
Abstract
It is well established that lactate can be used as an energy substrate by the brain by conversion to pyruvate and a subsequent oxidation in the mitochondria. Knowing the need for readily metabolizable substrates directly after ischemia and the protective effect of lactate after excitotoxicity, the aim of this study was to investigate whether lactate administration directly after ischemia could be neuroprotective. In vitro, the addition of 4 mmol/L
Keywords
Introduction
Stroke is the first cause of disability and the third cause of death in the world (Feigin et al, 2003; Murray and Lopez, 1997). Approximately 80% of strokes are ischemic and most of the infarcts involve the territory of the middle cerebral artery. During an ischemic stroke, glucose and oxygen supply to the brain decreases, leading to a cascade of damaging mechanisms. Disruption of brain metabolism is clearly a key element in stroke; a better knowledge of its cellular and molecular determinants may lead to novel therapeutic targets.
Glucose was believed to be the only energy substrate for neurons until Schurr et al showed 20 years ago that lactate, as a sole energy substrate, can support normal synaptic function in rat hippocampal slices (Schurr, 2006; Schurr et al, 1988), opening the possibility that lactate produced during ischemia might not just be an end product, but might also be used as a substrate for energy metabolism. A few years later, Pellerin and Magistretti (1994) proposed their astrocyte–neuron lactate shuttle hypothesis, in line with the idea that lactate can be used by neurons.
On the basis of this hypothesis, several studies have tested whether lactate is beneficial to neurons under pathologic conditions, such as ischemia, glutamate excitotoxicity, and traumatic brain injury, and showed promising results even if the use of lactate as an energy substrate by neurons is still a source of controversy (Hertz, 2008). In vitro, on hippocampal slices, it has been shown that lactate, either endogenously produced during hypoxia or applied exogenously, can be used after hypoxia, is preferential to glucose during the reoxygenation period (Cater et al, 2003; Schurr et al, 1997b), and is protective against glutamate excitotoxicity (Schurr et al, 1999). In vivo, lactate administered to cortical superfusate during and after global ischemia in rat, allows a better recovery of the electrocorticogram during reperfusion and reduces amino-acid release (Phillis et al, 1999). When perfused in combination with glutamate, lactate has been shown to protect against glutamate excitotoxicity and to reduce lesion size (Ros et al, 2001). Another study using an inhibitor of lactate transport, which also inhibits the entry of pyruvate into the mitochondria, showed an important neuroprotective effect of endogenously produced lactate during global ischemia (Schurr et al, 2001). Furthermore, lactate administration in patients after traumatic brain injury improved neurologic outcome (Ichai et al, 2009).
There is growing evidence showing that lactate is required to sustain neuronal recovery directly after ischemia. However, the effect of lactate administration after reperfusion has never been tested. Therefore, the aim of this study was to test a potential neuroprotective effect of lactate administration at the end of ischemia or later.
Materials and methods
All animal experiments were conducted in accordance with the guidelines of the cantonal veterinary office.
Organotypic Hippocampal Slice Cultures
Hippocampal slice cultures obtained from P12 rats were subjected to in vitro ischemia by exposure to reduced oxygen and glucose concentrations (oxygen and glucose deprivation, OGD), as described previously (Hirt et al, 2004). Briefly, 350-μm-thick coronal hippocampal slices were cultured on porous membranes (Millicell; Millipore, Billerica, MA, USA) in a medium containing 25% horse serum (Oxoid, Hampshire, UK), 50% minimal essential medium supplemented with HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) and sodium bicarbonate (MEM; Gibco, Paisley, UK), 25% HBSS (Hanks' balanced salt solution; Gibco), 2 mmol/L
Transient Middle Cerebral Artery Occlusion in the Mouse
A total of 13 male ICR-CD1 mice (weighing 24 to 34 g, Charles River, L'Arbresle, France) were anesthetized by an intraperitoneal injection of 8 mg/kg xylazine (Rompun 2%, Bayer, Zürich, Switzerland)+100 mg/kg ketamine (Ketanarkon 100, Streuli Pharma AG). A second dose of 4 mg/kg xylazine+50 mg/kg ketamine was added after 40 mins of anesthesia. A total of 26 male ICR-CD1 mice (22 to 32 g, Charles River) were anesthetized and maintained under 1.5% to 2% isoflurane in 30% oxygen and 70% nitrous oxide using a face mask. At 0 h, ischemia was induced by inserting an 11-mm silicone-coated 8-0 filament through the left common carotid artery into the internal carotid artery (see Wiegler et al (2008) and references therein). The filament was withdrawn after 30 (for the mice anesthetized with xylazine–ketamine) or 60 mins (for the mice anesthetized with isoflurane), allowing reperfusion. Regional cerebral blood flow was measured by LDF (laser-Doppler flowmetry) (Periflux 5000, Perimed, Stockholm, Sweden) with a flexible probe fixed on the skull, 1 mm posteriorly and 6 mm laterally from the bregma. Throughout the entire operation, regional cerebral blood flow was monitored and maintained under 20% of the baseline level during ischemia and above 50% of the baseline level after reperfusion. Throughout surgery and until awaking, rectal temperature was maintained at 37±0.5°C using a temperature control unit (FHC, Bowdoinham, ME, USA).
Mice were administered 0.025 mg/kg of buprenorphine subcutaneously for analgesia at the end of the operation. Once the animals were awake, they were housed in an incubator at 31°C. The mice were killed 48 h after ischemia.
To mimic the in vitro condition, we estimated that the intracerebroventricular injection of 2 μL of 100 mmol/L
Determination of Ischemic Lesion Volumes
Animals were killed at 48 h after the onset of focal ischemia and 20-μm-thick, 720-μm-apart, coronal cryostat sections were stained with cresyl violet for histologic determination of lesion size. A digitalized image of the Nissl-stained tissue was obtained under a light stereomicroscope (Leica MZ16FA, Leica, Heerbrugg, Switzerland). The lesion area was determined by an examiner blinded for the treatment group using ImageJ software (NIH, Bethesda, MD, USA, http://rsb.info.nih.gov/ij/) on stained sections. Direct infarct volume was calculated by multiplying the sum of the infarct areas on each section by the spacing distance. To avoid bias due to edema, an indirect lesion size was calculated as follows: indirect lesion=Volumecontralateral−(Volumeipsilateral−direct infarct volume) (Swanson et al, 1990).
Neurologic Deficits
A neuroscore composed of three tests was assessed 48 h after ischemia for mice operated under isoflurane anesthesia. In this neuroscore, each test has a maximum of three points. The first test evaluates the neurologic deficit and is graded for severity, as described previously (Hirt et al, 2004) (0: no observable neurologic deficit; 1: failure to extend the right forepaw; 2: circling to the contralateral side, and 3: loss of walking or righting reflex). The second test is a beam walking test (Carter et al, 2001) (0: mice walk directly to the end of the beam; 1: mice can walk along the beam, but slip a few times; 2: mice cannot walk more than a few steps. 3: mice do not move). The third test is the Rotarod treadmill (UgoBasile, Comerio, Italy) (Carter et al, 2001) test. The mice were placed on the rotating drum, set to accelerate uniformly from 4 to 40 r.p.m., and their latency to fall from the drum was recorded. The animals were trained on 2 different days before surgery, with two trials in each training session. The test was then performed 2 days after middle cerebral artery occlusion (MCAO), with two consecutive trials for each animal. The better of the two trials was selected. Points were attributed on the basis of performances expressed as a percentage of the best performance before ischemia (0:90% to 100% and then 0.5 point for each 15% decrease).
In Vivo Magnetic Resonance Studies of Lactate-Injected Mice
To evaluate the distribution of the intracerebroventricularly injected lactate, six ICR-CD1 mice (weighing 29 to 33 g) were measured by magnetic resonance (MR) imaging and spectroscopy before and after an intracerebroventricular injection of 2 μL of 100 mmol/L
All MR studies were carried out in a horizontal, 14.1-T/26-cm magnet (Magnex Scientific, Abingdon, UK), as described previously (Lei et al, 2009).
Throughout the entire MR studies, including the intracerebroventricular injection, the animals were maintained in an anesthetized state with 1% to 2% isoflurane mixed with O2 and stereotaxically fixed with two ear pieces and a bite piece in a holder (RAPID Biomedical GmbH, Rimpar, Gemany). During the mean time, the animal was simultaneously monitored for breathing and temperature using an MR-compatible monitor system (Model 1025, SA Instruments, Stony Brook, NY, USA), and rectal temperature was maintained at ∼37.0°C by circulating warm water.
Briefly, multislice T2-weighted images were acquired using the fast spin-echo technique (Hennig, 1988), with TEeff (effective echo time) =50 msecs and TR (repetition time) 6,000 msecs to locate the volume of interest in the left striatum (6 to 8 μL) or cortex (2.2 to 2.5 μL). Thereafter, all first- and second-order shim terms over the volume of interest were altered accordingly, using the echo-planar version of FASTMAP (see Mlynarik et al (2006) and references therein), which resulted in a water line width within 25 Hz. Localized 1H-MR spectrum was obtained using the SPECIAL techniques, TE/TR=2.8/4,000 msec in combination with outer volume suppression and VAPOR water suppression (see Mlynarik et al (2006) and references therein). To sustain sufficient signal-to-noise ratios from such microliter volumes, 240 and 480 of such spectra were acquired for the striatum and cortex, respectively.
The in vivo1H-MR spectra obtained from each study were processed as in Tkac et al, (2007), frequency drift was corrected, summed and eddy current was compensated using the water signal from the same volume of interest. Thereafter, absolute quantification was obtained using a linear combination analysis method, LCModel, assuming 80% tissue water content (see Tkac et al (2007) and references therein).
Statistics
All data are presented as mean±s.d. Statistical analyses were carried out using nonparametric tests: the Kruskal–Wallis test followed by Dunn's multiple-comparison test for in vitro experiments and the Mann–Whitney test for in vivo experiments (one-tailed P-value for in vivo experiments).
Results
In an in vitro model of ischemia consisting of OGD in rat organotypic hippocampal slices, the administration of 4 mmol/L
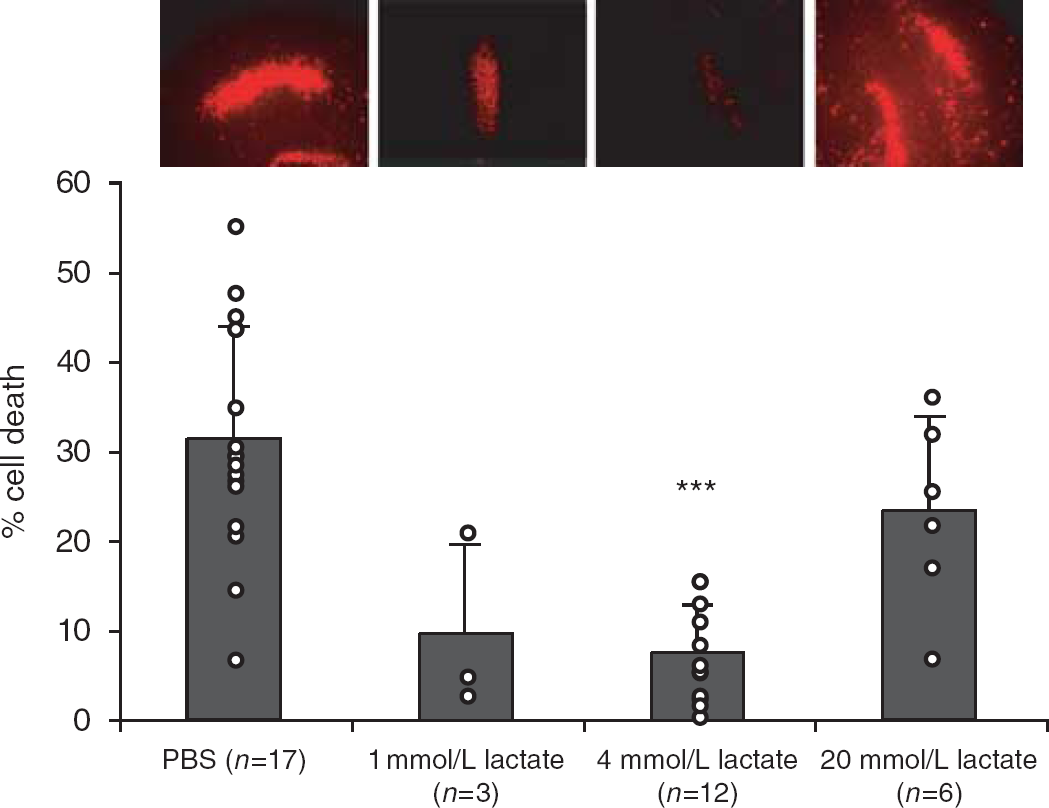
Administration of 4 mmol/L
On the basis of this apparent neuroprotective effect of lactate in vitro, we studied the potential beneficial effect of lactate administration in vivo after 30 mins of MCAO under xylazine–ketamine anesthesia. A volume of 2 μL of 100 mmol/L
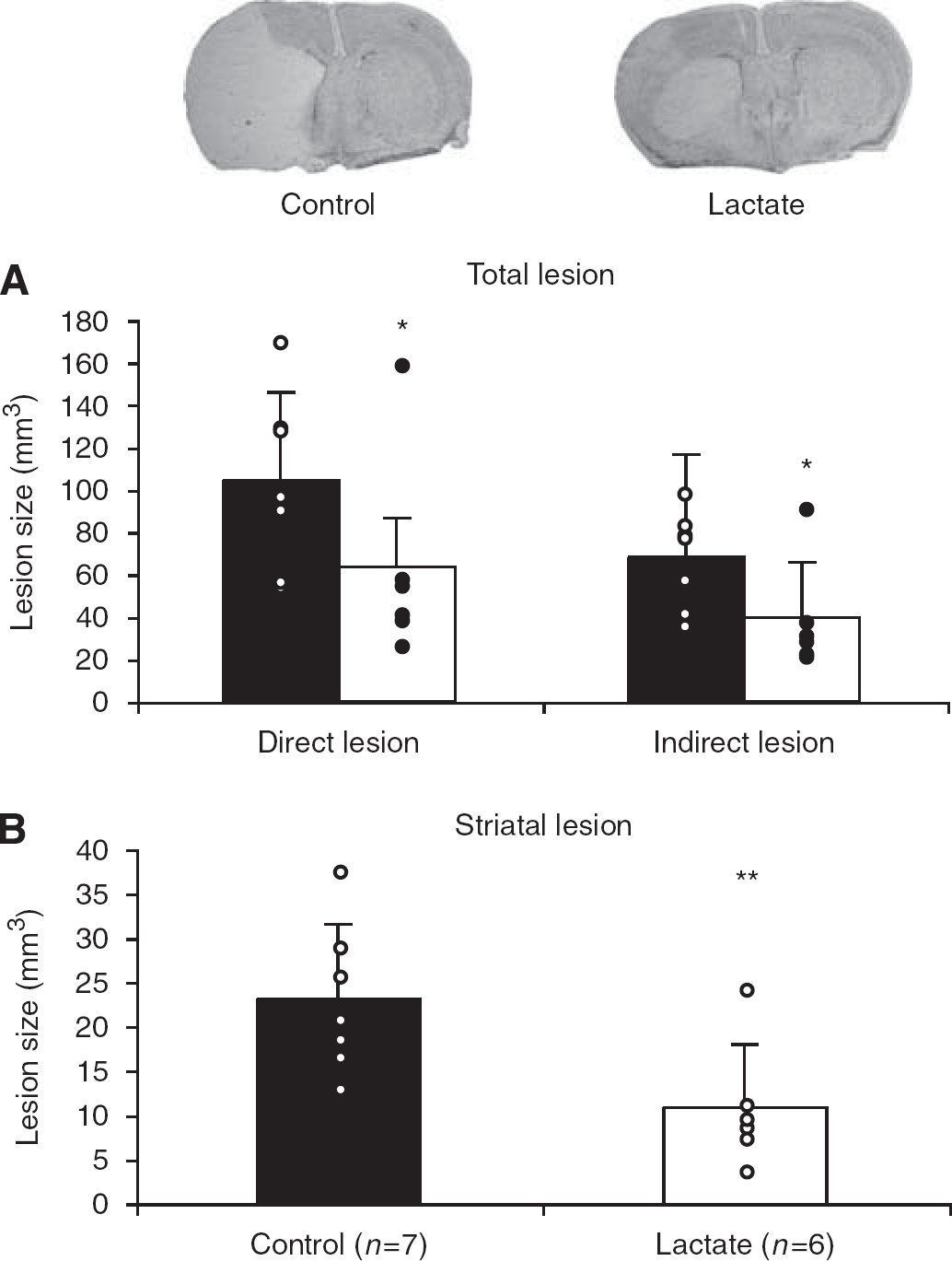
Injection of 2 μL of 100 mmol/L
To determine whether the neuroprotective effect of lactate in vivo is present under different anesthetic conditions and longer ischemia duration, we carried out a second set of experiments with 60 mins MCAO in mice under isoflurane anesthesia. This set of experiments was blinded and was carried out randomly. In addition, behavior was assessed. In these conditions, 48 h after ischemia, lesions of control mice were similar to those produced by the aforementioned 30 mins MCAO under xylazine–ketamine anesthesia. Lactate administration had a small, nonsignificant (P=0.06), protective effect (from 112±30 mm3 for control mice to 94±18 mm3 for lactate-injected mice) (Figure 3A). Once again, when locating the lesion among the cortex, striatum, and hippocampus, no protection after lactate treatment was observed in the cortex and hippocampus. However, lactate induced a significant reduction in lesion size in the striatum (from 19±3 mm3 for control mice to 14±4 mm3 for lactate-injected mice, P=0.036) (Figure 3B). Lactate administration significantly improved the neuroscore at 48 h from 5.3±1.7 for control mice to 3.6±1.3 for lactate-treated mice (P=0.046) (Figure 3C).
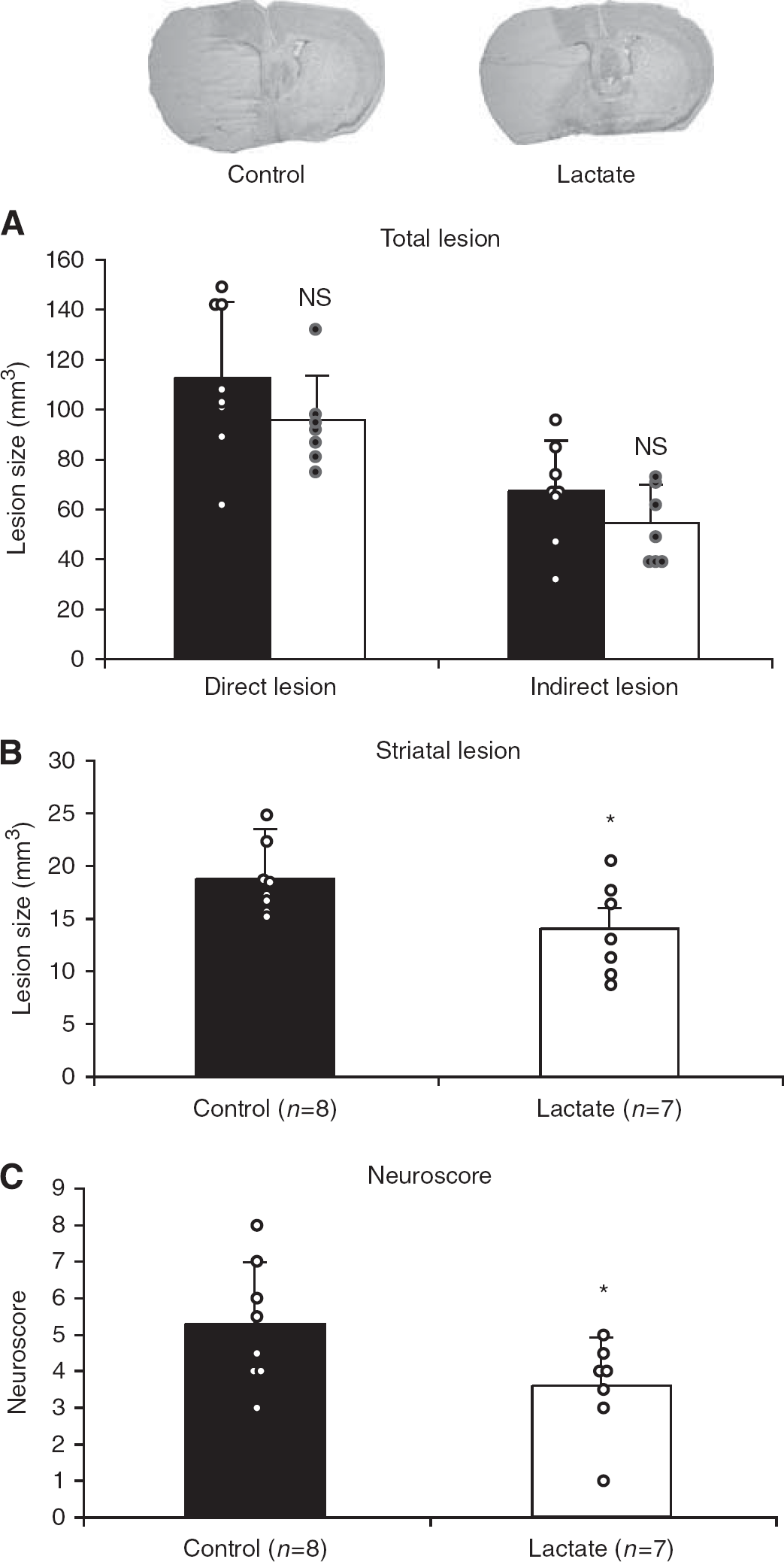
Injection of 2 μL of 100 mmol/L
In an attempt to delineate the therapeutic window of lactate administration, 11 mice were injected intracerebroventricularly with either PBS or 100 mmol/L lactate (2 μL of each) 60 mins after the end of 60 mins MCAO under isoflurane anesthesia and were killed at 48 h. The secondary anesthesia required for the intracerebroventricular injection with xylazine–ketamine by itself had a protective effect, resulting in lesions of 78±22 mm3 compared with those of 112±30 mm3 as described in Figure 3A. Lactate injection at this later time point was not significantly neuroprotective, neither on the total lesion (Figure 4A) nor in the striatum (Figure 4B). However, it had a striking effect on the behavior of these mice, decreasing their neuroscores very significantly (P=0.009) at 48 h, from 4.9±2.2 in control mice to 1.4±0.9 in lactate-injected mice (Figure 4C). Moreover, Table 1 shows for this set of mice that control mice lost significantly more weight (P=0.04) than did lactate-injected mice, reflecting a better neurologic outcome in treated animals.
Effect of anesthesia and lactate on physiological parameters
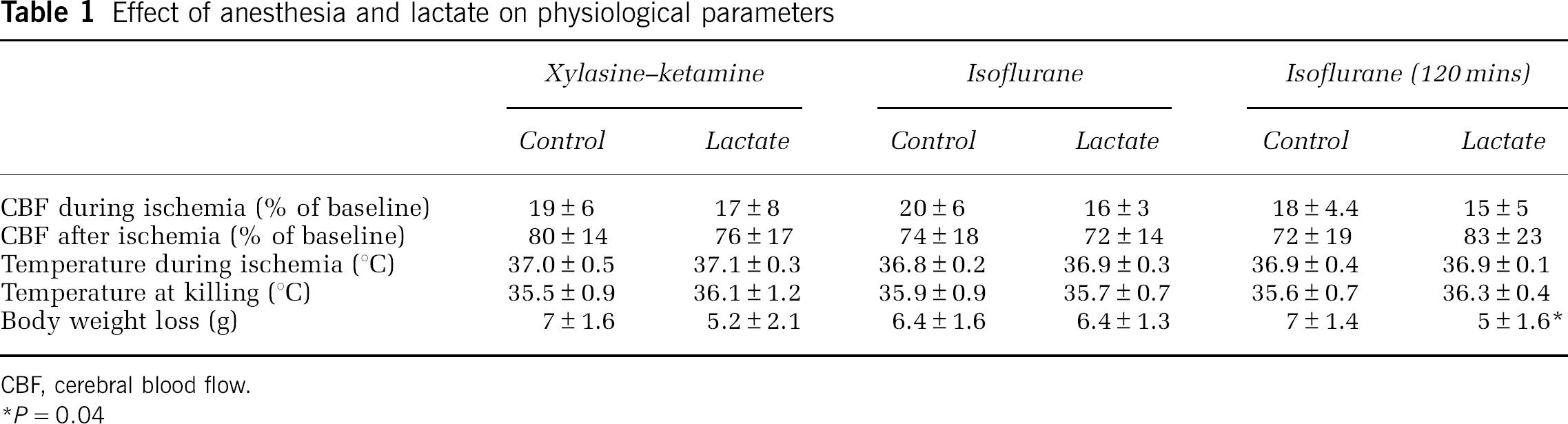
CBF, cerebral blood flow.
P=0.04
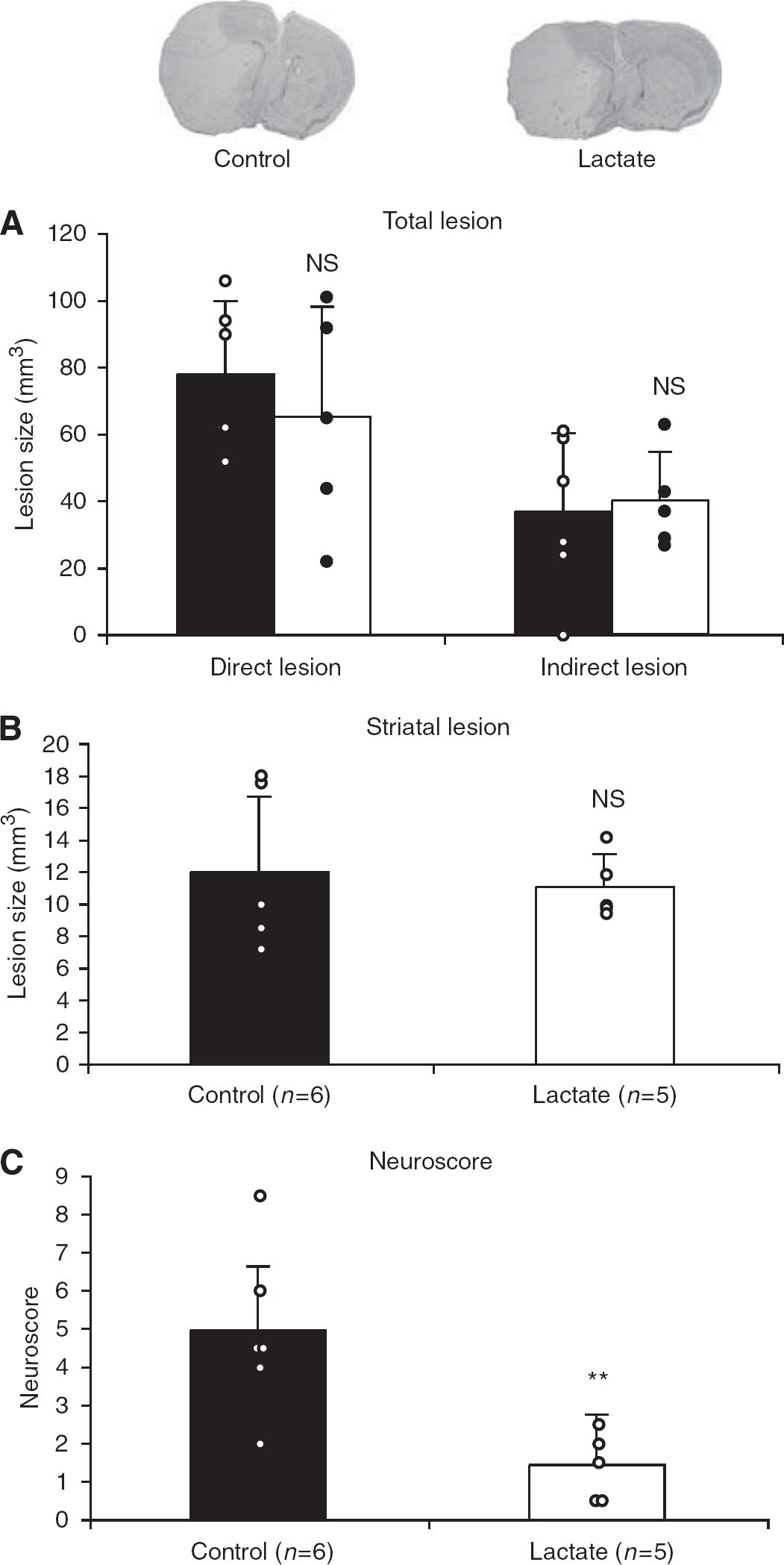
Injection of 2 μL of 100 mmol/L
With other neuroprotective agents tested after ischemia, in the majority of cases, protection appears in the cortex, which is less severely injured by MCAO. In an attempt to understand why lactate protected mainly the striatum in this study, we measured lactate concentrations in the striatum and cortex of healthy mice by MR spectroscopy before and after injecting 2 μL of 100 mmol/L
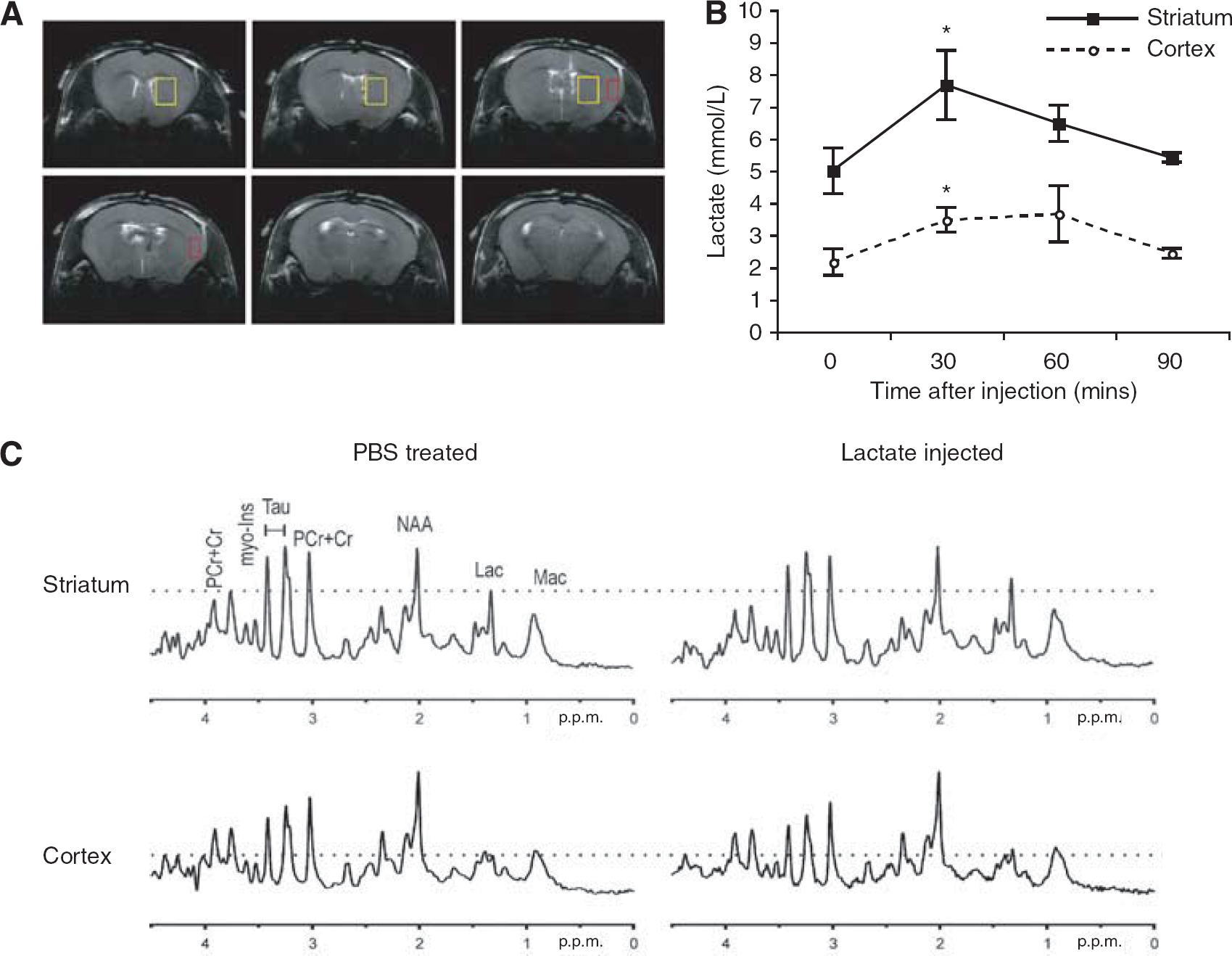
Intracerebroventricular injection of 2 μL of 100 mmol/L
Discussion
The results presented in this study show, using three different models, that lactate administered at early time points after ischemia can indeed protect against cell death, decrease lesion size, and improve neurologic outcome.
In vitro, the addition of 4 mmol/L Na
The conditions we used for MCAO in this study induced very large lesions covering more than half of the left hemisphere. In these conditions, the penumbra is reduced to a small rim around the ischemic core and neuroprotection might be difficult to obtain (as reviewed in Hossmann (2008)). The 40% reduction in lesion size that was observed under xylazine–ketamine anesthesia is thus very impressive. With isoflurane anesthesia (Sakai et al, 2007), a longer duration of ischemia resulted in a similar lesion size, which was ascribed to isoflurane protection (Sakai et al, 2007). Under these conditions, in which isoflurane protects, an additional beneficial effect of lactate may be more difficult to show. Moreover, isoflurane neuroprotection might already involve lactate, because lactate has been shown to increase in the medium of cells treated with isoflurane (Brabec et al, 1984). Although the total lesion size was not affected by lactate treatment under isoflurane anesthesia, lactate significantly reduced the striatal lesion with both types of anesthesia, when administered at reperfusion. Striatal protection was unexpected, as this structure is the first to be damaged after a milder ischemia (Lei et al, 2009), probably because it is less well irrigated by collateral circulation. Therefore, we speculated whether the intracerebroventricularly injected lactate reached the striatum and cortex evenly. The results presented in Figure 5 show that lactate was distributed in both structures. However, basal concentrations were significantly different between these two regions as already observed by other groups (Tkac et al, 2004). Thus, the 1 mmol/L increase observed in the cortex might not be sufficient enough for protection, consistent with in vitro experiments (Figure 1), whereas the striatal increase between 2 and 4 mmol/L might be more efficient. However, at 48 h, the cortical lesion may still be evolving in our model and thus a protection of the cortex by lactate may become apparent only at later time points.
The protective effect of lactate treatment observed in this study might be surprising, because endogenous lactate is known to increase dramatically after ischemia (Malisza et al, 1998). Without lactate injection, lactate has been shown to increase during ischemia because of anaerobic glycolysis, normalizing within 15 mins after reperfusion in the rat after 2 h MCAO (Higuchi et al, 1996) and strongly increasing from 1 h after reperfusion, from 12 mins global ischemia or 2 to 3 h MCAO in the rat (Michaelis et al, 1999; Thoren et al, 2006). One possible explanation for a lactate decrease at reperfusion could be the preferential consumption of lactate when adenosine triphosphate stores deplete. Indeed, adenosine triphosphate is required for the phosphorylation of glucose to produce glucose-6-phosphate for glycolysis, whereas lactate can directly be metabolized without an investment of adenosine triphosphate (Schurr, 2006). Moreover, Schurr et al (1997a, 1997b, 1997c) showed that, at least in vitro, lactate produced by astrocytes can be used after hypoxia and is even preferred to glucose, which our results support. At later time points, increased lactate is probably not because of a lack of blood supply and anaerobic metabolism, as glucose levels in the ischemic core are already normalized 2 h 30 mins after reperfusion (Lei et al, 2009), but might be due to the metabolism of macrophages infiltrating into the brain after ischemia (Petroff et al, 1992) or may possibly reflect the fact that lactate transporters (Kuhr et al, 1988; Walz and Mukerji, 1988) have changed, so that the lactate clearance rate is altered relative to the synthesis rate. On the basis of our in vitro experiments, lactate seems to be neuroprotective at low doses and toxic at higher concentrations. Thus, lactate increases induced by ischemia might protect in the beginning and become toxic later when it reaches higher concentrations. After 1 h of reperfusion in the rat, lactate increases between 30 (Michaelis et al, 1999) and 300% (Thoren et al, 2006), depending on the model. Owing to technical limitations, we did not measure lactate concentration 1 h after reperfusion in our model. However, under isoflurane anesthesia, a 30-min MCAO induced a 3 mmol/L increase in lactate 2 h after reperfusion compared with that in control animals (Lei et al, 2009), which is similar to the increase observed after lactate injection (Figure 5). As lactate has been shown to increase rapidly during the first 20 mins of ischemia, but to be stable thereafter (Hopwood et al, 2005), the concentration of lactate at reperfusion should be similar in our 30- and 60-min models of MCAO. It might thus be possible that endogenous lactate is already increased at 1 h of reperfusion and available in a sufficient amount to fulfill metabolic requirements; therefore, lactate administration is not beneficial compared with endogenous lactate. This might explain why it influenced neither the total nor the striatal lesion. Conversely, Higuchi et al (1996) showed a decrease in lactate concentration from the end of ischemia until 15 mins of reperfusion, but no change thereafter until 1 h after reperfusion from 1 h of ischemia in the rat. If the lactate evolution in our model is similar, endogenous lactate concentration should be higher at reperfusion than 1 h later; thus, the difference in protection observed between the two time points of lactate injection could not be explained. However, these are only speculations and we will need to measure the exact early evolution of lactate after ischemia in our model and control plasma glucose to better understand a possible relationship between endogenous and injected lactate. Conversely, the expression of MCTs (monocarboxylate transporters), which are necessary for lactate internalization, has been shown to be influenced by ischemia, decreasing in some cells and increasing in others (reviewed in Pierre and Pellerin (2005)). However, MCT 1 and 2 seem to be stably expressed after ischemia, at least for 4 h (Zovein et al, 2004), as well as lactate dehydrogenase, which might even increase (Calvert et al, 2006). Thus, neither a reduction in MCTs nor a reduction in lactate dehydrogenase should be the reason for the lack of effect of lactate injected 60 mins after reperfusion in our experiments. However, the short therapeutic window we observe with lactate in this severe ischemic model is not surprising, because the length of the therapeutic window of other neuroprotective agents has been shown to decrease with severity of ischemia (e.g., D-JNKI1; Hirt et al (2004)).
In view of a potential clinical application, the substantial improvement in neurologic outcomes is extremely interesting. In our study, we observed a significant improvement in the neuroscore assessed at 48 h after ischemia when lactate was administered at reperfusion, and even more, when it was administered 1 h after reperfusion. In addition, with this later administration, mice lost significantly less weight than did control animals, which is another indication of improvement of the animal's health status (Modo et al, 2000). However, there is a discrepancy between the lesion volume and the neurologic outcome of these mice (injected at 120 mins after the onset of ischemia). This could be explained if lactate had different effects depending on the moment of administration. When injected at reperfusion, it might be metabolized and might promote the survival of neurons, as reflected in the observed decrease in lesion size. There is no effect on lesion volume 1 h after reperfusion, although lactate supply could possibly improve the performances of suffering, but surviving, neurons. However, its beneficial effect on neurologic outcome suggests that it could influence other mechanisms that still need to be discovered; this hypothetical action is beyond the scope of this study.
Knowing that lactate can be used as an energy substrate by neurons (Schurr et al, 1988), and that it can be used even when adenosine triphosphate stores are depleted, we believe that at least a part of the beneficial effect of lactate treatment might be due to its metabolic utilization. However, we have no experimental data to support this hypothesis and we cannot exclude the presence of another mechanism. For example, lactate might influence osmolarity, which could have beneficial effects on edema when moderate (with 4 mmol/L lactate), but could become toxic with higher concentrations of lactate. Another explanation for the in vivo results could be the lactate effects on CBF. Shackford et al (1994) showed a decrease in cerebral vascular resistance leading to increased CBF after lactate infusion. Moreover, peri-infarct depolarizations after ischemia, which are increased with hypoglycemia, have been shown to lead to vasoconstriction (Strong et al, 2007). Therefore, addition of lactate may not have a direct survival effect, but may give energy to reduce the number of peri-infarct depolarizations and reduce the associated vasoconstriction. Our in vitro data, however, show that the neuroprotection observed after lactate treatment is not only due to CBF changes. In any case, understanding the mechanism underlying the protective effect of lactate injection is of great interest and will be a subject for further studies.
In conclusion, lactate administration directly to the brain after reperfusion can effectively protect against ischemia-induced cell death and disability. In view of a potential clinical application, the next step will be to test whether lactate can have the same beneficial effects when injected intravenously, which can be expected knowing that blood lactate is an important energy source for the human brain (van Hall et al, 2009).
Footnotes
Acknowledgements
The authors thank Dr Melanie Price for proofreading the manuscript, Aurélie Calame for her help, and Dr Luc Pellerin for helpful advice.
The authors declare no conflict of interest.