Gmax = 1/[1 + exp(Vm–V1/2)/Vc], where Gmax was the maximum conductance at + 70 mV, V1/2 was the membrane voltage at which the current amplitude was half-maximum, and Vc was the slope factor at V1/2. The steady-state inactivation properties of IA were determined by measuring the current availability with a testing pulse of + 70 mV following 2 seconds prepulse between −120 mV and 0 mV The plot of mean normalized peak currents as a function of prepulse voltage was fitted with a normalized Boltzmann distribution: I/Imax = 1/[1 + exp(V1/2–Vm)/Vc], where Imax was the maximum current at + 70 mV To study the removal of inactivation, IA was inactivated by depolarizing to +70 mV for 100 milliseconds from a holding potential of −60 mV, and then inactivation was removed by hyperpolarizing to −120 mV for different periods of time before a test pulse of + 70 mV The plot of normalized peak currents as a function of hyperpolarization duration was fitted with a biexponential function.
In current-clamp recording, fast I-clamp mode was used. The first spike latency was measured from the onset of the current injection to the peak of the action potential evoked by a threshold current (100 milliseconds).
Drug Application
Phorbol-12,13-dibutyrate, chelerythrine, GF109203X, and 4α-phorbol were purchased from Sigma. Drugs were prepared as concentrated stocks and stored at −20°C. Working solutions were prepared immediately before use.
Immunohistochemistry
The following primary antibodies were used: mouse anti-protein kinase Cα (anti-PKCα) (1:50; Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse anti-PKCγ (1:500; Sigma), and rabbit anti-ChAT (anti-choline acetyltransferase) (1:250; Chemicon, Temecula, CA, USA). Animals were anesthetized and perfused through the ascending aorta with a solution of phosphate-buffered saline (PBS) (0.01 mol/L, pH 7.4) for ∼5 minutes, followed by 4% paraformaldehyde in PBS for 20 to 30 minutes. Brains were removed and postfixed in 4% paraformaldehyde at 4°C overnight. Coronal sections containing striatum were cut (50 μm) with a vibratome and collected in PBS. Sections were blocked with normal serum (10% in PBS containing 0.5% Triton X-100) for 2 hours at room temperature and incubated in a solution contaisning primary antibody of PKC isoform for 18 hours at 4°C. Then, sections were incubated in a solution containing fluorescein-conjugated secondary antibody (1:100; Vector Laboratories) for 2 hours at room temperature. After repeated washes in PBS, the stained sections were incubated with rabbit anti-ChAT at 4°C overnight, followed by rhodamine-conjugated secondary antibody (1:100; Vector Laboratories) for 4 hours at room temperature. The labeled neurons were examined with a microscope (BX50; Olympus Optical) equipped with reflected light fluorescence attachment (BX-FLA; Olympus Optical). Fluorescent images were acquired with a digital camera coupled to a software (DP70-BSW; Olympus Optical) at × 40 magnification. The settings were kept constant throughout all experiments.
Western Blotting
Brain tissues were lysed with ice-cold RIPA buffer (50 mmol/L Tris, pH 7.4, 150 mmol/L NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate; Boston BioProducts, Worcester, MA, USA) supplemented with a protease inhibitor cocktail (Roche) and incubated an additional 30 minutes on ice. After brief sonication on ice, cell lysates were centrifuged at 12,000 g for 20 minutes at 4°C to pellet nuclei and debris, and the resulting supernatants were collected for analysis. Protein concentration was determined by bicinchoninic acid (BCA) protein assay (Bio-Rad, Hercules, CA, USA). Protein samples were boiled in 2 × sodium dodecyl sulfate gel-loading buffer (Invitrogen) before sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins (20 μg) were separated on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and transferred to nitrocellulose membranes (Millipore, Bedford, MA, USA). The membranes were rinsed with distilled water, blocked with 1% bovine serum albumin (Sigma) in Tris-buffer saline (TBS)-0.1% Tween 20 (TBS with 0.1% Tween 20) for 1 hour, and then incubated with primary antibodies overnight in blocking buffer at 4°C. We used rabbit polyclonal anti-Kv4.2 or Kv1.4 (1:1,000; Chemicon) or mouse monoclonal anti-β-actin antibodies (1:20,000; Sigma). The membranes were washed with TBST, and incubated at room temperature for 1 hour with horseradish peroxidase-conjugated anti-rabbit (1:5,000; Chemicon) or anti-mouse secondary antibodies (1:20,000; Chemicon). Bands were detected by the enhanced chemiluminescence (ECL; Amersham, Piscataway, NJ, USA) and visualized by exposing the membrane to X-ray films (Fuji, Tokyo, Japan).
Data Analysis
The values were presented as mean ± s.e.m. Unless stated otherwise, analysis of variance followed by post hoc Scheffe's test was used for statistical analysis (StatView 5.0; Abacus Concepts, Berkeley, CA, USA). Changes were considered significant if P < 0.05.
Results
Increase of A-Type Potassium Current in Large Aspiny Neurons After Transient Forebrain Ischemia
Whole-cell patch-clamp recording was performed on visually identified MS and LA neurons in brain slices prepared before ischemia and at different intervals after reperfusion. Because MS neurons start to degenerate at 6 to 8 hours after transient forebrain ischemia and most of them die by 24 hours following reperfusion (Pulsinelli et al, 1982), we recorded IA from MS neurons at 4 hours and 8 hours after ischemia. The changes of IA in LA neurons were examined at 6 and 24 hours after ischemia, since these neurons are resistant to ischemic insults. Large aspiny neurons (> 20 μm in diameter) were distinguishable from MS neurons (10 to 20 μm) based on their somatic size and electrophysiological properties (Deng et al, 2007; Kawaguchi et al, 1995).
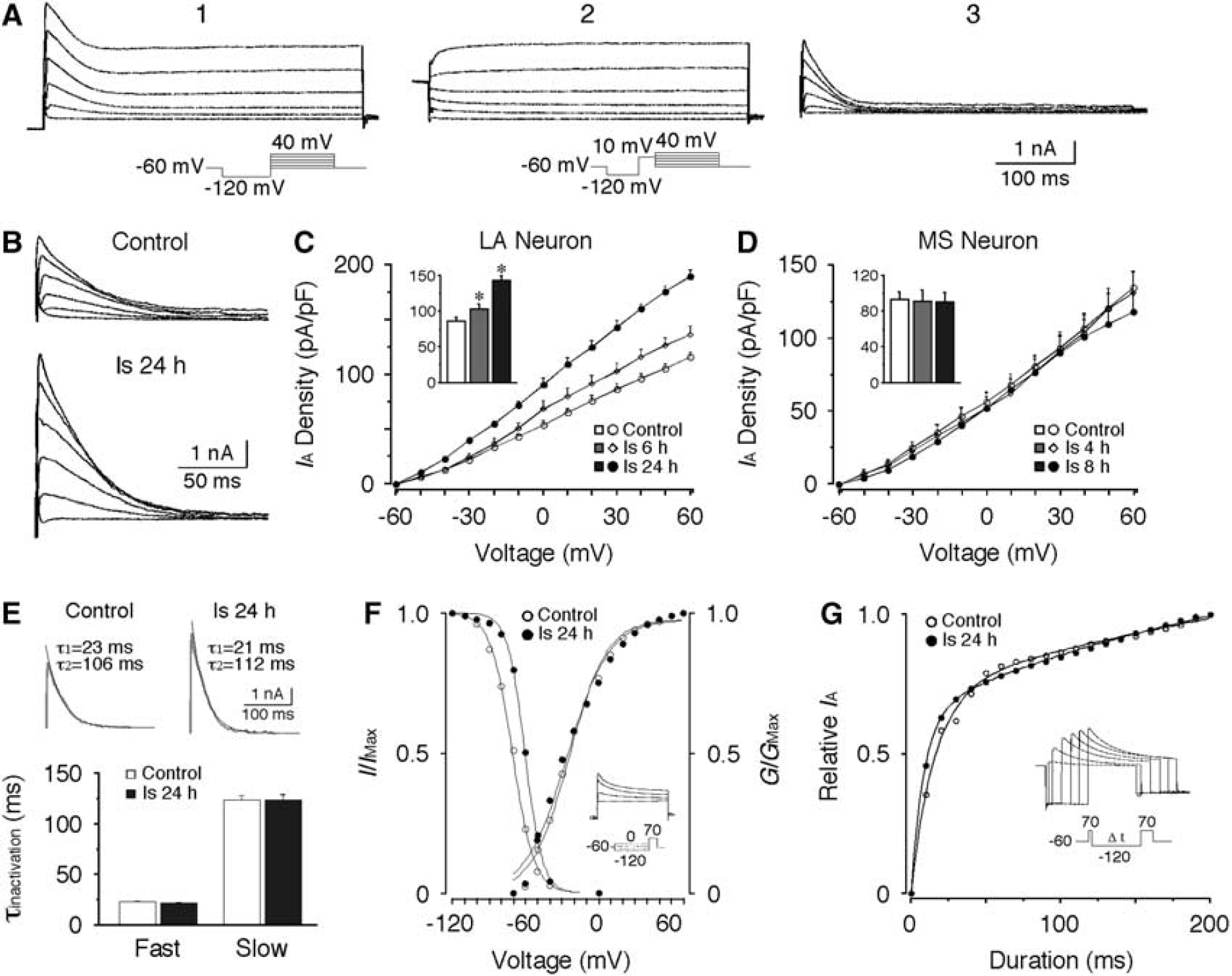
IA was isolated using protocols as shown in Figure 1A. In LA neurons, the current density of IA was significantly increased after ischemia (Figures 1B and 1C). At a depolarizing step of +30mV, the current density of IA was 86.5 ± 4.7 pA/pF (n = 18) in control neurons and dramatically increased to 142.6 ± 6.2 pA/pF at 24 hours after ischemia (n = 14, P < 0.01). However, no significant change in IA was detected in MS neurons after ischemia (Figure 1D). When evoked at + 30 mV, the current density of IA in MS neurons was 93.3 ± 8.2 pA/pF (n = 15) in control, and 90.1 ± 10.7 pA/pF (n = 7) at 8 hours after ischemia. These data indicate that the current densities of IA in LA neurons and MS neurons are similar in control animals, but are differentially altered after ischemia.
Increase of A-type potassium current (IA) in large aspiny (LA) neurons after transient forebrain ischemia. (A) Representative recordings showing the isolation of IA. (A1) Currents evoked by a series of depolarizing steps from −60 mV to +40 mV following a hyperpolarizing step of −120 mV. (A2) Currents evoked by the same voltage steps with a prepulse of +10 mV to inactivate IA. (A3) The IA was isolated by subtracting the currents from A1 by that from A2. (B) Representative traces of IA recorded from control and ischemic LA neurons. (C) Pooled data showing the postischemic increase of the current density of IA in LA neurons. (D) No significant change in the current density of IA was detected in medium spiny (MS) neurons after ischemia. The inserts in (C, D) show the current densities of IA evoked at +30 mV. (E) Decay time constants of IA in LA neurons. (Upper panel) Representative fittings of the decay phase of IA (evoked at +30 mV) with a biexponential function. (Lower panel) Both of the fast and slow decay time constant of IA remained unchanged after ischemia. (F) Voltage dependence of the activation and inactivation of IA in LA neurons. No significant change in activation curves was observed. However, the inactivation curve significantly shifted in a depolarizing direction after ischemia. The insert shows a representative recording of the steady-state inactivation of IA and the protocol. (G) Removal of inactivation of IA in LA neurons. The removal of inactivation curves were fitted with a biexponential function. While the slow component showed no significant change, the fast time constant was decreased after ischemia. The insert shows a representative recording and the protocol. *P < 0.01.
To further characterize the changes of IA in LA neurons, the decay kinetics and the voltage dependence of activation and inactivation of IA were investigated. The decay phase of IA could be well fitted by a biexponential function, yielding fast and slow decay time constants (c). As shown in Figure 1E, neither τfast nor τslow displayed any significant change after ischemia. The voltage-dependent activation of IA in control LA neurons was fitted by a single Boltzmann distribution with a V1/2 of-24.5 ± 2.3 mV and a Vc of 14.7 ± 0.9 mV (n = 11). No significant changes in V1/2 and Vc were detected 24 hours after ischemia (V1/2: −23.1 ± 2.0 mV; Vc: 15.6 ± 0.6 mV; n = 11). For steady-state inactivation, the V1/2 of the inactivation curve shifted significantly from −71.5 ± 3.4 mV (n = 11) of control to −57.5 ± 1.1 mV (n = 10, P < 0.01) at 24 hours after ischemia without obvious change in the Vc (Figure 1F). The removal of inactivation of IA was also examined. In control LA neurons, the fast and slow time constants of the removal of inactivation were 18.5 ± 1.9 milliseconds and 458.3 ± 92.3 milliseconds (n = 7), respectively. Although the fast time constant was decreased 24 hours after ischemia (9.8 ± 1.4 milliseconds, n = 8, P < 0.01), the slow time constant remained at similar levels (444.3 ± 68.7 milliseconds, n = 8; Figure 1G). Hence, most of the biophysical features of IA remain unchanged after ischemia.
Up-regulation of A-Type Potassium Current by Protein Kinase C in Large Aspiny Neurons After Ischemia
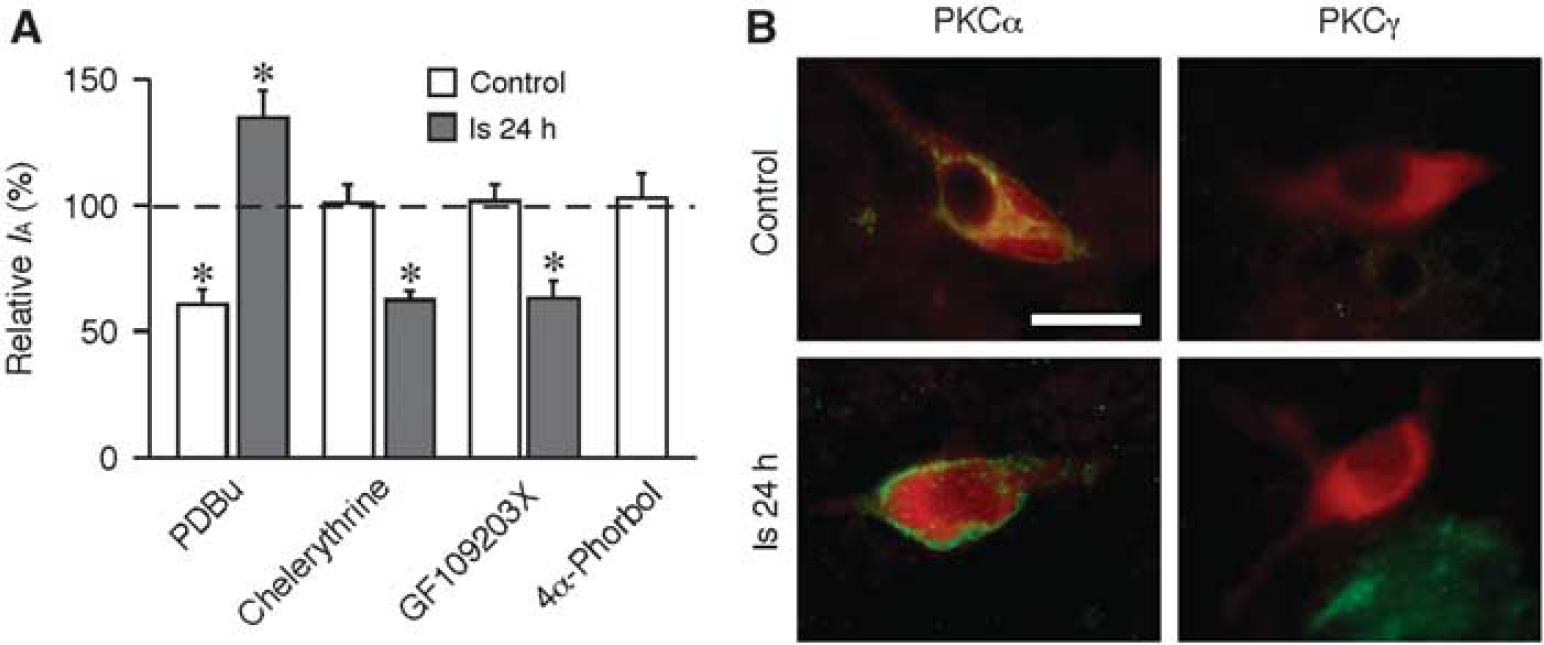
Previous studies have demonstrated that PKC modulates IA in neurons (Yuan et al, 2002). To reveal the PKC effects on IA, the IA in LA neurons was compared before and after application of PKC activators or inhibitors into the bath solutions of brain slices. In control LA neurons, application of phorbol-12,13-dibutyrate (1 μmol/L), an activator of PKC, reduced the amplitude of IA by 41.1% ± 5.9% (n = 9, P < 0.01). However, inhibition of PKC with chelerythrine (10 μmol/L, n = 7) or GF109203X (1 μmol/L, n = 6) had no effects on IA (Figure 2A), which indicates that there is no tonic PKC regulation of IA in control neurons. In contrast, application of phorbol-12,13-dibutyrate on LA neurons 24 hours after ischemia significantly enhanced IA by 33.6% ± 3.7% (n = 6, P < 0.01). Furthermore, application of chelerythrine (n = 7) or GF109203X (n = 5) dramatically decreased IA by 40.1% ± 3.2% and 38.0% ± 6.7%, respectively (P < 0.01; Figure 2A). Parallel studies on effects of cAMP-dependent protein kinase (PKA) and protein tyrosine kinase did not yield similar results (data not shown). These results indicate that PKC has a condition-specific up-regulation of IA after ischemia. The up-regulation of IA by PKC might have a role in the postischemic increase of IA in LA neurons.
Protein kinase C (PKC) is involved in the up-regulation of A-type potassium current (IA) after ischemia. (A) Application of PKC activator (phorbol-12,13-dibutyrate (PDBu)) inhibited the amplitude of IA in control neurons but enhanced IA after ischemia. PKC inhibitors, either chelerythrine or GF109203X, had no effect on IA in control neurons but decreased IA after ischemia. The inactive form of phorbol ester (4α-phorbol) had no effect on IA. The currents were evoked at +30 mV. The amplitudes of currents recorded in the presence of PKC activator or inhibitor were normalized to those before application of these drugs, respectively. (B) Expression of PKCα in large aspiny (LA) neurons. LA neurons were identified using antibodies against choline acetyltransferase (ChAT) (red). PKCα immunoreactivity (green) was detected in both control and postischemic LA neurons. No immunoreactivity of PKCγ (green) was detected in LA neurons before and after ischemia. Scale bar: 20 μm. *P < 0.01.
To identify the PKC isoforms involved in the up-regulation of IA, the expression of PKC isoforms in LA neurons was examined. Previous studies have shown that PKCα and PKCγ are expressed in striatal neurons (Yoshihara et al, 1991). Immunostaining showed that the PKCα mainly located in the perikarya of the control LA neurons (Deng et al, 2009). However, at 24 hours after ischemia, most of the PKCα was found in the plasma membrane, indicating the activation of PKC (Figure 2B). Immunoreactivity for PKCγ was not detectable in LA neurons (Figure 2B). These results suggest that activation of PKCα might be associated with the postischemic up-regulation of IA in LA neurons.
Protection of Neurons Against Ischemic Insults by Enhancement of A-Type Potassium Current
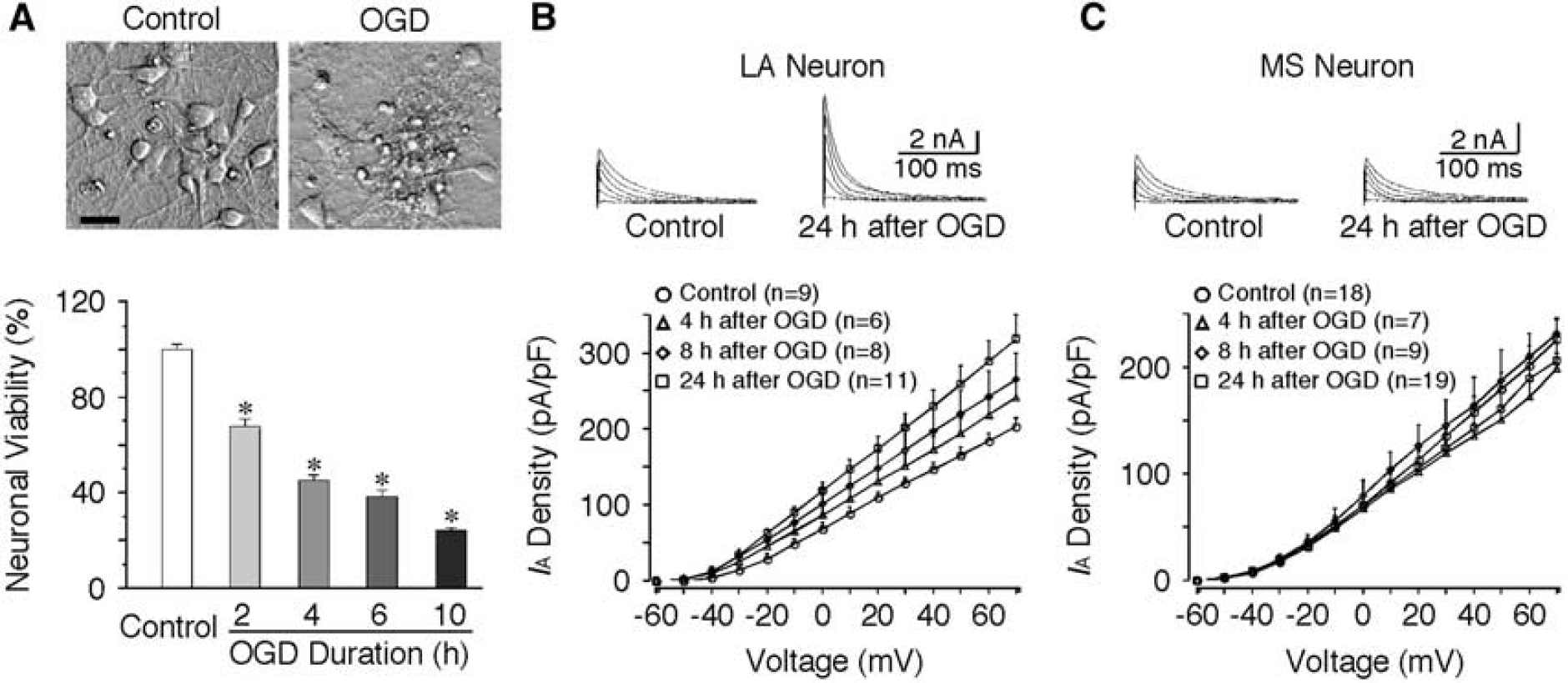
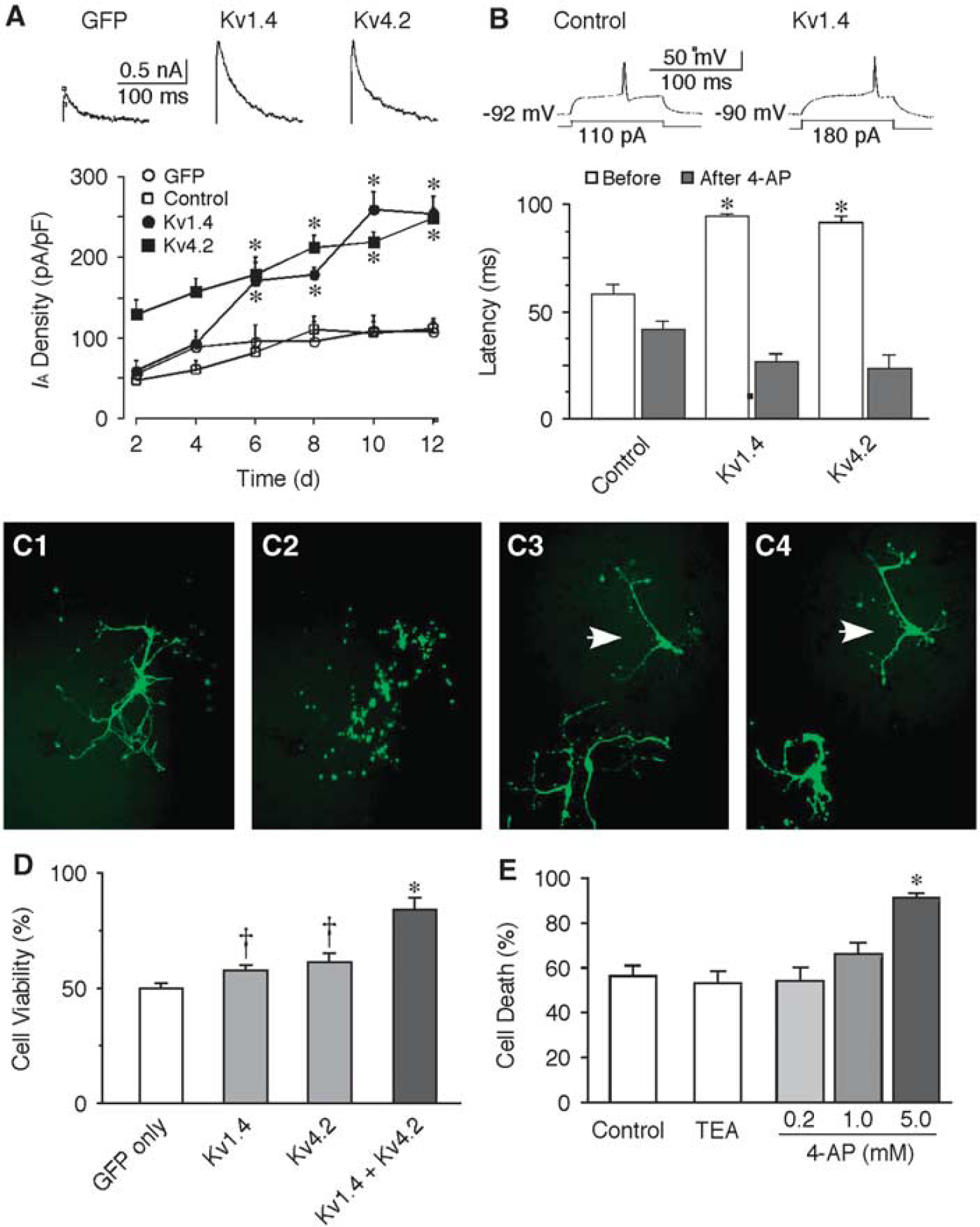
If an increase of IA is neuroprotective against ischemic challenge, overexpression of IA may improve the survival of MS neurons. To test this hypothesis, we used primary neuronal cultures from striatum because they compose of >90% of MS neurons. Ischemia in vitro was mimicked by OGD for different durations in cultures at 10 DIV. Neuronal viability was evaluated at 24 hours after OGD using MTT assay. The extent of cell death was more severe with the increase of OGD duration (Figure 3A). Oxygen/glucose deprivation for 4 hours induced ∼50% of cell death (45.0% ± 1.9%, n = 6, P < 0.01), and was selected as the experimental paradigm in the following studies. To verify if the alteration of IA in cultures after OGD is consistent with the in vivo results, the IA in cultured neurons was compared before and after OGD. The IA was increased in cultured LA neurons but remained unchanged in cultured MS neurons following OGD. The current density of IA in LA neurons was 128.2 ± 8.1 pA/pF (n = 9, at +30 mV) in control and 201.7 ± 18.5 pA/pF (n = 11, P < 0.01) at 24 hours following 4 hours OGD (Figure 3B). However, no significant change of IA was observed in MS neurons after OGD (135.8 ± 11.6 pA/pF of control versus 124.9 ± 9.7 pA/pF 24 hours after OGD; Figure 3C). These results are consistent with the above in vivo data. Kv1.4 and Kv4.2 encode A-type potassium channels and they express abundantly in the brain. Recombinant expression of either Kv1.4 or Kv4.2 allowed an increase of IA by more than twofold in cultured neurons (Figure 4A). Recombinant Kv has no significant effect on resting membrane potentials (control: −63.6 ± 1.3 mV, n = 13; Kv1.4: −64.6 ± 1.6 mV, n = 5; Kv4.2: −60.8 ± 1.9 mV, n = 6). To compare the membrane excitability of control and transfected neurons, the response to a threshold depolarizing current was examined with the holding potential maintained at around −90 mV. Compared with that of control neurons (118.2 ± 12.9 pA, n = 13), the threshold current for evoking first spike was significantly increased in neurons transfected with Kv1.4 (185.6 ± 22.1 pA, n = 5, P < 0.01) or Kv4.2 (185.3 ± 17.3 pA, n = 6, P < 0.01). Consistently, the input resistance was dramatically decreased in neurons transfected with Kv channels (control: 459.8 ± 62.4 MΩ, n = 13; Kv1.4: 275.4 ± 30.3 MΩ, n = 5; Kv4.2: 261.4 ± 27.9 MΩ, n = 6; P < 0.05). In addition, the half-width of action potential was significantly reduced from 1.12 ± 0.05 milliseconds of control (n = 13) to 0.73 ± 0.04 milliseconds of Kv1.4 (n = 5, P < 0.01) and 0.69 ± 0.04 milliseconds Kv4.2 (n = 6, P < 0.01). Moreover, the increase of IA by either Kv1.4 or Kv4.2 resulted in nearly doubling of first firing latency (Figure 4B). These results indicate that the membrane excitability in neurons infected with recombinant Kv is significantly decreased.
Differential changes of A-type potassium current (IA) in cultured striatal neurons after oxygen/glucose deprivation (OGD). The large aspiny (LA) neurons in culture were identified based on their large cell bodies (> 30 μm) as compared with the medium spiny (MS) neurons (10 to 20 μm). (A) Effects of OGD on neuronal viability. (Upper panel) Representative photomicrographs showing the neuronal damage 24 hours after 4 hours OGD. Scale bar: 20 μm. (Lower panel) Pooled data showing the neuronal viability at 24 hours after OGD of different durations. The neuronal damage was significantly increased with increasing duration of OGD (n = 12 per group). The neuronal viability was evaluated using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. (B) Increase of IA in cultured LA neurons after OGD. (Upper panel) Representative traces of IA recorded before and 24 hours after 4 hours OGD. (Lower panel) Pooled data showing the significant increase of current density of IA after OGD. (C) The current density of IA in cultured MS neurons remained about the same after OGD. (Upper panel) Representative traces of IA before and 24 hours after 4 hours OGD. (Lower panel) Pooled data showing that no significant change in the current density of IA was observed after OGD. *P < 0.01.
Up-regulation of A-type potassium current (IA) protects striatal neurons from ischemic insults. (A) The IA is enhanced in neurons transfected with Kv1.4 or Kv4.2 channels. (Upper panel) Representative traces of IA evoked at +30 mV in striatal neurons transfected with green fluorescent protein (GFP), Kv1.4 or Kv4.2 channel, respectively. (Lower panel) Temporal changes of IA in striatal neurons after transfection. At 8 days after transfection (10 days in vitro (DIV)), the current density of IA in neurons transfected with GFP alone was 81.8 ± 16.2 pA/pF (n = 6), and was 178.8 ± 8.1 pA/pF (n = 8, P < 0.01) and 212.9 ± 4.6 pA/pF (n = 7, P < 0.01) in neurons transfected with Kv1.4 or Kv4.2, respectively. (B) Transfection with Kv1.4 or Kv4.2 channels decreased the neuronal excitability. Neurons with recombinant Kv had a significantly longer latency to the first spike than the control ones (control: 56.5 ± 8.3 milliseconds, n = 13; Kv1.4: 94.8 ± 1.3 milliseconds, n = 5, P < 0.01; Kv4.2: 91.7 ± 2.9 milliseconds, n = 6, P < 0.01). These changes were completely reversed by application of 10 mmol/L 4-aminopyridine (4-AP) (n = 4 in each group, P < 0.01). (C) Representative photomicrographs showing neurons transfected with GFP alone before (C1) and after oxygen/glucose deprivation (OGD) (C2), and neurons transfected with Kv1.4 plus Kv4.2 channels before (C3) and after OGD (C4) in an identical field. Neurons transfected GFP alone died after OGD, resulting in fragments in the same location (C2), whereas most of the neurons transfected with GFP plus Kv1.4 and Kv4.2 remained intact (arrows, C4). (D) In comparison to neurons transfected with GFP alone, the cell viability after 4 hours OGD was significantly increased in neurons transfected with Kv1.4 or Kv4.2 channels. Cotransfection of Kv1.4 and Kv4.2 channels further increased the neuronal survival after OGD. (E) Application of IA channel blocker 4-AP, but not other potassium channel blocker tetraethylammonium (TEA) (5 mmol/L), into the medium increased neuronal death after 4 hours OGD in a dose-dependent manner. †P < 0.05, *P < 0.01. Kv, voltage-dependent potassium current.
The neuronal viability after OGD was compared between the control cultures transfected with GFP alone and the cultures transfected with recombinant Kv (plus GFP) by counting the number of intact eurons in the identical fields at 24 hours before and 24 hours after 4 hours OGD (Figure 4C). Compared with the control cultures, the percentage of cell survival following OGD was significantly increased from 50% ± 2.16% (n = 59 wells) to 57.6% ± 2.4% (n = 59 wells, P < 0.05) or 61.4% ± 3.8% (n = 31 wells, P < 0.05) when transfected with Kv1.4 or Kv4.2, respectively (Figure 4D). Cotransfection of Kv1.4 and Kv4.2 further increased the viability to 84.1% ± 5.3% (n = 33 wells, P < 0.01) after OGD. In contrast, application of 4-AP at concentrations that block the IA during OGD led to an increase of neuronal death in a dose-dependent manner (Figure 4E). In neurons treated with 5 mmol/L 4-AP, 4 hours OGD induced 91.6% ± 1.5% of cell death, which was significantly greater than that of control neurons (56.4% ± 4.3%, n = 9, P < 0.01). Moreover, application of 5 mmol/L TEA had no significant effect on the cell death after 4 hours OGD (53.3% ± 3.9%, n = 8; Figure 4E), indicating that 4-AP-sensitive IA, rather than other potassium currents, has neuroprotective effects. These data demonstrate that the increase of IA in MS neurons by recombinant expression of Kv1.4 or Kv4.2 channels conferred an increased resistance against ischemic insult.
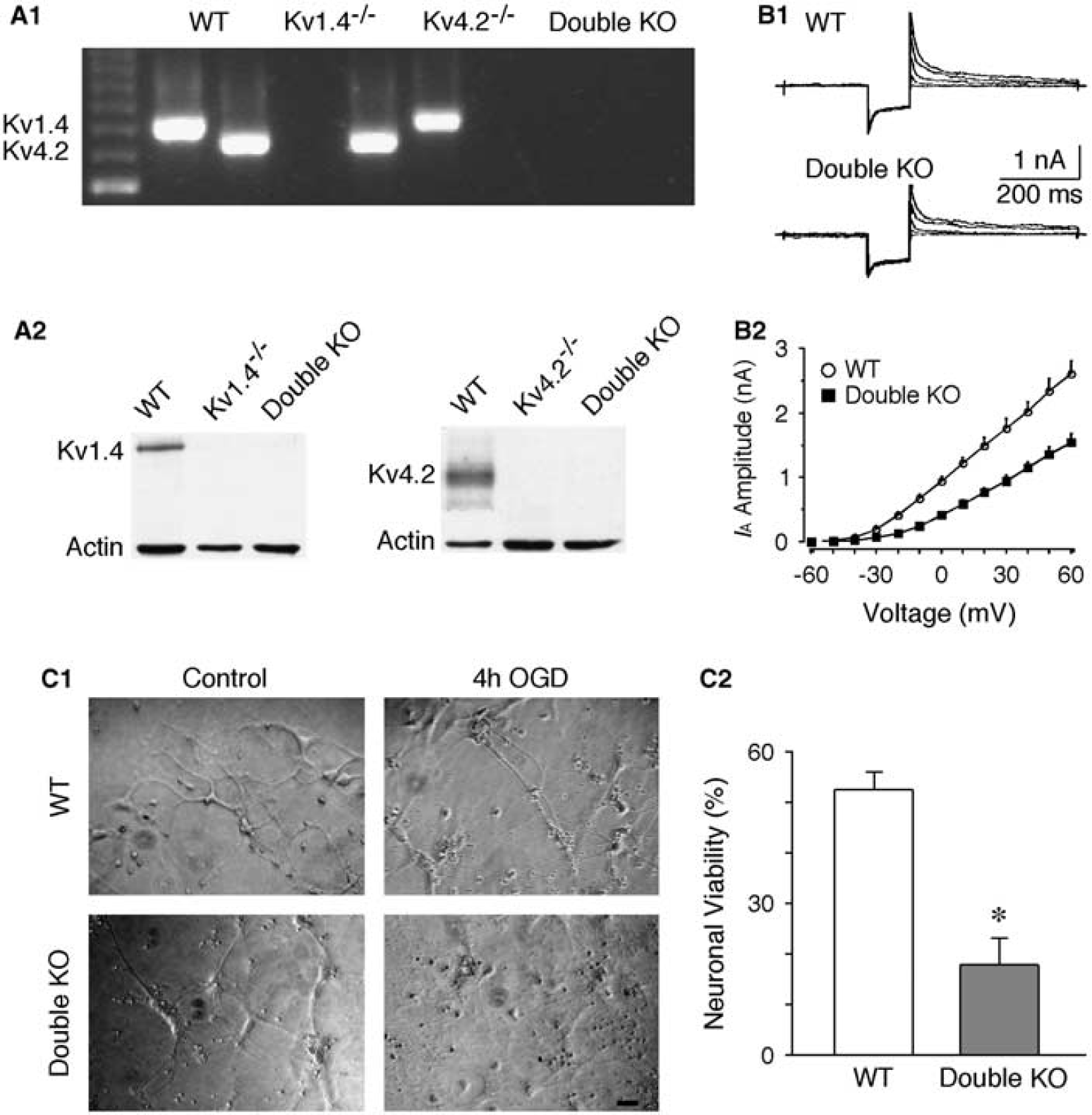
Increased Neuronal Damage in Neurons Lacking Kv1.4 and Kv4.2
We further tested the roles of IA in ischemic neuronal death in another ischemia-vulnerable brain region, cerebral cortex, using double KO mice lacking Kv1.4 and Kv4.2. Cortical neurons express prominent IA mediated largely by Kv1.4, Kv4.2, and Kv4.3 (Norris and Nerbonne, 2010). Recordings from cultured cortical neuron revealed that the amplitude of IA in neurons obtained from mice lacking Kv1.4 and Kv4.2 subunits was significantly reduced, compared with that of WT mice. When evoked at a voltage of +30mV, the IA amplitudes were 1.77 ± 0.14 nA (n = 7) and 0.95 ± 0.08 nA (n = 8) in neurons from WT and double KO mice, respectively (P < 0.01; Figures 5A and 5B). Moreover, neurons lacking Kv1.4 and Kv4.2 exhibited a much higher sensitivity to ischemic insults. The cell survival at 24 hours after OGD (4 hours) was 52.6% ± 3.3% in WT neurons, and 17.9% ± 5.3% in double KO neurons (n = 10 in each group, P < 0.01; Figure 5C). These findings indicate that IA has an important role in neuronal protection after cerebral ischemia, and that the neuroprotective roles of IA is not only restricted in the striatum.
Double knockout (KO) of Kv1.4 and Kv4.2 increases neuronal vulnerability to oxygen/glucose deprivation (OGD). (A) Genotyping of KO mice. PCR genotyping of genomic mouse tail DNA samples was shown in (A1). The primers amplify a 783-bp fragment from the Kv1.4 locus and a 712-bp fragment from the Kv4.2 locus. (A2) Western blotting confirmed the absence of Kv1.4 protein (Mr ∼ 95 kDa) in the brain extracts of Kv1.4−/- and double KO mice, and the absence of Kv4.2 protein (Mr ∼ 75 kDa) in the brain extracts of Kv4.2−/- and double KO mice. β-Actin is used as a protein loading control. (B) Decreased A-type potassium current (IA) in cortical neurons lacking both Kv1.4 and Kv4.2. Traces were evoked with voltages from −30 mV to 50 mV (in 20 mV increments). (B1) Representative recording traces. (B2) Grouped data of IA amplitude. (C) Cortical neurons cultured from double KO mice exhibited an increased cell death 24 hours after OGD (4 hours). (C1) Photomicrographs showing the culture neurons from wild-type (WT) and double KO mice in control and 4 hours OGD-treated group. (C2) Histogram showing the neuronal viability significantly reduced in culture neurons from double KO mice after OGD. Scale bar: 50 μm. *P < 0.01. Kv, voltage-dependent potassium current.
Neuroprotective Roles of A-Type Potassium Current Against Ischemia In Vivo
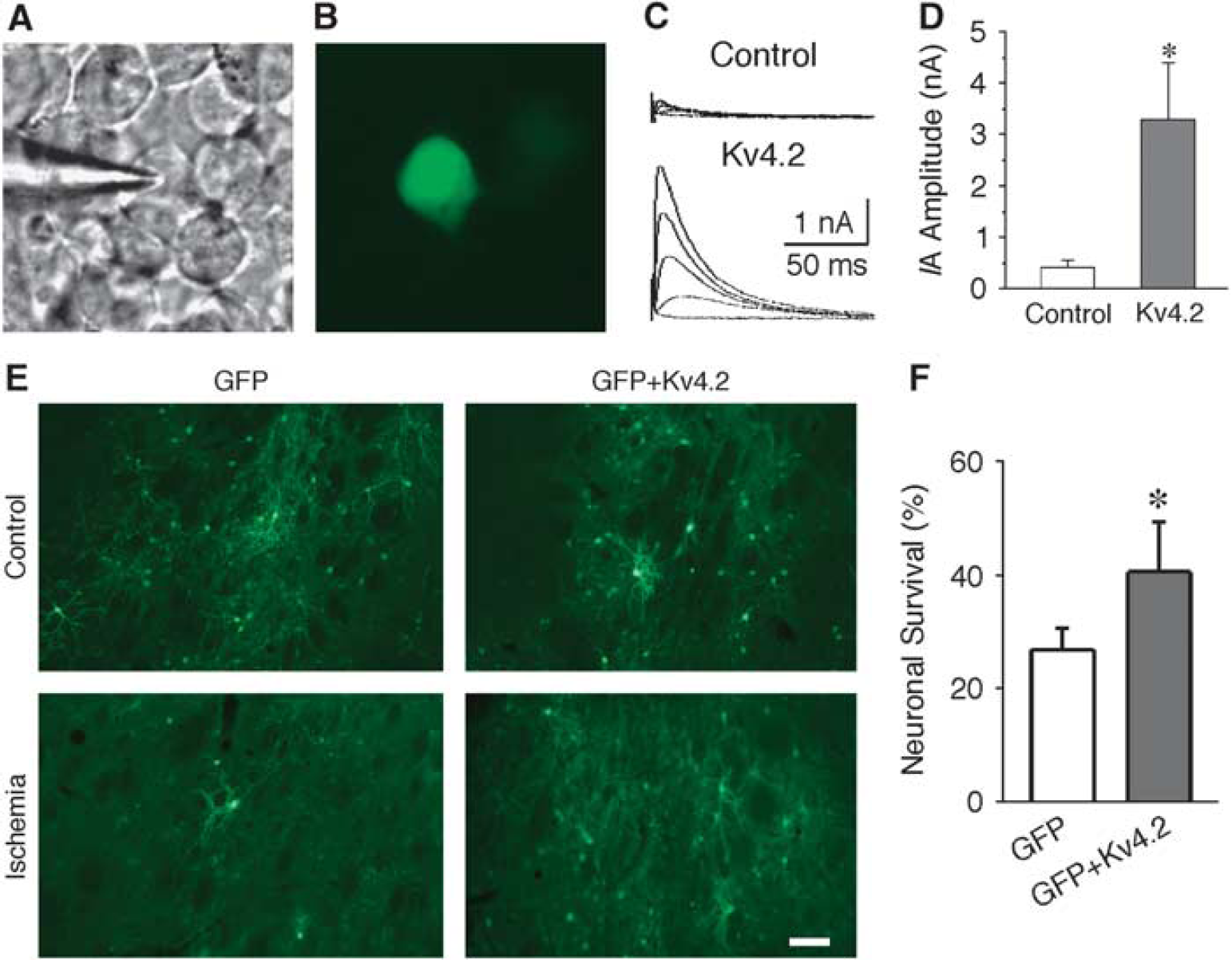
The protective effects of IA in striatal neurons were further examined using in vivo preparation. A-type potassium current was up-regulated in striatal neurons by overexpression of Kv4.2 subunit with AAV1-Kv4.2. To visualize the infected neurons, AAV1-GFP was also delivered together with AAV1-Kv4.2. We first examined the efficacy of AAV1-Kv4.2 infection. As shown in Figures 6A and 6B, the HEK cells that have little endogenous potassium current were infected with AAV1-GFP plus AAV1-Kv4.2. The amplitude of IA in GFP-positive cells (3.28 ± 1.12 nA, n = 3) was significantly increased as compared with that of GFP-negative cells (0.41 ± 0.15 nA, n = 3, P < 0.01; Figures 6C and 6D). To obtain the optimal experimental conditions in vivo, different dosages of AAV1-GFP were delivered into the striatum and the animals survived for different durations. Based on these preliminary studies (data not shown), 8 μL AAV1-GFP and survival time of 14 days were chosen as our experimental paradigms. AAV1-GFP (4 μL) plus AAV1-Kv4.2 (4 μL) were delivered into the right side of the striatum. For comparison, AAV1-GFP alone was delivered into the left side of the striatum of the same animal. The animals were then subjected to transient forebrain ischemia 13 days after infection, and GFP-positive neurons were counted 24 hours after reperfusion. As shown in Figure 6, the neuronal viability was significantly increased in neurons infected with Kv4.2 (neuronal survival in AAV1-GFP group: 26.9% ± 3.8%; neuronal survival in AAV1-GFP plus AAV1-Kv4.2 group: 40.5% ± 9.1%; P < 0.01, χ2 test; n = 8 to 12 animals in each group). The above results indicate that overexpression of IA protects striatal neurons against ischemic insults in vivo.
Increased neuronal viability in Kv4.2-transfected striatal neurons after transient forebrain ischemia. (A, B) Photomicrographs of the HEK cells in the same area under bright field (A) and florescence microscope (B), showing the green fluorescent protein (GFP)-positive and -negative cells 1 day after infection of Kv4.2 and GFP. (C) Representative traces showing the A-type potassium current (IA) recorded from GFP-negative (Control) and GFP-positive cells (Kv4.2 infected). (D) Pool data indicated that the IA was significantly increased in cells coinfected with AAV1-Kv4.2 and AAV1-GFP. (E) Representative photomicrographs showing striatal neurons infected with AAV1-GFP or AAV1-GFP plus AAV1-Kv4.2 with or without ischemia. Scale bar: 100 μm. (F) Pooled data showing the number of GFP-positive neurons in the striatum significantly increase in animals infected with AAV1-GFP plus AAV1-Kv4.2 at 24 hours after ischemia. *P < 0.01. AAV, adeno-associated virus, Kv, voltage-dependent potassium current.
Discussion
The present study has demonstrated that IA is up-regulated in ischemia-resistant LA neurons but remains unchanged in ischemia-vulnerable MS neurons after transient forebrain ischemia. The up-regulation is accompanied by activation of PKCα, which is a plausible pathway physiologically responsible to the enhancement of IA in LA neurons after ischemia. Genetic deletion of Kv1.4 and Kv4.2 increases ischemic neuronal death, whereas elevation of IA in striatal neurons by transfection of Kv1.4 and Kv4.2 increases the neuronal viability. Taken together, the evidence argues for the causal effects of enhancement of IA for neuronal protection following cerebral ischemia and an important role of post-translational modification.
Previous reports have shown that IA is altered after ischemia/hypoxia (Chi and Xu, 2000), yet the functional significance of these changes remains unclear. Our results indicate that the enhancement of IA protects neurons against cerebral ischemia. The up-regulation of IA by PKCα, probably through both Kv1.4 and Kv4.2, might contribute to the increase of IA in LA neurons after ischemia. It has been demonstrated that IA in LA neurons is mainly contributed by Kv4.2, whereas Kv1.4 is expressed in a subset of LA neurons (Song et al, 1998). Although Kv1.4 subunits in intact brains are found predominantly located at axon bundles and nerve terminals (Song et al, 1998; Trimmer and Rhodes, 2004), these subunits might be increased in the cell bodies of LA neurons after ischemia. One of the possible stimuli for such a dynamic poll of Kv1.4 is the loss of afferent and efferent innervations as suggested by the hippocampal neurons in culture (Maletic-Savatic et al, 1995). It has been shown that pyramidal neurons in the cerebral cortex (the major afferents to LA neurons) and MS neurons in the striatum (the targets of LA neurons) die in 24 hours after transient forebrain ischemia (Pulsinelli et al, 1982). As a result, LA neurons lost their afferents and targets at 24 hours after ischemia, which might cause the spatial changes of Kv1.4. Another possible mechanism for the expression of Kv1.4 in the cell bodies after ischemia might be the changes of neuronal activity. Studies have demonstrated that the localization of Kv channels can be modified by the increase of neuronal activity (Misonou et al, 2004).
The present study has demonstrated that the activation of PKC may have a role in up-regulating IA in LA neurons after ischemia. Previous evidence suggests that activation of PKC down-regulates IA in naive neurons, which is probably mediated via an ERK (extracellular-regulated kinases) pathway (Yuan et al, 2002). It is unlikely that the up-regulation of IA after ischemia is due to the depression of PKC action on ERK pathway, because inhibition of PKC failed to potentiate IA in LA neurons after ischemia. The mechanisms underlying the PKC up-regulation of IA in postischemic neurons need to be further investigated. PKC may have effects on both Kv1.4 and Kv4.2. In the present study, IA is not up-regulated in MS neurons after ischemia. Intriguingly, recombinant expression of Kv1.4 (and/or Kv4.2) in cultured MS neurons was sufficient to confer neuroprotection to OGD. However, overexpression of Kv1.4 alone only has mild protection in MS neurons. Stronger protection was observed after overexpression of both Kv1.4 and Kv4.2. The reason why it needs overexpression of both Kv4.2 and 1.4 channels probably is due to the lack of PKCα expression in MS neurons (Yoshihara et al, 1991). Therefore, both up-regulation of Kv channels and PKCα activation might be required for in vivo neuroprotection against ischemia.
Changes of neuronal excitability may have important roles in ischemic cell death. It has been shown that, in the striatum, ischemia-sensitive spiny neurons are depolarized, whereas ischemia-resistant interneurons are hyperpolarized (Calabresi et al, 1997; Centonze et al, 2001). Potassium currents are essential for the proper function of neurons by maintaining the resting membrane potentials and regulating the spike threshold, action potential repolarization, and membrane excitability (Storm, 1990). Alterations of potassium currents modulate neuronal excitability and ionic homeostasis, which might affect the excitotoxic cell death. A-type potassium current and Ikd are the major voltage-dependent potassium currents in neurons. It has been reported that the excessive potassium efflux triggers apoptotic cell death in several experimental settings including those mimicking ischemic conditions (Yu et al, 1997, 1999a). Although sustained activation of Ikd has been implicated to contribute to the excessive potassium efflux and induce apoptosis (Yu et al, 1999b), a transient increase of Ikd may be protective by decreasing neuronal excitability (Deng et al, 2005). A-type potassium current is a crucial modulator of information processing and neuronal excitability. For example, the increase of excitability in CA1 neurons due to the decrease of IA contributes to the temporal lobe epilepsy (Bernard et al, 2004). An enhancement of IA leads to a suppression of neuronal excitability (Varga et al, 2004), thus may ameliorate excitotoxic cell death following cerebral ischemia.
Footnotes
The authors declare no conflict of interest.
References
1.
BernardCAndersonABeckerAPoolosNPBeckHJohnstonD (2004) Acquired dendritic channelopathy in temporal lobe epilepsy. Science305:532–5
2.
CalabresiPAsconeCMCentonzeDPisaniASancesarioGD'AngeloVBernardiG (1997) Opposite membrane potential changes induced by glucose deprivation in striatal spiny neurons and in large aspiny interneurons. J Neurosci17:1940–9
3.
CentonzeDMarfiaGAPisaniAPicconiBGiacominiPBernardiGCalabresiP (2001) Ionic mechanisms underlying differential vulnerability to ischemia in striatal neurons. Prog Neurobiol63:687–96
4.
ChesseletMFGonzalesCLinCSPolskyKJinBK (1990) Ischemic damage in the striatum of adult gerbils: relative sparing of somatostatinergic and cholinergic interneurons contrasts with loss of efferent neurons. Exp Neurol110:209–18
5.
ChiXXXuZC (2000) Differential changes of potassium currents in CA1 pyramidal neurons after transient forebrain ischemia. J Neurophysiol84:2834–43
6.
ChoiDWRothmanSM (1990) The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci13:171–82
7.
DengPPangZZhangYXuZC (2004) Developmental changes of transient potassium currents in large aspiny neurons in the neostriatum. Brain Res Dev Brain Res153:97–107
8.
DengPPangZPLeiZXuZC (2009) Excitatory roles of protein kinase C in striatal cholinergic interneurons. J Neurophysiol102:2453–61
9.
DengPPangZPZhangYXuZC (2005) Increase of delayed rectifier potassium currents in large aspiny neurons in the neostriatum following transient forebrain ischemia. Neuroscience131:135–46
10.
DengPZhangYXuZC (2007) Involvement of I(h) in dopamine modulation of tonic firing in striatal cholinergic interneurons. J Neurosci27:3148–56
11.
GribkoffVKStarrettJEJrDworetzkySIHewawasamPBoissardCGCookDAFrantzSWHemanKHibbardJRHustonKJohnsonGKrishnanBSKinneyGGLombardoLAMeanwellNAMolinoffPBMyersRAMoonSLOrtizAPajorLPieschlRLPost-MunsonDJSignorLJSrinivasNTaberMTThalodyGTrojnackiJTWienerHYeleswaramKYeolaSW (2001) Targeting acute ischemic stroke with a calcium-sensitive opener of maxi-K potassium channels. Nat Med7:471–7
12.
HeurteauxCBertainaVWidmannCLazdunskiM (1993) K+ channel openers prevent global ischemia-induced expression of c-fos, c-jun, heat shock protein, and amyloid beta-protein precursor genes and neuronal death in rat hippocampus. Proc Natl Acad Sci USA90:9431–5
13.
HowardDBPowersKWangYHarveyBK (2008) Tropism and toxicity of adeno-associated viral vector serotypes 1, 2, 5, 6, 7, 8, and 9 in rat neurons and glia in vitro. Virology372:24–34
14.
JonasEAKaczmarekLK (1996) Regulation of potassium channels by protein kinases. Curr Opin Neurobiol6:318–23
LondonBWangDWHillJABennettPB (1998) The transient outward current in mice lacking the potassium channel gene Kv1.4. J Physiol509(Part 1):171–82
17.
LoweryRLZhangYKellyEALamantiaCEHarveyBKMajewskaAK (2009) Rapid, long-term labeling of cells in the developing and adult rodent visual cortex using double-stranded adeno-associated viral vectors. Dev Neurobiol69:674–88
18.
Maletic-SavaticMLennNJTrimmerJS (1995) Differential spatiotemporal expression of K+ channel polypeptides in rat hippocampal neurons developing in situ and in vitro. J Neurosci15:3840–51
19.
MattsonMPBargerSWBegleyJGMarkRJ (1995) Calcium, free radicals, and excitotoxic neuronal death in primary cell culture. Methods Cell Biol46: 187–216
20.
MisonouHMohapatraDPParkEWLeungVZhenDMisonouKAndersonAETrimmerJS (2004) Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci7:711–8
21.
NorrisAJNerbonneJM (2010) Molecular dissection of I(A) in cortical pyramidal neurons reveals three distinct components encoded by Kv4.2, Kv4.3, and Kv1.4 alpha-subunits. J Neurosci30:5092–101
22.
PangZPDengPRuanYWXuZC (2002) Depression of fast excitatory synaptic transmission in large aspiny neurons of the neostriatum after transient forebrain ischemia. J Neurosci22:10948–57
23.
PulsinelliWABrierleyJB (1979) A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke10:267–72
24.
PulsinelliWABrierleyJBPlumF (1982) Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol11:491–8
25.
RedmanPTHeKHartnettKAJeffersonBSHuLRosenbergPALevitanESAizenmanE (2007) Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1. Proc Natl Acad Sci USA104:3568–73
26.
RenYLiXXuZC (1997) Asymmetrical protection of neostriatal neurons against transient forebrain ischemia by unilateral dopamine depletion. Exp Neurol146:250–7
27.
RothmanSMOlneyJW (1986) Glutamate and the pathophysiology of hypoxic–ischemic brain damage. Ann Neurol19:105–11
28.
SongWJTkatchTBaranauskasGIchinoheNKitaiSTSurmeierDJ (1998) Somatodendritic depolarization-activated potassium currents in rat neostriatal cholinergic interneurons are predominantly of the A type and attributable to coexpression of Kv4.2 and Kv4.1 subunits. J Neurosci18:3124–37
TanakaK (2001) Alteration of second messengers during acute cerebral ischemia - adenylate cyclase, cyclic AMP-dependent protein kinase, and cyclic AMP response element binding protein. Prog Neurobiol65:173–207
31.
TrimmerJSRhodesKJ (2004) Localization of voltage-gated ion channels in mammalian brain. Annu Rev Physiol66:477–519
32.
VargaAWYuanLLAndersonAESchraderLAWuGYGatchelJRJohnstonDSweattJD (2004) Calcium-calmodulin-dependent kinase II modulates Kv4.2 channel expression and upregulates neuronal A-type potassium currents. J Neurosci24:3643–54
33.
WeiLYuSPGottronFSniderBJZipfelGJChoiDW (2003) Potassium channel blockers attenuate hypoxia-and ischemia-induced neuronal death in vitro and in vivo. Stroke34:1281–6
34.
YoshiharaCSaitoNTaniyamaKTanakaC (1991) Differential localization of four subspecies of protein kinase C in the rat striatum and substantia nigra. J Neurosci11:690–700
35.
YuSPYehCStrasserUTianMChoiDW (1999a) NMDA receptor-mediated K+ efflux and neuronal apoptosis. Science284:336–9
36.
YuSPYehCHGottronFWangXGrabbMCChoiDW (1999b) Role of the outward delayed rectifier K+ current in ceramide-induced caspase activation and apoptosis in cultured cortical neurons. J Neurochem73:933–41
37.
YuSPYehCHSensiSLGwagBJCanzonieroLMFarhangraziZSYingHSTianMDuganLLChoiDW (1997) Mediation of neuronal apoptosis by enhancement of outward potassium current. Science278:114–7
38.
YuanLLAdamsJPSwankMSweattJDJohnstonD (2002) Protein kinase modulation of dendritic K+ channels in hippocampus involves a mitogen-activated protein kinase pathway. J Neurosci22:4860–8
39.
ZouBLiYDengPXuZC (2005) Alterations of potassium currents in ischemia-vulnerable and ischemia-resistant neurons in the hippocampus after ischemia. Brain Res1033:78–89