
Abstract
Brain N-acetylaspartate (NAA) can be quantified by in vivo proton magnetic resonance spectroscopy (1H-MRS) and is used in clinical settings as a marker of neuronal density. It is, however, uncertain whether the change in brain NAA content in acute stroke is reliably measured by 1H-MRS and how NAA is distributed within the ischemic area. Rats were exposed to middle cerebral artery occlusion. Preischemic values of [NAA] in striatum were 11 mmol/L by 1H-MRS and 8 mmol/kg by HPLC. The methods showed a comparable reduction during the 8 hours of ischemia. The interstitial level of [NAA] ([NAA]e) was determined by microdialysis using [3H]NAA to assess in vivo recovery. After induction of ischemia, [NAA]e increased linearly from 70 µmol/L to a peak level of 2 mmol/L after 2 to 3 hours before declining to 0.7 mmol/L at 7 hours. For comparison, [NAA]e was measured in striatum during global ischemia, revealing that [NAA]e increased linearly to 4 mmol/L after 3 hours and this level was maintained for the next 4 h. From the change in in vivo recovery of the interstitial space volume marker [14C]mannitol, the relative amount of NAA distributed in the interstitial space was calculated to be 0.2% of the total brain NAA during normal conditions and only 2 to 6% during ischemia. It was concluded that the majority of brain NAA is intracellularly located during ischemia despite large increases of interstitial [NAA]. Thus, MR quantification of NAA during acute ischemia reflects primarily changes in intracellular levels of NAA.
Keywords
N-Acetylaspartate (NAA) in the brain can be measured noninvasively by proton magnetic resonance spectroscopy (1H-MRS). As NAA in the mature brain is located almost exclusively within the nerve cells (Nadler and Cooper, 1972; Urenjak et al., 1992, 1993), the decreased NAA level observed, e.g., in cerebral infarcts, Alzheimer's disease, and Parkinson's disease, is believed to demonstrate a corresponding neuronal loss (for review see Tsai and Coyle, 1995). There are two main prerequisites for the use of NAA as a marker of neuronal density in clinical settings: The content of NAA in brain tissue always corresponds to the decrease in neuronal density, and the NAA level is reliably measured by the MR technique. This is certainly not always the case. In acute focal cerebral ischemia, there is an almost complete loss of viable neurons within the infarct core a few hours after start of ischemia (Garcia et al., 1993; Dereski et al., 1993), whereas the NAA level is reduced by only 20 to 30% (Sager et al., 1995; Gyngell et al., 1995). After 24 hours the reduction amounts to 80 to 90%, which is more in line with the extent of neuronal damage (Monsein et al., 1993; Sager et al., 1995). Thus, in conditions of acute cerebral injury, the use of NAA as a neuronal marker gives a false low estimate of the extent of neuronal damage. The reason is obviously that the disappearance of NAA is retarded in the developing infarct, but it is unknown whether NAA is retained in the non-viable neurons or how it is distributed in other compartments of the brain. This is important not only for understanding the dynamic changes in NAA during acute brain ischemia but also for interpreting quantitation of NAA by 1H-MRS.
The level of NAA determined by proton spectroscopy is bound to yield too high values because the technique includes other compounds such as glutamate, glutamine, and N-acetylaspartylglutamate (Howe et al., 1993; Henriksen, 1995). It is, however, uncertain whether the ischemic decrease of brain NAA level with time, measured by 1H-MRS, is comparable with the decrease determined by more specific, but invasive, whole-tissue measurements by use of HPLC.
In the present work, we determined the tissue content of NAA in the developing infarct in rat brain after middle cerebral artery occlusion (MCAO) by biochemical means and by localized in vivo 1H-MRS. In addition, the NAA level was measured in the interstitial space using the microdialysis technique to assess the NAA distribution within the infarct.
MATERIALS AND METHODS
General animal handling
Male Sprague-Dawley rats (350 to 410 g) were used. Anesthesia was, in the microdialysis experiments, induced by intraperitoneal pentobarbital (Mebumal, 50 mg/kg) and in 1H-MRS experiments with a 2-mL subcutaneous injection of a mixture of atropine (9%), Hypnorm (11%), Dormicum (23%), and distilled water (57%). A polyethene catheter was placed in the tail artery for blood pressure measurement and blood sampling. Arterial blood gases and pH were measured regularly to ensure that Pa
Induction of brain ischemia
We used the monofilament occlusion of the MCA as described by Koizumi et al. (1986) and Memezawa et al. (1992) to induce focal cerebral ischemia. The rats were placed in the supine position, and a lateral longitudinal incision extending from the clavicula to the head was made. The adipose tissue and muscles were gently retracted, and the common (CCA), external (ECA), and internal (ICA) carotid arteries were exposed. The CCA was ligated proximally and the ECA distally to the bifurcation by 4-0 sutures. The ICA was reversibly closed by a vascular clip just proximal to the point where it divides into the ICA and the pterygopalatine artery (PA). A loose suture was placed around the CCA, and a small incision was made in the CCA to insert the catheter (o.d. 0.61 mm) containing the monofilament (diameter 0.28 mm) to enter the ICA. The catheter-monofilament unit was advanced through the ICA, past the PA, to the basis of the skull. From this point the monofilament was introduced ~1 cm into the brain, reaching and hence occluding the MCA. This produces large infarcts, which comprise the medial and lateral portions of the striatum and most of the neocortex. Sham-operated rats underwent the same procedure except for the final MCAO.
Permanent global ischemia was induced by cardiac arrest by infusing 1 mL 150 mmol/L KC1 via a catheter (o.d. 0.96 mm) inserted into the jugular vein.
Synthesis of [3H]NAA
[3H]NAA was synthesized and purified by the following procedure:
Microdialysis
In vitro calibration. The microdialysis probes (Carnegie; 3 mm concentric, o.d. 0.5 mm) were calibrated prior to animal experiments by determining the relative loss (RL) of either [14C]mannitol (0.5 to 1 µCi, specific activity 58 mCi/mmol; Amersham Life Science, U.K.), 3H]NAA (l to 3 µCi, specific activity 250 mCi/mmol), or NAA (50 to 100 µmol/L). The probes were placed in Dulbecco's phosphate-buffered saline (PBS; Gibco) at 37°C, solidified by 0.5% agar (Bacto-Agar; Difco Laboratories, U.S.A.), and perfused with Ringer's solution (in mmol/L: 145 NaCl, 3 kCl, 1 MgCl2, 1.2 CaCl2) at a flow rate of 1 µL/min. The RL was calculated as follows: RL = (Ci − Cout)/Ci, where Ci is the inlet concentration of NAA and Cout the outlet concentration of NAA.
Only probes that exhibited RLs of 25 to 30% were used in the in vivo experiments. To measure [14C]mannitol and [3H]NAA in dialysate samples, 3 ml Pico Aqua (Packard) was added to each sample and the radioactivity was counted on a Packard liquid scintillation counter. Dialysate NAA was detected as described by Sager et al. (1997).
In vivo studies. Following anesthesia, the head was fixed in a stereotaxic frame. The skull was exposed and rinsed by 0.1% H2O2 before a trephine opening was drilled in the parietal bone. The dura was carefully incised by a 23G cannula, and the guide cannula was positioned in the brain using a micromanipulator (coordinates: 0.4 mm anterior and 3 mm lateral to bregma and 0.2 mm ventral to the brain surface; Paxinos and Watson, 1982) and fixed to the skull by use of dental cement (Simplex Rapid). The microdialysis probe was guided to the right striatum (6.2 mm ventral to brain surface) while perfused with Ringer's solution at 1 µL/min. Samples of 20 µL were collected and used for NAA determination (n = 4) or measurements of the RL of [3H]NAA (1 to 3 µCi) and the RL of [14C]mannitol (0.5 to 1 µCi) (n = 4-5). Following a 2-hour stabilization period, MCAO or global ischemia was induced as described above and maintained for 7 hours. To minimize heat loss, the head and body were wrapped in aluminum foil and covered by isolating Styrofoam while the body temperature was maintained by the servo-controlled heating pad.
After the ischemic period, the rats subjected to focal ischemia were then killed by an overdose of Mebumal. The chest was opened, and a cannula was inserted into the exposed aorta to perfuse the brains with a saline solution containing 10% 2,3,5-triphenyltetrazolium chloride (TTC) followed by a saline solution with 0.1% malachite green. The brains were removed and proper MCAO was verified by lack of malachite green staining in the vascular bed and the size of infarction from the tissue not colored by the TTC (Liszczak et al., 1984; Hatfield et al., 1991). Animals in which the filament caused bleeding were easily identified and discarded. The brains were immersion fixed in buffered formalin (pH 7.4) for 3 to 4 days before being embedded in a 3% agar solution and subsequently sliced in 1-mm-thick sections. The sections were graphed by a color camera into a Macintosh computer by use of the NIH Image shareware program. The hemispheric and infarct volumes were calculated by multiplying the respective areas with the slice thickness.
Procedure for determining NAA tissue levels
The rats were anesthetized and subjected to proximal MCAO as described above. After endotracheal intubation, the rats were placed in a circularly polarized transmit/receive head coil. The animals were fixed using an integrated stereotaxic head holder. The rectal temperature was monitored and automatically kept at 37°C by circulation of warm water on which the rat was placed. Within the scanner, the rats were mechanically ventilated with N2O/O2 (0.4:0.2 L/min). The halothane percentage (0.5 to 1.5%) was gradually increased as the initial anesthesia subsided, keeping the blood pressure between 80 and 100 mm Hg.
The NAA concentration ([NAA]) in the infarct was quantified in the striatum by 1H-MRS as well as by HPLC in sham-operated rats (n = 3) and after 3 hours (n = 6), 5 hours (n = 7), and 8 hours (n = 6) of MCAO. The infarct volumes were determined from diffusion-weighted imaging (DWI) hyperintensity. The metabolite concentration in the corresponding contralateral region in each rat served as control. The rats were decapitated and the brains frozen in dry ice. The brains were divided by a frontal cut through the middle of the infarct. Ten-micron-thick frozen sections were cut from the caudal part and stained with hematoxylin/eosin. The cranial part was used to quantify NAA in normal and infarcted tissue by HPLC. Tissue samples for HPLC analysis were chosen from the corresponding location of the spectroscopic voxel on the DWI, dissolved in 0.1 mol/L perchloric acid, extracted, and measured by HPLC as described by Sager et al. (1995). The NAA content in each sample was expressed as millimoles per kilogram of tissue wet weight.
MR instrumentation
The MR experiments were performed using a Sisco 4.7T nuclear MR system, equipped with a 12.5-cm Oxford insert gradient system, having a maximum gradient of 100 mT/m.
A T1-weighted sagittal spin echo scout image [field of view 3 × 6 cm2, matrix 128 × 128, echo time (TE) 16 milliseconds, repetition time (TR) 400 milliseconds, 2 averages) was obtained and used as a localizer for multislice diffusion-weighted images. The cranial and caudal boundaries on the sagittal T1-weighted image were selected (range 1.65 mm), giving 12 coronal slices with a slice thickness of 1.0 mm and a separation of 1.5 mm between the centers of each slice. Diffusion-weighted images were obtained by the following parameters: field of view 4 × 4 cm2, matrix 128 × 128, zero filed to 256 × 256, TE 80 milliseconds, TR 1.2 s, 2 averages. The measurements were performed with a diffusion gradient (d) of 10 milliseconds, a delay between the two diffusion-weighted gradients (D) of 36 milliseconds, and three different gradient amplitudes (b values of 0, 1,000, and 2,000 s/mm2).
With use of the diffusion-weighted images, a proper slice was selected so that a hyperintense area (ischemic territory) was present in both the preceding and the succeeding slide. For localized 1H-MRS, a 3 × 3 × 3-mm3 (27-µL) voxel was placed in the infarct or in the corresponding region of the contralateral hemisphere. The field homogeneity within the volume of interest was optimized with a resulting water spectrum line width of ~10 to 12 Hz.
For water-suppressed localized proton spectroscopy, in the contralateral and ipsilateral hemispheres, a localized point resolved spectroscopy (double spin echo) pulse sequence was used (Moonen et al., 1989) with the water suppression enhanced through effects (WET) water suppression module as described by Ogg et al. (1994). Numerically optimized waveforms were used for both the initial slice-selective 90° radiofrequency pulse and the slice-selective 180° radiofrequency pulse. The duration of the radiofrequency pulses was 2.5 milliseconds. The sequence was preceded by a six-pulse WET module using numerically optimized pulses. A crusher gradient duration of 5 to 37 milliseconds with a gradient strength of 5 mT/m was used. Eight and 128 acquisitions were used for water and metabolite spectra, respectively (TR 2.0 seconds). For metabolite and water spectra, the TE was 30, 50, 80, 136, and 272 milliseconds and 30, 50, 70, 100, and 136 milliseconds, respectively, giving a total scan time of ~25 minutes.
Proton spectra were phased and analyzed by an in-house-designed line-fitting computer program. The T2 decay of water and NAA in each experiment was fitted, assuming a single exponential decay of T2, to estimate the signal amplitudes at TE = 0. The concentration of total brain NAA was estimated using brain water content as an internal standard (Michaelis et al., 1993; Christiansen et al., 1993). The intensity of the unsuppressed water signal was related to the intensity of the NAA signal, assuming a 80% water content (≈44 mol/L) in the brain (Ernst et al., 1993).
Calculations and statistics
The interstitial [NAA] was determined from the in vivo RL of [3H]NAA according to the internal reference technique described by Scheller and Kolb (1991):
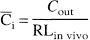
where Cout is the dialysate concentration of NAA and RLin vivo the relative loss of [3H]NAA in the brain and Ci, the interstitial NAA concentration.
The total tissue concentration of NAA in MR experiments was calculated using water as a internal standard by following equation (Christiansen et al., 1993):
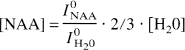
where IONAA and IOH2O are the intensity of NAA and water peaks at 0 TE, ⅔ is the fraction between the number of protons responsible for the MR signal in each compound, and [NAA] and [H2O] are the concentrations of NAA and water, respectively.
The relative distribution of NAA in the interstitial space was calculated as follows:
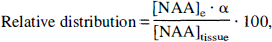
where α is the interstitial volume fraction and [NAA]e and [NAA]tissue are the interstitial and tissue NAA concentrations, respectively. The changes in α were derived from the changes in RL of the interstitial space marker mannitol, assuming that the RL of mannitol at basal conditions equals an α of 20% (Nicholson and Phillips, 1981; Lundbæk and Hansen, 1992).
Significant differences in dialysate and interstitial [NAA] from control samples (sample no. 6) were determined by repeated measures analysis of variance with Dunnett's multiple comparison posttest. Significant differences in temporal changes in tissue [NAA] changes were analyzed by unpaired t test. Differences in NAA reduction between methods were determined by paired t test. Differences in infarct volumes determined from DWI and TTC staining were determined by unpaired t test. All data are presented as means ± SD.
RESULTS
Location and volumes of infarcts
Figure 1 shows the correlation between the delineation of the ischemic area in a rat subjected to MCAO as determined by DWI and from hematoxylin/eosin staining of the same brain. This is in line with other studies showing a close correlation between the regional extent of infarcts depicted by MRI and histology (Allegrini and Sauer, 1992; Minematsu et al., 1992; Pierpaoli et al., 1993; Hoehn-Berlage et al., 1995). Thus, the fine correlation between the two methods ensured that proper tissue samples could be collected for biochemical measurement of NAA.
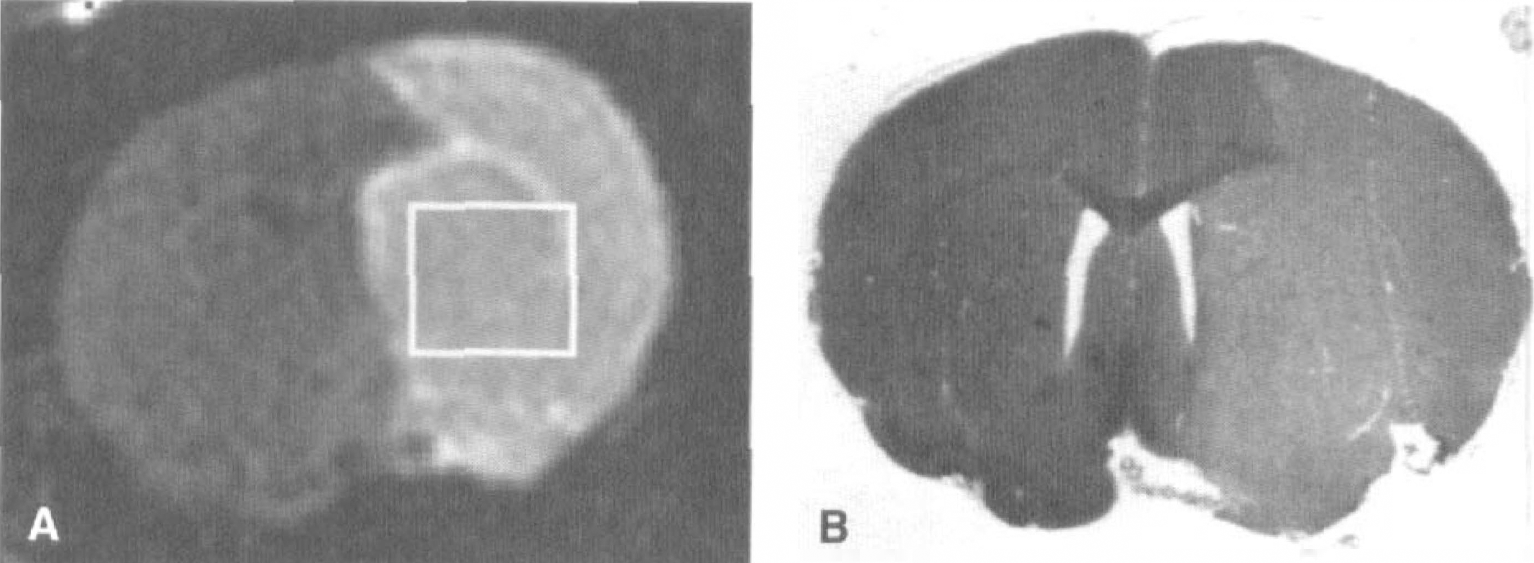
Location of the ischemic area produced by MCAO, as determined by DWI
The infarct sizes were determined from both DWI and from TTC-stained brains. In the microdialysis experiments, the size of the infarcts after 7 hours of MCAO as measured from the TTC staining was 246 ± 36 mm3. This was not different from the infarct volumes as determined from DWI after 8 hours of ischemia: 260 ± 50 mm3 (P > 0.05).
Time course of changes in tissue NAA concentration
Tissue [NAA] was determined by localized 1H-MRS and HPLC in the same animal. Basal striatal [NAA] measured in the nonoccluded side was 7.96 ± 1.43 mmol/kg (HPLC, n = 20) and 11.09 ± 1.27 mmol/L (1H-MRS, n = 22) (P < 0.0001, paired t test). Figure 2 shows representative proton spectra in the contralateral and ipsilateral side of a rat after 3, 5, and 8 hours of MCAO. In the control side of the brain, peaks of NAA, choline, creatine, and myo-inositol are clearly seen. After 3 hours of MCAO, there was a pronounced signal amplitude elevation of lactate that was present for the rest of the ischemic period. At this moment there was a significant [NAA] reduction compared with the sham-operated rats (Fig. 3), whether determined by 1H-MRS or HPLC (P < 0.01 and P < 0.05, respectively). The reduction became more prominent during the experimental period. There was no difference at any time during the ischemic period in the relative reduction in [NAA] determined by either HPLC and localized 1H-MRS, although the initial concentrations were significantly different.
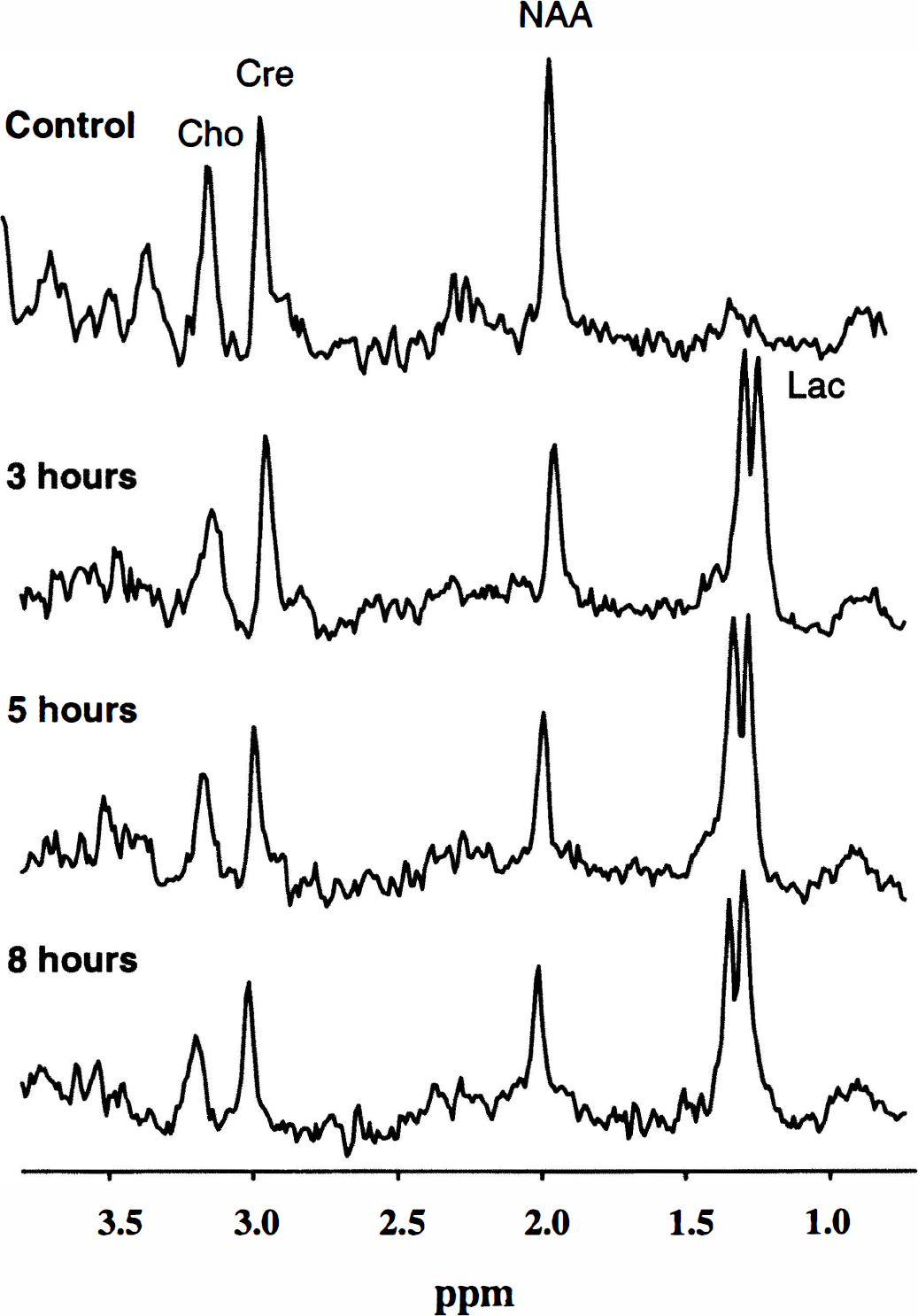
Representative water-suppressed proton spectra (TE 30 msec) obtained from a 27-µL voxel in contralateral (control) and ipsilateral striatum after 3, 5, and 8 hours of MCAO. NAA, creatine (Cre), and choline (Cho) are clearly detected in all spectra. Note the decline in NAA signal amplitude with time as well as the pronounced increase in lactate (Lac) after 3 hours of ischemia and later.
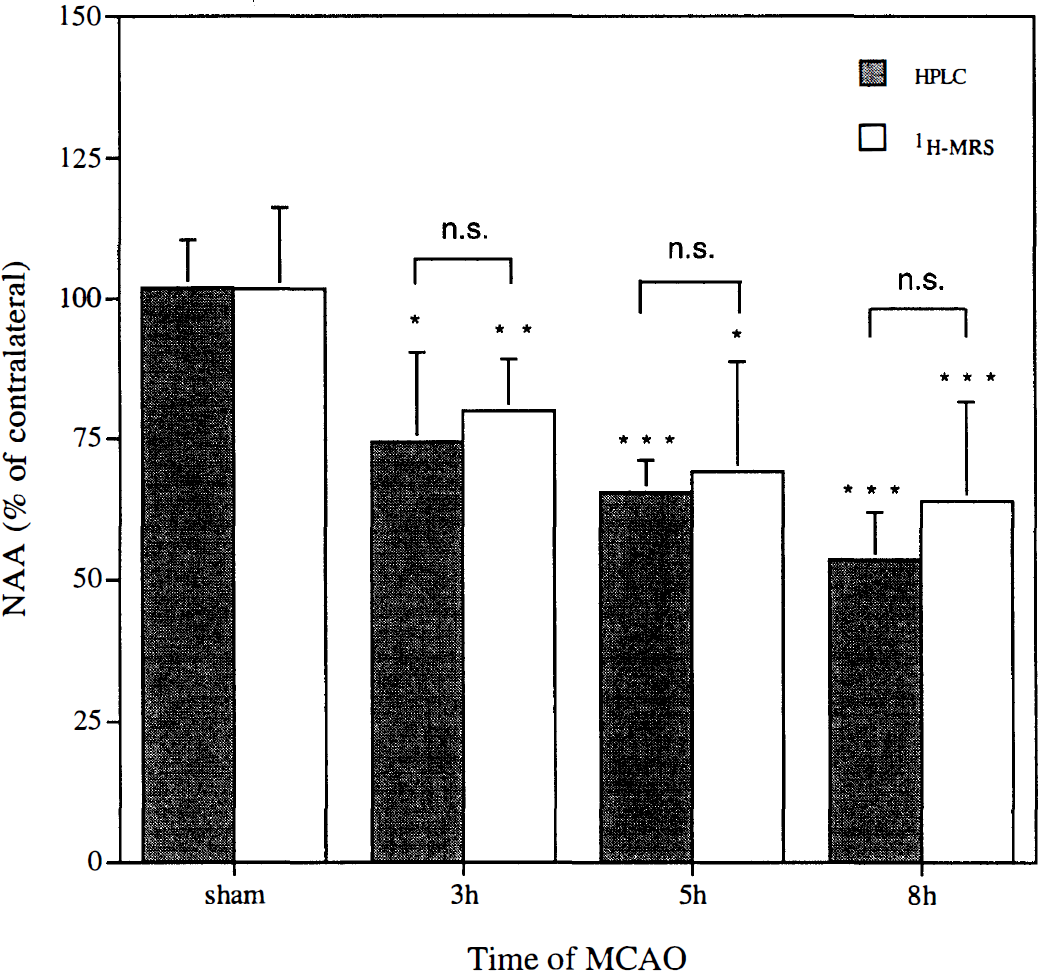
Temporal decline in infarct [NAA] in MCAO rats. The groups were as follows: sham-operated rats (n = 3), 3 hours (n = 6), 5 hours (n = 7), and 8 hours (n = 6). NAA was quantified using water as an internal reference (see Materials and Methods). The reduction in infarct [NAA] is expressed relative to the contralateral [NAA] at each time point. Significant NAA reduction was observed, with both methods, already after 3 hours of ischemia. Significant differences for each method compared with the corresponding group of sham-operated rats are denoted as follows: *P < 0.05, **P < 0.01, ***P < 0.001. There was no difference between HPLC (filled columns) and 1H-MRS (open columns) estimated NAA reduction at any time point (NS).
Microdialysis during focal ischemia
Dialysate [NAA]. Each dialysate sample was collected over a 20-minute period. The insertion of the microdialysis probe caused a transient increase in [NAA]d, which subsided after 1 hour. The value from the last sample before ischemia (i.e., sample no. 6; Fig. 4A) of 6.33 ± 3.84 µmol/L was considered basal [NAA]d. The start of focal ischemia induced a continuous increase in [NAA]d, which peaked at 65 µmol/L after ~2 hours. This level was maintained for another 2 hours after which [NAA]d steadily declined to reach 30 µmol/L after 7 hours.
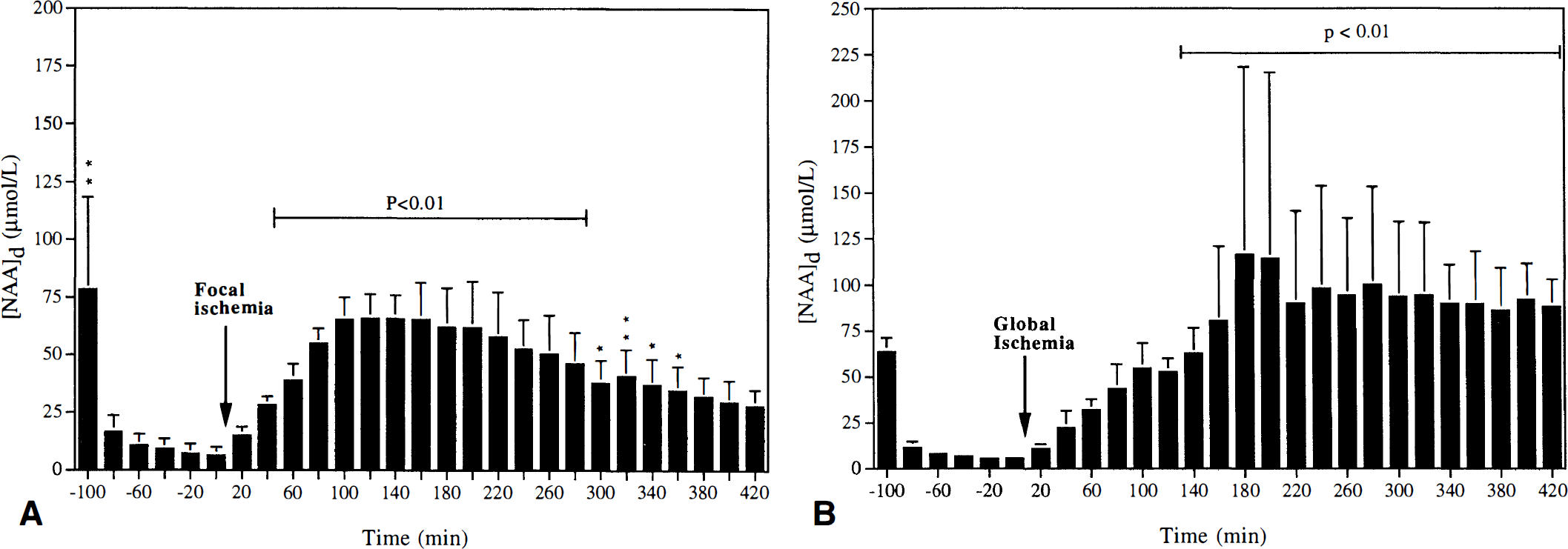
Change in dialysate NAA concentrations ([NAA]d) before and during 7 hours of focal (
Relative loss of [14C]mannitol and [3H]NAA. Changes in RL of [14C]mannitol (interstitial volume marker) and [3H]NAA (to determine the in vivo recovery) during focal ischemia are shown in Fig. 5A. During the initial 2 hours following probe implantation, RL of [3H]NAA and [14C]mannitol ranged from 9 to 13 and from 7 to 9%, respectively. Immediately following induction of focal ischemia, the RL of each compound was halved. The RL of [14C]mannitol and [3H]NAA remained reduced throughout the ischemic period, although with variations between 1.5 and 4.5% ([14C]mannitol) and 3 and 6% (3H]NAA). The RL of [3H]NAA was somewhat higher than the RL of [14C]mannitol throughout the ischemic period.
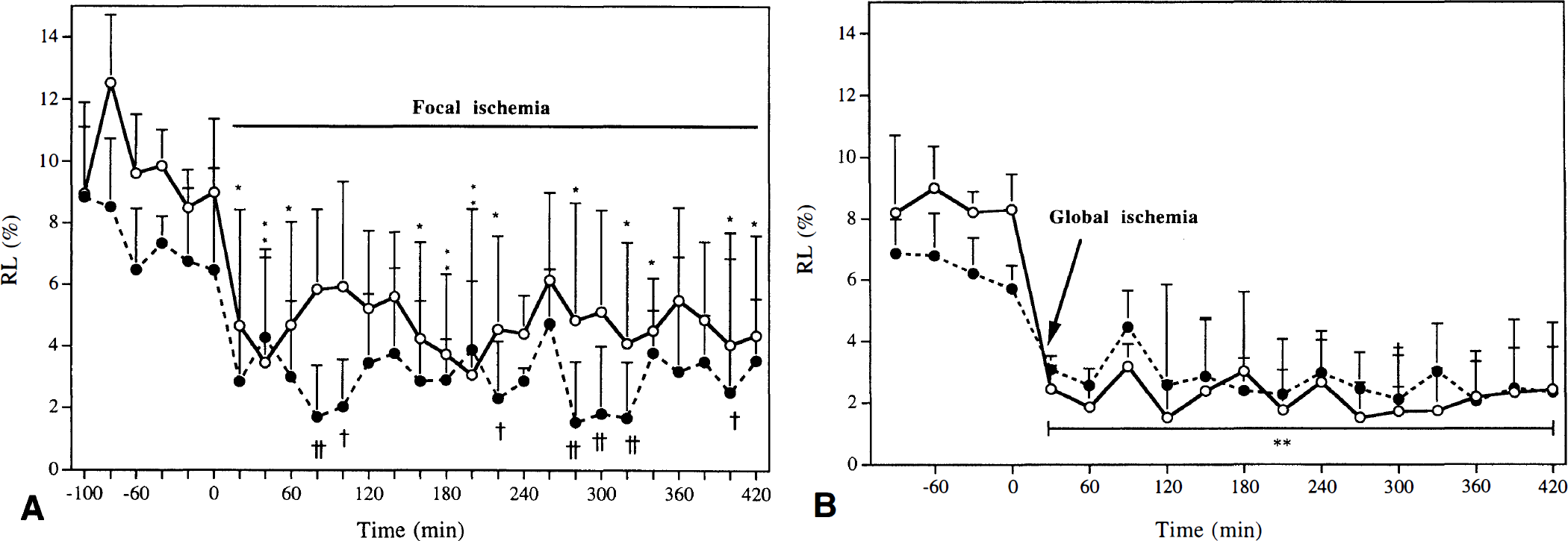
Time course of change in RL of [3H]NAA (open circles) and [14C]mannitol (filled circles) before and during 7 hours of focal
Interstitial [NAA]. We estimated, using Eq. 1, [NAA]e to be 70 µmol/L during control conditions (Fig. 6A). After induction of MCAO, [NAA]e increased steadily and significantly over 3 to 4 hours. After 40 minutes of ischemia, the value was ~800 µmol/L and after 3.5 hours ~2,000 µmol/L (P < 0.01). Thereafter, the [NAA]e declined steadily and reached a value of 650 µmol/L after 7 hours (P < 0.05).
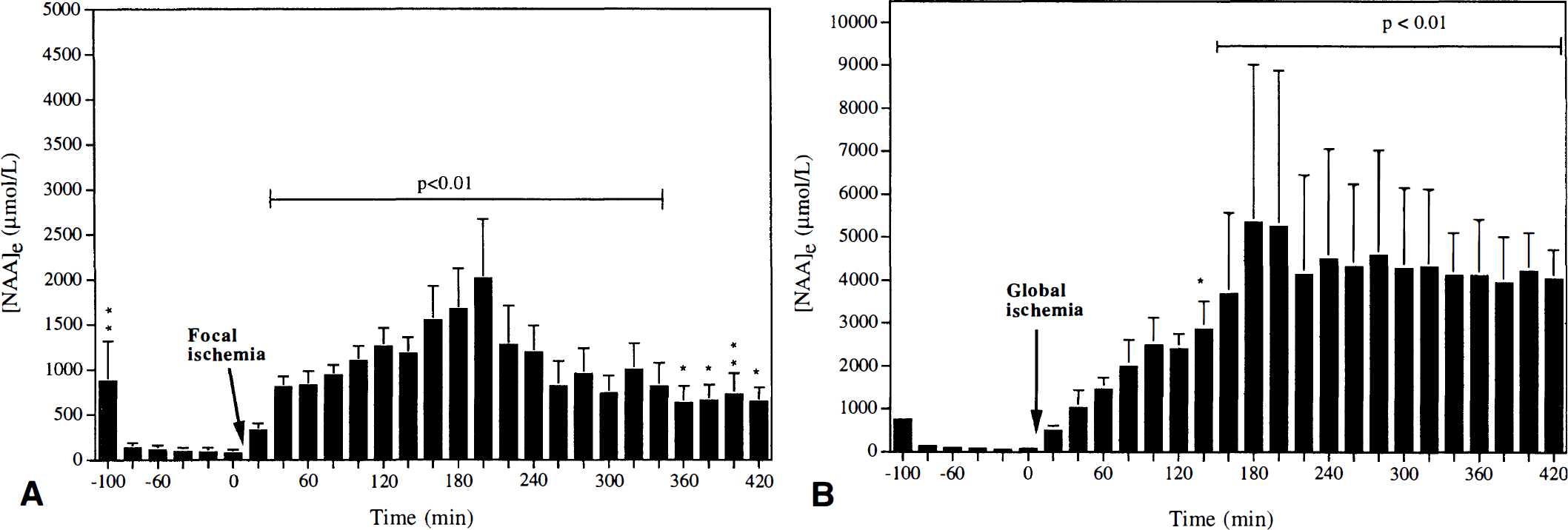
Calculated interstitial NAA concentration ([NAA]e) during focal
Microdialysis during global ischemia
Dialysate [NAA]. Each dialysate sample was collected over a 20-minute period. The basal [NAA]d was 5.6 ± 0.8 µmol/L. After cardiac arrest, [NAA]d exhibited a linear rate of increase to reach a level of ~100 µmol/L after 3 hours. In the remaining 4-hour period, [NAA]d remained close to this level (Fig. 4B).
Relative loss of [14C]mannitol and [3H]NAA. The dialysate samples were collected over a 30-minute period. As seen during focal ischemia, the RL of both [14C]mannitol and [3H]NAA declined during global ischemia (Fig. 5B). During normal condition, RL of [3H]NAA was 8 to 9%, whereas the RL of [14C]mannitol was 6 to 7%. Immediately following cardiac arrest, RL of [14C]mannitol was halved, whereas the RL of [3H]NAA was reduced by two-thirds to 3% (P < 0.01).
Interstitial [NAA]. A basal [NAA]e of 67 µmol/L was estimated from the RL of [3H]NAA (Eq. 1). After only 1 hour of ischemia, the level rose 20-fold and after 3 hours 70-fold to stabilize at a concentration of ~4 mmol/L. This level was maintained throughout the experiment (Fig. 6B). For calculation of interstitial [NAA] during normal and ischemic conditions, we used the RL average values of [3H]NAA of 8.4 and 2.1%, respectively.
DISCUSSION
Tissue NAA quantification by HPLC and 1H-MRS
The basal [NAA] as determined by 1H-MRS was significantly higher (11 mmol/L) than the concentration measured by HPLC (8 mmol/kg), which is in accordance with data from several other studies (Burri et al., 1990; Urenjak et al., 1992; Barker et al., 1993; Barker et al., 1994; Hüppi et al., 1995). The difference could arise from inherent problems with the MR signal assigned to NAA or the mode by which quantification of brain substances is performed by MR. Part of the assigned NAA peak in proton spectra originates from signal overlap from several different acetyl groups. 1H-MRS does not distinguish between N-acetyl resonance frequencies and other N-acetylated amino acids, such as N-acetylaspartylglutamate, N-acetylglutamate, and the N-acetyl moieties present on neuraminic acids, or glutamine/glutamate resonances (Frahm et al., 1989; Hennig et al., 1992; Howe et al., 1993; Henriksen, 1995). Together these compounds contribute ~3 mmol/L (Frahm et al., 1989), which could explain the difference between basal brain [NAA] determined by HPLC and 1H-MRS.
Quantitation of brain metabolites, by localized 1H-MRS, introduces a number of potential errors during focal ischemia. The MR quantitation was performed by relating the signal in question to the unsuppressed water signal (Barker et al., 1994; Henriksen, 1995; Christiansen et al., 1993; Hajek, 1995), assuming a constant brain water concentration of 800 g/L (80%) (Ernst et al., 1993). This is not valid in focal cerebral ischemia as focal water content increases from 82 to 86% (Henriksen, 1995; Dickinson and Betz, 1992). The change, however, is rather small compared with the observed changes in [NAA] and would also affect the quantitation by HPLC. The cerebral edema (of cytotoxic origin) may change the T2 relaxation time of NAA and hence reduce the NAA signal intensity. As we estimate the NAA signal intensity at TE = 0, any presumable T2 effect is eliminated. In addition, van der Toorn et al. (1995) showed the T2 relaxation time of NAA to be fairly constant in the first day after induction of focal ischemia in the rat. The sensitivity of the transmitter/receiver system, i.e., coil loading (B1 effects), may also be influenced. However, the use of the internal reference method alleviates this problem, as coil load effects impact all resonances equally. In the PRESS sequence, we used a TR of 2 seconds, which introduces saturation, because the T1 relaxation time of water is 1.9 seconds and that of NAA is 2 seconds (van der Toorn et al., 1995). Ischemia might change the T1 relaxation time to produce a false decrease in peak amplitude. But, as shown by van der Toorn et al. (1995), the T1 relaxation times of water and NAA were relatively stable during 3 to 6 hours of ischemia and hence changes in saturation effects are not important in this context. Finally, alterations in the local magnetic field, i.e., susceptibility changes, could affect the resonance frequency of NAA. Such alterations originate primarily from changes in the ratio of oxy- and deoxyhemoglobin, causing a broadening of the line width and thus reducing the NAA signal amplitudes. We observed no change in the line width of NAA, which remained between 10 and 12 Hz in all experiments, in agreement with van der Toorn et al. (1994).
The observed ischemic reduction in [NAA] agrees with our previous data using HPLC (Sager et al., 1995) and data obtained by 1H-MRS on focal ischemic rats (Gyngell et al., 1995; Higuchi et al., 1996) and cats (van der Toorn et al., 1994). The relative reduction in [NAA], as determined by the two methods, was not significantly different for the whole ischemic period, although the reduction at each time point, as determined by 1H-MRS, was somewhat lower (Fig. 2). This could be explained if the non-NAA compounds that are included in the NAA assigned peak in the spectra were more resistant to ischemia than NAA, but no data on this issue are available. Nevertheless, other data strongly suggest that the reduction of NAA content in ischemic brain tissue can be determined temporarily with 1H-MRS (van der Toorn et al., 1994; Gyngell et al., 1995; Higuchi et al., 1996). In accordance with this view, a significant correlation between the ipsilateral/contralateral ratios of NAA determined by either HPLC or 1H-MRS has also been shown in rats subjected to quinolinic acid-induced neuronal injury (Strauss et al., 1997).
Interstitial NAA in striatum
Control conditions. The microdialysis technique was employed to determine the interstitial levels of NAA ([NAA]e) in striatum. We measured the RL of [3H]NAA to determine in vivo recovery and the RL of the proper interstitial marker [14C]mannitol (Sisson and Oldendorf, 1971) to assess changes of interstitial space volume fraction, as described by Sager et al. (1997). The higher RL value of NAA demonstrates that NAA has a larger distribution volume than mannitol, which could be explained by either degradation of NAA in the interstitial space, cellular uptake, or clearance by capillaries. The fact that Sager et al. (unpublished observations) have demonstrated a specific NAA carrier in astrocytes suggests that glial uptake and subsequent degradation by the enzyme acylase II (Goldstein, 1976) is important. The basal [NAA]e of 67 to 70 µmol/L in the striatum is in concert with previous reports from Taylor et al. (1994) and Sager et al. (1997).
Ischemic conditions. The interstitial level of NAA reflects the balance between release from neurons and subsequent transport/degradation. During global ischemia, no clearance is possible via capillaries and diffusional transport is very limited, but there are still processes operating to degrade NAA (Sager et al., 1995). After induction of global ischemia, [NAA]e increased linearly and reached a maximal value of 4 to 5 mmol/L after 3 hours. This level was maintained for the rest of the observation period (Fig. 6B). The corresponding tissue concentration is expected to be 7.4 mmol/kg after 3 hours and 6.4 mmol/kg after 7 hours, as brain NAA in global ischemia is degraded at a rate of 0.2 mmol/kg/h, with a corresponding increase of aspartate content (Sager et al., 1995). It is noteworthy that the interstitial level after 3 hours (and later) of ischemia is less than the corresponding tissue concentration. This observation suggests that after 3 hours of global ischemia, a steady-state condition prevails in which there is a concentration gradient of NAA from neurons via the interstitial space to glial intracellular space in which NAA presumably is degraded.
Following experimental focal ischemia in the rat, the dorsolateral striatum undergoes complete infarction due to cessation of blood flow (Memezawa et al., 1992). It is therefore not unexpected that [NAA]e increased with a comparable time course as in global ischemia. The fact that no plateau phase was observed but that the peak level of 1 to 2 mmol/L was followed by a steady decline that reached 0.7 mmol/L after 7 hours (see Fig. 6A) shows that other mechanisms are active. As in global ischemia, [NAA]e is smaller than the corresponding tissue concentration, suggesting that an equilibrium is not reached. It is possible that a fraction of the total intracellular NAA may be compartmentalized (McIntosh and Cooper, 1965) and this NAA pool is not available for exchange. The attenuated change of [NAA]e could be due to a reduced neuronal release of NAA but also to the fact that diffusional transport out of the ischemic territory via the interstitial space and capillaries is now possible. In the surrounding tissue, viable glia cells are able to take up and subsequently degrade NAA. These processes maintain a concentration gradient that is important for the removal of NAA and explain why the rate of disappearance of NAA is faster in focal than in global ischemia (Sager et al., 1995).
The RL of [14C]mannitol declined approximately twofold during ischemia, demonstrating the well known reduction of the interstitial space volume fraction (Lundbæk and Hansen, 1992; van der Toorn et al., 1996a). Surprisingly, RL remained low throughout the experimental period both in global and in focal ischemia. This observation shows that mannitol was confined to the interstitial space and suggests that the cell membrane retains its low passive permeability during 7 hours of cerebral ischemia, which it eventually is bound to lose. Further experiments using other techniques, e.g., the tetramethylammonium microelectrode technique (Nicholson and Phillips, 1981), are needed to substantiate the present observations.
Distributional changes in brain NAA during ischemia
We have earlier shown that NAA cannot be used as a marker of viable neurons in acute ischemia due to the fact that a significant amount of NAA is still present in the evolving infarct (Sager et al., 1995). The present results strongly suggest that NAA mainly is confined to neurons in the developing infarct even though neurons, by unknown mechanisms, release NAA to the interstitial space. Despite the fact that [NAA]e reached the millimolar range during ischemia, this compartment represents an insignificant part (2 to 6%) of total brain NAA (see Table 1). Our data therefore support the assumption that the observed reduction of the apparent diffusion coefficient of NAA during focal ischemia is due mainly to changes in the intracellular diffusion characteristics of NAA (Wick et al., 1995; van der Toorn et al., 1996b). Whatever the mechanism responsible for this change in diffusion of NAA is, our results show that changes in the distribution of NAA during focal ischemia, presumably accompanied by decreased NAA apparent diffusion coefficient value, do not affect the quantitation of NAA by localized 1H-MRS.
Relative contribution of interstitial NAA to total brain in rats subjected to focal or global ischemia
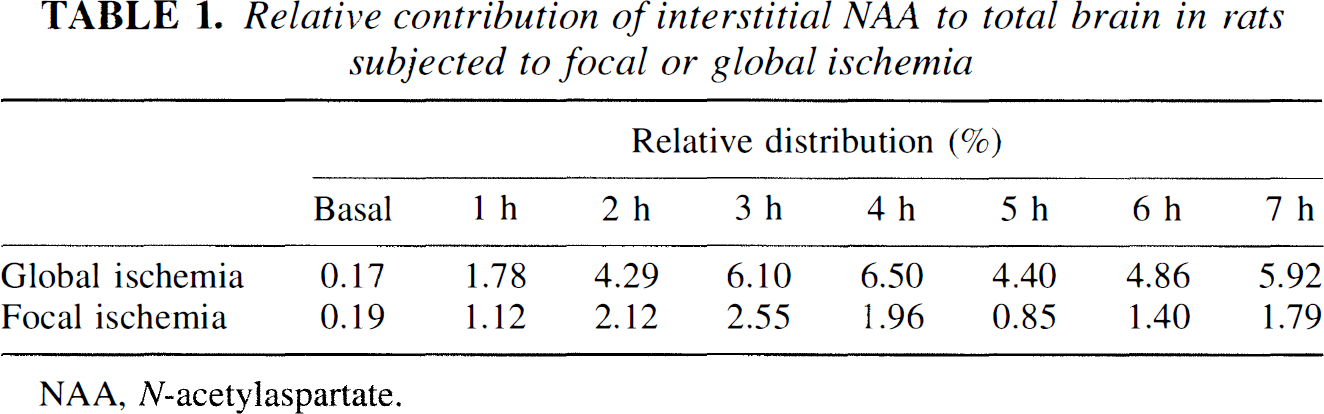
NAA, N-acetylaspartate.
Footnotes
Acknowledgment
The authors thank Ms. Helle Simonsen, Danish Research Center for Magnetic Resonance, for technical assistance.