
Abstract
Hyperglycemia after aneurysmal subarachnoid hemorrhage (aSAH) occurs frequently and is associated with delayed cerebral ischemia (DCI) and poor clinical outcome. In this review, we highlight the mechanisms that cause hyperglycemia after aSAH, and we discuss how hyperglycemia may contribute to poor clinical outcome in these patients. As hyperglycemia is potentially modifiable with intensive insulin therapy (IIT), we systematically reviewed the literature on IIT in aSAH patients. In these patients, IIT seems to be difficult to achieve in terms of lowering blood glucose levels substantially without an increased risk of (serious) hypoglycemia. Therefore, before initiating a large-scale randomized trial to investigate the clinical benefit of IIT, phase II studies, possibly with the help of cerebral blood glucose monitoring by microdialysis, will first have to improve this therapy in terms of both safety and adequacy.
Introduction
Aneurysmal subarachnoid hemorrhage (aSAH) is a devastating disease with high case morbidity and case fatality rates (van Gijn et al, 2007). Although these rates have decreased in the last decades, still 35% of aSAH patients die within the first month after the hemorrhage (Nieuwkamp et al, 2009). Prevention and treatment of neurologic complications such as rebleeding, hydrocephalus, and delayed cerebral ischemia (DCI) are obvious targets to improve prognosis. Besides neurologic complications, patients with aSAH often have cardiopulmonary and general medical complications that can influence outcome. Hyperglycemia in aSAH patients is common and associated with poor clinical outcome (Badjatia et al, 2005; Charpentier et al, 1999; Dorhout Mees et al, 2003; Frontera et al, 2006; Kruyt et al, 2008, 2009; Lanzino et al, 1993). As treatment for hyperglycemia is available, it has attracted increasing attention as a target for intervention, although adequate and safe glycemic control is difficult to achieve in patients with aSAH.
As hyperglycemia seems to be implicated in the pathway from aSAH to poor clinical outcome, insight into these mechanisms may reveal new treatment options. We undertook a nonsystematic literature search to provide an overview of the potential causes and consequences of hyperglycemia in aSAH patients and to address the pathophysiological mechanisms that might link hyperglycemia to poor clinical outcome. Furthermore, we performed a systematic literature search to review studies on glycemic control in aSAH patients. Finally, we provide directions for further trials and clinical management concerning glycemic control in this patient group.
Mechanisms Leading to Hyperglycemia After Aneurysmal Subarachnoid Hemorrhage
In patients admitted within 72 hours from aSAH, mean admission glucose is around 9 mmol/L, and around 3 out of 4 patients with aSAH are hyperglycemic on admission (Kruyt et al, 2009). Studies that report on glucose levels during the first 1 or 2 weeks of the clinical course report that hyperglycemia persists with levels exceeding 7 to 8mmol/L (Badjatia et al, 2005; Dorhout Mees et al, 2003; Frontera et al, 2006; Kruyt et al, 2008; Lanzino et al, 1993).
In ischemic stroke, hyperglycemia on admission is also frequent (Capes et al, 2001). This is, at least in part, attributed to (unrecognized) abnormalities of glucose metabolism such as diabetes mellitus (DM) that was existent before the stroke in around one third of patients (Gray et al, 2004; Kernan et al, 2005; Vancheri et al, 2005). As DM is not a risk factor for aSAH (Adams Jr et al, 1984; Feigin et al, 2005) and as patients with SAH are relatively young, the proportion of aSAH patients with preexistent abnormalities of glucose metabolism is much smaller than in patients with ischemic stroke.
There are several explanations for the increased glucose levels in acute aSAH patients. First, aSAH is accompanied by the activation of the hypothalamic–pituitary–adrenal axis and the activation of the sympathetic autonomic nervous system (Gauna et al, 2005). This activation results in an increase in the levels of stress hormones such as cortisol and catecholamines up to day 10 after the aSAH (Naredi et al, 2000; Vergouwen et al, 2010). These hormones enhance glycogenolysis, gluconeogenesis, proteolysis, and lipolysis, all resulting in excessive glucose production (Barth et al, 2007; Seematter et al, 2004). Moreover, catecholamines inhibit glucose transport by inhibition of insulin binding, resulting in insulin resistance with hyperinsulinemia (Gearhart and Parbhoo, 2006; Hunt and Ivy, 2002). Indeed, increased levels of cortisol are associated with increased levels of blood glucose in aSAH patients (Vergouwen et al, 2010). Second, aSAH is accompanied by an increased inflammatory response with release of cytokines (Hendryk et al, 2004). Cytokines, in turn, have been linked directly to hyperglycemia and insulin resistance (Hendryk et al, 2004; Hotamisligil and Spiegelman, 1994; Plomgaard et al, 2005; Rask-Madsen et al, 2003). In addition, cytokines stimulate the hypothalamic–pituitary–adrenal axis, further increasing the stress response. Interestingly, an altered glucose metabolism and hyperglycemia can in turn also stimulate the inflammatory response, raising the question of what is the cause and what is the effect (Esposito et al, 2002).
Third, besides a central role in the stress reaction, the hypothalamus also has an important role in maintaining glucose homeostasis by reducing hepatic gluconeogenesis and increasing insulin sensitivity (Obici et al, 2002; Schwartz and Porte, 2005). Neuropathological studies show that the majority of acute aSAH patients have hypothalamic lesions (Crompton, 1963). Moreover, a recent study reported hypothalamic dysfunction in the acute phase of aSAH (Klose et al, 2010), whereas the same study could not confirm earlier findings of hypothalamic dysfunction at longer (years) follow-up (Klose et al, 2010; Schneider et al, 2007). Whether acute hypothalamic dysfunction indeed contributes to hyperglycemia in aSAH patients is currently unclear.
In conclusion, increased stress and inflammatory responses seem to be the most important contributors to hyperglycemia after aSAH. Preexistent abnormalities of glucose metabolism do not seem to have a prominent role in aSAH patients, and the role of hypothalamic dysfunction and impaired glucose homeostasis remains uncertain (Figure 1, upper panel).
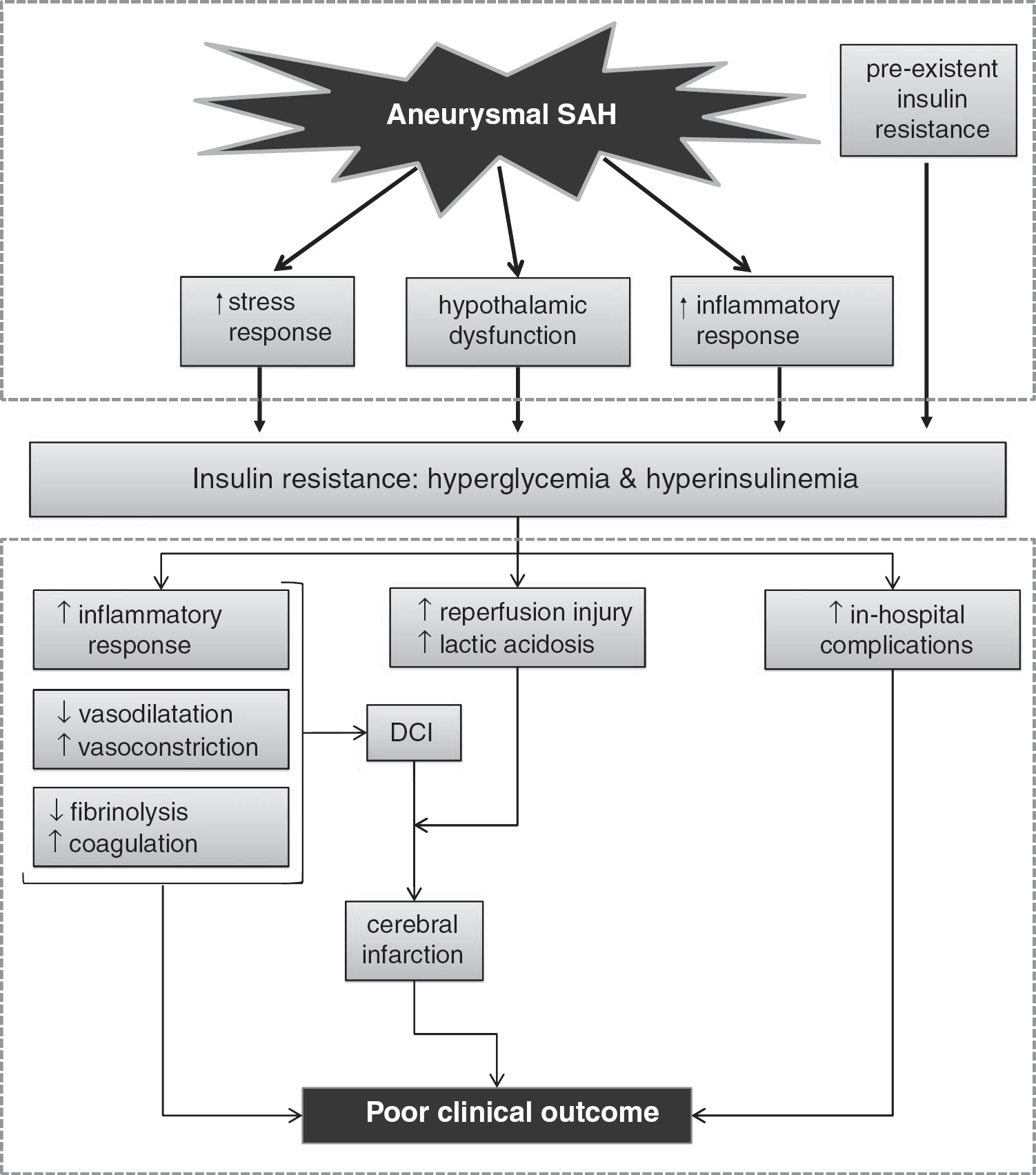
Figure with hypothetical mechanisms: leading to insulin resistance in patients with aneurysmal subarachnoid hemorrhage (upper panel); explaining a detrimental effect of hyperglycemia on clinical outcome (lower panel). DCI, delayed cerebral ischemia; SAH, subarachnoid hemorrhage.
Hyperglycemia and Clinical Outcome After Aneurysmal Subarachnoid Hemorrhage
Patients with hyperglycemia have an approximately threefold increased risk for poor outcome, and this seems unrelated to the different cutoff levels used to define hyperglycemia (Kruyt et al, 2009). The association between high levels of blood glucose and poor clinical outcome is more pronounced with persistent hyperglycemia than with hyperglycemia on admission (Badjatia et al, 2005; Frontera et al, 2006; Kruyt et al, 2008; Lanzino et al, 1993). A recent study showed that aSAH patients with persistent hyperglycemia are seven times more likely to have poor outcome than patients with normoglycemia, whereas in the same study, isolated hyperglycemic events throughout the clinical course were not predictive of poor outcome (McGirt et al, 2007).
How does Hyperglycemia Affect Outcome After Aneurysmal Subarachnoid Hemorrhage?
Although several studies found that high levels of blood glucose in the acute phase of aSAH are an independent risk factor for poor clinical outcome (Badjatia et al, 2005; Charpentier et al, 1999; Frontera et al, 2006; Kruyt et al, 2008), this does not prove causality of that relation. Elevated levels of admission blood glucose for example could constitute no more than a marker of SAH severity rather than a causative factor leading to secondary damage and consequently poorer clinical outcome. Indeed, high levels of admission glucose are associated with more severe SAH (Dorhout Mees et al, 2003; Kruyt et al, 2008). On the other hand, several experimental and imaging studies, as well as clinical observations, have highlighted mechanisms through which hyperglycemia may affect clinical outcome after aSAH (Figure 1, lower panel).
In critical illness in general, hyperglycemia on admission and during the clinical course is associated with various in-hospital complications such as respiratory failure, nosocomial infections, and impaired wound healing, all of which are contributors to poor outcome (Frontera et al, 2006; van den Berghe et al, 2001). Several trials, including trials in patients with aSAH (Bilotta et al, 2007) showed that IIT lowered these in-hospital complications (van den Berghe et al, 2005, 2001).
Another factor that could link hyperglycemia to poor outcome is DCI. The DCI occurs in approximately one third of aSAH patients, mostly between days 4 and 10 (Roos et al, 2000) and can progress to irreversible cerebral infarction with subsequent poor clinical outcome (Rosengart et al, 2007). Patients with hyperglycemia on admission or during the clinical course develop DCI and cerebral infarction more often than patients with normal blood glucose levels (Badjatia et al, 2005; Charpentier et al, 1999; Frontera et al, 2006; Juvela et al, 2005; Kruyt et al, 2008; Lanzino, 2005). The association between admission hyperglycemia and DCI was weaker than that of persistent hyperglycemia and DCI, and the association between admission glucose and DCI did not persist after multivariable assessment in several studies (Badjatia et al, 2005; Frontera et al, 2006; Juvela et al, 2005; Kruyt et al, 2008). The fact that aSAH patients with preexistent DM are at an increased risk of DCI compared with aSAH patients without preexistent DM lends further support of a link between abnormalities of glucose metabolism and DCI in aSAH patients (Dumont et al, 2009; Fergusen and Macdonald, 2007).
The cause of DCI in aSAH patients remains unclear, but various mechanisms have been proposed that may be influenced by hyperglycemia. One of these mechanisms is vasospasm, which is frequently seen in aSAH patients (Fergusen and Macdonald, 2007).
Although vasospasm is associated with the occurrence of DCI, it does not fully explain DCI (Dankbaar et al, 2009). Several other mechanisms, of which microthrombosis and inflammation are the most prominent, seem to contribute to DCI (Vergouwen et al, 2008). Hyperglycemia is closely linked to increased coagulation and decreased fibrinolysis, both of which enhance thrombin formation. In studies in healthy individuals, hyperglycemia stimulates coagulation by increasing platelet activation through thrombin–antithrombin complexes and the tissue factor pathway (Gentile et al, 2007; Rao et al, 1999; Stegenga et al, 2006; Vaidyula et al, 2006), and hyperinsulinemia decreases fibrinolytic activity by increasing plasminogen activator inhibitor (Festa et al, 1999; Meigs et al, 2000; Pandolfi et al, 2001; Stegenga et al, 2006).
Besides interference with coagulation and fibrinolysis, hyperglycemia is also associated with an increase in proinflammatory transcription factors and proinflammatory cytokines (Bemeur et al, 2007; Esposito et al, 2002; Garg et al, 2006; Martini and Kent, 2007). In aSAH patients, several studies have linked markers of increased inflammation to the development of DCI (Esposito et al, 2002; Hirashima et al, 1997; Osuka et al, 1998).
Once DCI is established in aSAH patients, the most imminent danger is that potentially reversible ischemic tissue progresses to irreversible infarcted tissue. Experimental and clinical imaging studies in ischemic stroke show that hyperglycemia is associated with this progression (Els et al, 2002; Parsons et al, 2002; Wagner et al, 1992).
Although restoration of blood flow to ischemic tissue is essential, reperfusion itself can also induce injury: so-called reperfusion injury (McCord, 1985). The mediators of reperfusion injury are inflammation and oxidative stress. Markers of inflammation are associated with the progression of cerebral ischemia to infarction (Chamorro and Hallenbeck, 2006; Wang et al, 2007) and, in experimental stroke, inhibition of the inflammatory response reduces infarct size (Arumugam et al, 2009). As outlined earlier, hyperglycemia increases the inflammatory response, and may therefore exacerbate reperfusion injury. The other mediator of reperfusion injury, oxidative stress, results from an imbalance between the production and neutralization of reactive oxygen species. Reactive oxygen species have been shown to increase neuronal death and infarct volume (Bemeur et al, 2005, 2007; Li et al, 2005; Muralikrishna and Hatcher, 2006; Suh et al, 2008). In experimental ischemic stroke, hyperglycemia increases the production of reactive oxygen species through the activation of protein kinase C and through increased NADP production, thus promoting oxidative stress.
In addition to increased reperfusion injury, hyperglycemia also promotes anaerobic glycolysis leading to the accumulation of lactic acid and a derangement in pH homeostasis. Both these processes have been proposed to contribute to increased brain injury (Anderson et al, 1999; Katsura et al, 1992; Siesjo, 1988). In support of a detrimental role of lactic acid is the finding that in patients with cerebral ischemia, hyperglycemia correlates with both an increased lactate production and with the progression from ischemic to infarcted tissue (Parsons et al, 2002).
In conclusion, a causal relation between hyperglycemia and poor outcome in aSAH patients remains elusive, but in the sequence of events that occur after aSAH, hyperglycemia may exert a detrimental effect by increasing secondary complications such as infection and cerebral ischemia, as well as by facilitating the progression from ischemia to irreversible infarction. Besides lowering of blood glucose levels with insulin, a more mechanistic approach with interventions directly aimed at the restoration of glucose homeostasis have potential clinical relevance. For example, pharmacological neutralization of cytokines was shown to revert insulin resistance (Xu et al, 2008), and inhibition of cortisol production prevented hyperglycemic aggravation of ischemic neuronal damage (Payne et al, 2003). These findings, however, are experimental and should not be extrapolated to the clinical setting.
Glucose Lowering Therapy
Studies in non-Aneurysmal Subarachnoid Hemorrhage Patients
Before addressing glucose lowering therapy in aSAH patients, we first summarize current insights on this topic in other groups of critically ill patients because this provides relevant background information.
In 2001, intensive insulin therapy (IIT) was implemented worldwide in intensive care unit (ICU) facilities after a landmark trial from Leuven demonstrated its clinical benefit in a surgical ICU (van den Berghe et al, 2001, 2006).
Several later trials could not confirm the previous positive findings (Annane et al, 2010; Brunkhorst et al, 2008; Finfer et al, 2009). In one of these trials, IIT was even associated with an increased risk of severe hypoglycemia (glucose < 2.2.mmol/L) and subsequent poor clinical outcome (Finfer et al, 2009). It remains unclear why these recent and earlier trials show conflicting results. One possible explanation is that in the time frame that separated these trials, the standards for hyperglycemia management in ‘regular care’ are more rigorous, thus decreasing the potential contrast with an IIT group in trials. Meanwhile, IIT is still recommended for patients admitted to an ICU (Griesdale et al, 2009), but the latter trials have emphasized the important drawback of IIT namely increased incidence of hypoglycemia.
In stroke other than aSAH, the UK Glucose Insulin in Stroke Trial failed to show a clinical benefit from glycemic control on clinical outcome (Gray et al, 2007). This trial, however, was stopped prematurely because of slow enrolment. Other limitations of the trial were a relatively short duration of treatment (24 hours) and a small contrast in mean glucose levels (0.57 mmol/L) between the intervention and the control group.
Studies in Aneurysmal Subarachnoid Hemorrhage Patients
We performed a systematic literature search on glucose lowering treatment in aSAH patients. MEDLINE was searched on published studies from 1966 to February 2010 written in English, German, French, or Spanish. We used the medical subject headings (MeSH) and search terms ‘blood glucose’ and ‘subarachnoid hemorrhage’ truncated text words ‘hyperglyc(a)emia,’ ‘glucose,’ ‘subarachnoid,’ ‘SAH,’ ‘h(a)emorrhage,’ and ‘bleeding’ in different combinations. Studies describing a consecutive series of aSAH patients who were treated with IIT and who reported glucose levels during IIT (i.e., not only admission glucose levels) could be included. When an article complied with the inclusion criteria but lacked information on parameters of hypoglycemia, we approached the authors to obtain these data. We hand searched the bibliographies of all included articles and abstract books of the European and American stroke congresses held from January 2008 to October 2009. The MEDLINE search yielded 92 articles. Most articles were excluded because patients were not given IIT or because glucose levels during the clinical course were not reported (58). Other reasons for exclusion were nonconsecutive series (20), same population described (2), and non-aSAH patients described (6).
No additional relevant studies were found by searching the bibliographies or abstract books. We identified six studies (listed in Table 1); five cohort studies, of which two compared glucose levels before and after the introduction of IIT and one small randomized controlled trial (Bell and Strong, 2005; Bilotta et al, 2007; Latorre et al, 2009; Naidech
Studies reporting on intensive insulin therapy in SAH patients
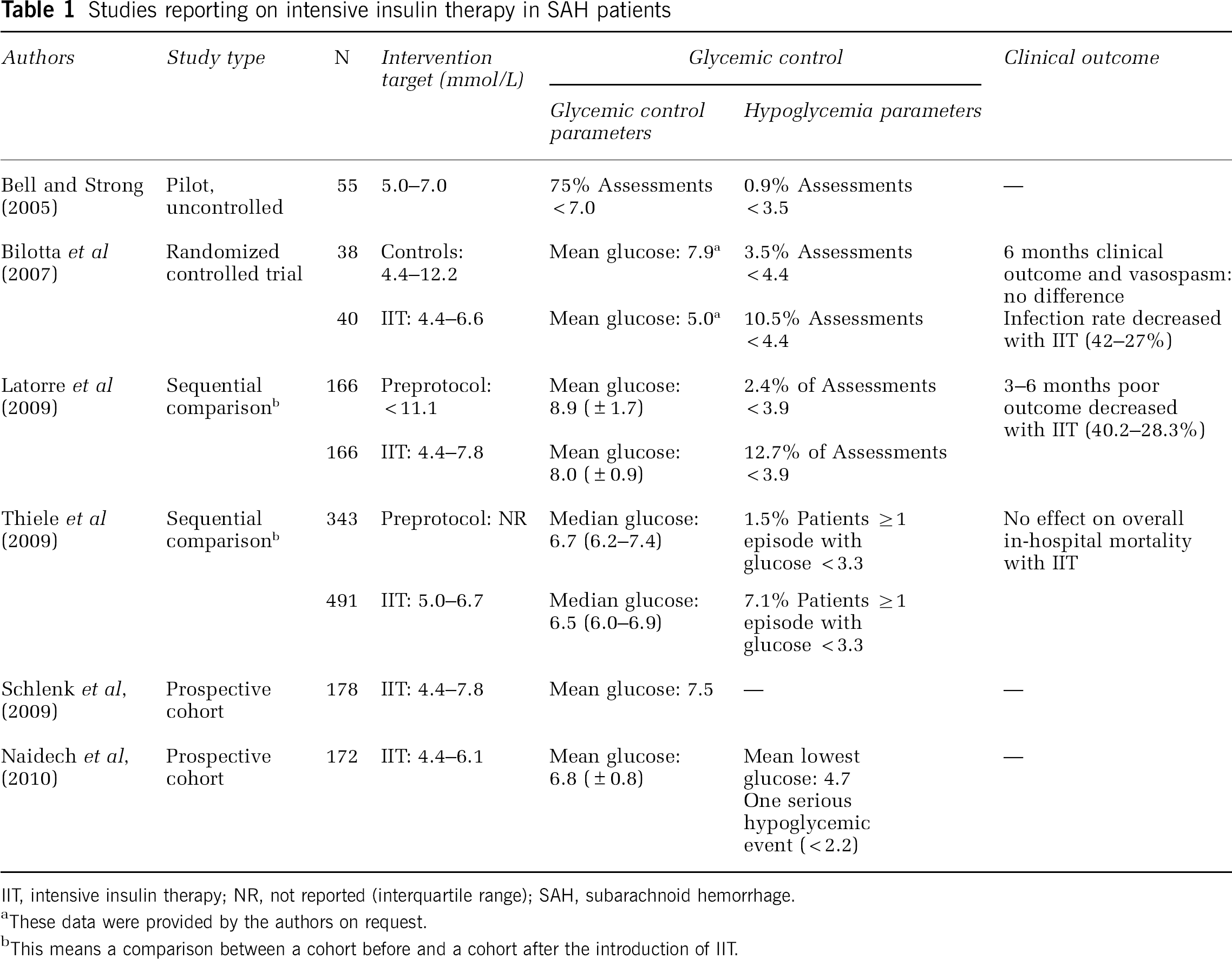
IIT, intensive insulin therapy; NR, not reported (interquartile range); SAH, subarachnoid hemorrhage.
These data were provided by the authors on request.
This means a comparison between a cohort before and a cohort after the introduction of IIT.
In all identified studies, patients were admitted to an ICU. Glucose levels targets varied somewhat between the studies with upper limits of glucose levels ranging from 6 to 7.8 mmol/L and lower limits ranging from 4.4 to 5.0 mmol/L. The cutoff level used to define hypoglycemia varied between studies (3.5 to 4.4 mmol/L). None of the studies reported neurologic deterioration during hypoglycemia; however, the warning symptoms of hypoglycemia might be less clear in patients with aSAH than in other patients, as these symptoms are similar to some stroke comorbidities (Hemmen et al, 2008).
One uncontrolled pilot study showed that IIT was feasible but was accompanied by hypoglycemic episodes (Bell and Strong, 2005). In the only randomized trial that was designed to test the applicability of IIT, 78 patients were enrolled. The IIT reduced the mean glucose from 8.0 to 5.0 mmol/L, but increased hypoglycemic episodes from 3.5% to 10.5% (Bilotta et al, 2007). The IIT reduced the infection rate from 42% to 27% (
In conclusion, currently there is no evidence that hyperglycemia in aSAH patients should be lowered actively. Moreover, IIT is accompanied by an increase in hypoglycemic episodes, which should raise concerns about the safety of this therapy.
Risk of Intensive Insulin Therapy: Hypoglycemia
As noted earlier, IIT is invariably accompanied by hypoglycemic episodes, which is posing a potential risk to aSAH patients subjected to this therapy. The brain relies on the continuous delivery of glucose through the blood to maintain normal metabolic function. Moreover, various studies indicate that under conditions of acute brain injury, glucose utilization is increased (Andersen and Marmarou, 1992; Bergsneider et al, 1997; Oddo et al, 2008). Thus, it is conceivable that hypoglycemia on top of preexisting cerebral damage and the increased glucose utilization is even more detrimental than for the healthy brain (double-hit theory) (de Courten-Meyers et al, 1994). Besides simple deprivation of energy, another interesting explanation for hypoglycemia to be detrimental to the brain is the occurrence of cortical spreading depression (CSD), a slowly moving wave that leads to intracellular calcium overload and depression of synaptic activity (Leão, 1944). Such depolarization waves occur frequently in patients with aSAH (Dreier et al, 2009; Strong et al, 2000). In focal ischemia of nonhuman primates, even mild hypoglycemia promoted the occurrence of CSDs (Dreier et al, 2009; Strong et al, 2000). More recently, in a patient with aSAH, a severe cluster of CSDs was associated with (IIT induced) hypoglycemia (Dreier et al, 2009; Strong et al, 2000). Interestingly, CSD can by itself also lead to the depletion of extracellular cerebral glucose levels thereby further jeopardizing brain tissue (Hashemi et al, 2009). Although IIT was never proven to be beneficial for neurologic critically ill patients, various IIT protocols, often with different glycemic targets, are being used on ICU's worldwide also for this particular patient group (see Table 1). This has driven various research groups to investigate the metabolic consequences of IIT in patients with brain injury with the use of microdialysis (Sarrafzadeh et al, 2004; Schlenk et al, 2008b, 2009; Vespa et al, 2006; Vespa, 2006). This technique, for which a microcatheter is inserted into the brain parenchyma, allows
Conclusions and Future Directions
For a long time, hyperglycemia was thought to be an adaptive beneficial response to critical illness to provide those organs that predominantly rely on glucose as metabolic substrate such as the brain. With the publication of the landmark Leuven trials, however, this concept has changed dramatically and IIT was implemented worldwide for all kinds of critically ill patients, including patients with brain injury. It is important, however, to realize that today IIT was proven only in monocenter studies and for a subgroup of critically ill patients. In addition, more recently a large multicenter study could not prove a beneficial effect from IIT and IIT was even associated with an increased risk of severe hypoglycemia and subsequent poor clinical outcome (Finfer et al, 2009). Therefore, although hyperglycemia, especially if persistent during the clinical course, clearly predicts poor clinical outcome in aSAH patients, care should be taken in extrapolating previous positive data to aSAH patients. The most recent guidelines for the treatment of aSAH state that it is imperative to avoid hyperglycemia (Bederson et al, 2009). However, it is unclear which glucose levels should be targeted and how this has to be accomplished. Only a large-scale randomized controlled trial investigating the clinical benefit of IIT in aSAH patients will be able to resolve this issue.
Before initiating such a trial, however, it is important to improve glycemic control in phase II studies with special reference to safety. In these studies, focus should be not only on the lowering of glucose levels, but, and perhaps even more so, on the prevention of hypoglycemia related to this therapy. As for the question which glucose targets should be aimed at, there is no unambiguous answer. On the one hand, when IIT does not achieve a substantial lowering of blood glucose levels compared with standard ICU therapy, this treatment is not likely to result in a clinical benefit (Gray et al, 2007; Griesdale et al, 2009). On the other hand, the most recent trials in non-aSAH patients have shown that IIT targeting blood glucose levels in a low physiological range (4.4 to 6.1 mmol/L) is associated with serious hypoglycemic episodes (glucose < 2.2 mmol/L) and subsequent worse clinical outcome (Brunkhorst et al, 2008; Finfer et al, 2009). A recent cohort study in aSAH patients emphasized this, by demonstrating that not high, but IIT-induced nadir levels of blood glucose, were associated with poor clinical outcome (Naidech
All studies we identified on IIT were performed in the ICU setting. Patients with clinically less severe aSAH are sometimes admitted to a stroke unit. In contrast to an ICU, a stroke unit often lacks the highly intensive treatment facilities available on the ICU. For instance, the lack of direct access to frequent arterial blood glucose monitoring and fewer personnel per patient to execute laborious treatment algorithms could render glycemic control even more challenging than on the ICU facility.
Considering this, if decided to actively lower hyperglycemia in aSAH patients as is recommended by the guidelines, the best advice seems to institute a multidisciplinary team, including stroke and diabetes experts to facilitate the implementation of glucose lowering protocols with the aim to lower blood glucose levels only if this can be performed without subsequent hypoglycemia. Such a protocol should probably include close monitoring of blood glucose levels with a high rate of insulin adjustments (i.e., every 1 or 2 hours). Continuous glucose monitoring devices that are now becoming available and the use of computerized treatment algorithms have the potential to facilitate this, but the safety of these devices will have to be investigated first (Vriesendorp et al, 2005).
In conclusion, target levels of blood glucose will remain arbitrary until a randomized control trial has investigated the clinical benefit of IIT. Before such a trial can start, focus should be on how IIT can be improved both in terms of efficacy but perhaps more important in terms of safety.
Search strategy and selection criteria
References for this review were identified by searches of PubMed from 1966 until February 2010 with the terms ‘blood glucose’ and ‘subarachnoid hemorrhage’ truncated text words ‘hyperglyc(a)emia,’ ‘glucose,’ ‘subarachnoid,’ ‘SAH,’ ‘h(a)emorrhage,’ and ‘bleeding.’ Articles were also identified through searches of the authors' own files. Only papers published in English, German, French, or Spanish were reviewed.
Footnotes
The authors declare no conflict of interest.