
Abstract
Emerging evidence supports that premature infants are susceptible to both cerebral white and gray matter injury. In a fetal rabbit model of placental insufficiency, preterm rabbits at embryonic day 22 (E22) exhibited histologic evidence of gray matter injury but minimal white matter injury after global hypoxia-ischemia (H-I). We hypothesized that the dissociation between susceptibility to gray and white matter injury at E22 was related to the timing of appearance of late oligodendrocyte progenitors (preOLs) that are particularly vulnerable in preterm human white matter lesions. During normal rabbit oligodendrocyte (OL) lineage progression, early OL progenitors predominated at E22. PreOL density increased between E24 and E25 in major forebrain white matter tracts. After H-I at E22 and E25, we observed a similar magnitude of cerebral H-I, assessed by cortical microvascular blood flow, and gray matter injury, assessed by caspase activation. However, the increased preOL density at E25 was accompanied by a significant increase in acute white matter injury after H-I that coincided with enhanced preOL degeneration. At E29, significant white matter atrophy developed after H-I at E25 but not E22. Thus, the timing of appearance of preOLs coincided with onset of a developmental window of enhanced white but not gray matter susceptibility to H-I.
Introduction
Periventricular white matter injury (PWMI) is the major form of brain injury and the leading cause of cerebral palsy and cognitive deficits in survivors of premature birth (Back, 2006; Volpe, 2009). Immature human cerebral white matter is particularly susceptible to oxidative damage of a magnitude consistent with hypoxia-ischemia (H-I) (Back et al, 2005; Haynes et al, 2003). Within white matter lesions, the oligodendrocyte (OL) lineage shows maturation-dependent vulnerability to cerebral H-I (Back et al, 2002; Riddle et al, 2006; Segovia et al, 2008). Four successive stages of the OL lineage are defined by the expression of OL lineage-specific markers (Back, 2006). These are OL progenitors (NG2+, Olig2+, O4–), preoligodendrocytes (preOLs) (O4+O1–), immature OLs (O4+O1+) and mature OLs (MBP+). The predominant expression of preOLs coincides with the high-risk period for PWMI (Back et al, 2001), and preOLs are selectively vulnerable to oxidative stress (Back et al, 1998; Baud et al, 2004). Nevertheless, it remains controversial whether injury from H-I would occur if white matter were deficient in susceptible OL precursors.
To address this fundamental question in the fetus, we used a rabbit placental insufficiency model that generates global fetal H-I. Compared with the postnatal rodent, the fetal rabbit brain is larger, has more prominent white matter tracts, myelinates in a similar perinatal time course to human, and maturation of OLs begins antenatally (Derrick et al, 2007; Drobyshevsky et al, 2005). After exposure to global H-I in utero at E22, newborn rabbits develop hypertonic motor deficits that resemble early cerebral palsy (Derrick et al, 2004; Drobyshevsky et al, 2007).
Here, we report that the susceptibility of preterm rabbit cerebral white matter to H-I varies markedly with developmental age. Pronounced resistance of white matter to injury at embryonic day 22 (E22) coincided with a paucity of preOLs. During a distinct window in cerebral development between E24 and E25, the density of preOLs was coordinately increased in major forebrain white matter tracts. This preOL expansion was accompanied by markedly enhanced susceptibility of white matter but not gray matter to injury from H-I at E25. White matter injury was defined by increased cell death that predominantly involved preOLs. Hence, the timing of the expansion of the preOL population in fetal white matter coincided with enhanced susceptibility of preterm cerebral white matter to injury from H-I.
Materials and methods
Our study was approved by the Animal Review Committee of the NorthShore University HealthSystem Research Institute. All animals received humane care in compliance with the Principles of Laboratory Care formulated by the National Society for Medical Research and with the National Institute of Health's Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences.
Animal Surgery
In vivo global H-I of fetuses at E22 or E25 was induced by uterine ischemia in timed-pregnant New Zealand white rabbits (Myrtle's Rabbits, Thompson Station, TN, USA) as described earlier (Derrick et al, 2004). Briefly, uterine ischemia, which resulted in fetal hypoxia, was induced in anesthetized pregnant dams with a 4F Fogarty arterial embolectomy catheter (Baxter Healthcare Corp., Santa Ana, CA, USA). The catheter was introduced into the left femoral artery, advanced 10 cm into the descending aorta to above the uterine and below the renal arteries, and the balloon was inflated with 300 μL of saline. Right lower extremity blood pressure was monitored by Doppler (Mini Dopplex D500; Huntleigh Technology, Luton, UK) to ensure continued ischemia. At the end of the procedure, the balloon was deflated and the catheter removed. The femoral artery was repaired with 7-0 sutures and the skin closed with 3-0 sutures. The mother was returned to her cage. The dams underwent hysterotomy at the conclusion of the studies.
Hypoxia-Ischemia Protocol
Preterm animals at E22 (67% to 70% gestation) or E25 (75% to 80% gestation) underwent sustained uterine ischemia for 40 mins. This was performed to model an insult of placental insufficiency to premature infants. This protocol results in global hypoxia to the fetus that is accompanied by immediate fetal bradycardia (from 180 to 80 b.p.m.) and an immediate drop in microvascular blood flow to the cerebral cortex, as determined by laser doppler measurements. In the control dams there were 23, 16, and 16 fetuses at E23, E26, or E29, respectively. A total of 48 live rabbit fetuses were studied after H-I at E22 and survival until E23, 33 live rabbit fetuses were studied after H-I at E25 and survival until E26, 7 rabbit fetuses were studied after H-I at E22 and survival until E29, and 11 rabbit fetuses from two dams were studied after H-I at E25 and survival until E29.
Quantification of Fetal Cortical Microvascular Blood Flow
Three fetuses from three different dams were investigated at each age. The surgical procedure was as described earlier (Derrick et al, 2004). Briefly, pregnant dams at E22 and E25 were anesthetized with intravenous fentanyl (75 μg/kg/h) and droperidol (3.75 mg/kg/h) and bag and mask ventilation provided to maintain normal arterial blood gases. After spinal anesthesia with 0.75% bupivacaine, the fentanyl and droperidol were reduced to enable the dam to breathe spontaneously. A Fogarty balloon catheter was inserted into the left femoral artery. An abdominal incision of 3 cm was made, the uterus exposed, and a ∼0.6 cm uterine incision made to expose the fetal scalp. A 27G needle was inserted at a point 1.5 mm lateral to the midline in the parietal cortical area. The incision site was enlarged with a 25G blunt needle avoiding any hemorrhage. Cortical microvascular blood flow was recorded with a PF 5010 laser Doppler perfusion monitor (Perimed AB, Stockholm, Sweden). A calibrated Perimed probe (415-230) with probe holder (PH07-4) was inserted to 1 mm depth. The needle holder was secured to the scalp with Loctite Super-Glue. Baseline values were obtained for 5 to 10 mins before inflating the balloon in the femoral catheter to initiate H-I. Measurements were continued until 30 mins after the conclusion of a 40 mins period of H-I.
Immunohistochemistry
Brains were immersion fixed in the skull in 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4) for ∼24 h at 4°C, and then stored in PBS. The forebrain was serially sectioned free floating (50 μm) in PBS with a Leica VTS-1000 vibrating microtome (Leica Microsystems Inc., Bannockburn, IL, USA). The detailed immunohistochemical protocols to visualize specific stages in the OL lineage were as described earlier (Craig et al, 2003). Fluorescent double labeling to distinguish preOLs (O4 + O1 –) and immature OLs (O4 + O1 +) used a biotinylated O4 and O1 antibodies (Back et al, 2001). OL progenitors were visualized with antibodies against the NG2 proteoglycan (Back et al, 2001; de Castro et al, 2005) and against the nuclear transcription factor Olig2 (Back et al, 2006). Microglia and macrophages were visualized with biotinylated Bandeiria griffonia isolectin-B4 (L2140; 1:100; Sigma, Saint Louis, MO, USA) (Back et al, 2002). Caspase-mediated cell death was visualized with an activated caspase-3 (AC-3) antibody (9661B; 1:250; Cell Signaling Technology Inc., Danvers, MA, USA).
Quantification of Oligodendrocyte Lineage Stages During Normal White Matter Maturation
Total density of O4- or O1-labeled cells was determined in tissue sections counterstained with Hoechst 33324 to define regional boundaries of the corpus callosum, corona radiata, internal capsule (IC), external capsule (EC), and anterior commissure from normal control animals at E22 (n = 2), E23 (n = 8), E24 (n = 6), E25 (n = 4), E29 (n = 10), and E32/term birth (n = 4). Cells were counted in a blinded manner in a minimum of three adjacent coronal sections (Craig et al, 2003). White matter tracts were analyzed at the level of the mid-septal nuclei and the anterior hippocampal formation. At least four fields were counted for each tract. Cell profiles that contained a nucleus, visualized with Hoechst 33324, were counted with a × 40 (0.0625 mm2/field) objective equipped with a counting grid, and were converted to cells/mm2. The profiles of both intact-appearing and degenerating OL-lineage cells were counted and together comprised the calculation of total cell density for each stage. Degenerating cells were confirmed to contain a pyknotic nucleus by Hoechst staining and typically had morphologic changes that included fragmented processes and shrunken cell bodies with both plasma membrane and intracellular staining (Back et al, 2002).
Quantification of the Density of Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling and Hoechst-Labeled Nuclei
Degenerating nuclei in four major white matter tracts at the level of the anterior septal nuclei (corpus callosum, corona radiata, IC, and EC) were visualized by a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) method for fluorescent in situ end labeling of double-stranded DNA fragmentation (Apoptag S7110; Millipore, Billerica, MA, USA). Sections were counterstained with Hoechst 33324 to delineate regional boundaries and to distinguish intact nuclei from degenerating nuclei that showed pyknosis or chromatin condensation (Back et al, 2002). Strategies used to systematically avoid the introduction of counting bias were described earlier (Riddle et al, 2006). Every field containing TUNEL-labeled nuclei was counted with a × 40 objective by an investigator blinded to experimental conditions. Between 5 and 12, images were counted in a minimum of three adjacent sections, as defined by the size of the white matter tract.
Quantification of Cerebral Cortical Injury
Cerebral cortical injury in both hemispheres was analyzed in animals that survived for 24 h after H-I at E23 (n = 13) and E26 (n = 6) relative to control animals at E23 (n = 6) and E26 (n = 6). Cortical lesions were identified in image montages of AC-3-labeled cells generated from digitized images acquired with a × 10 objective on a Leica DMIRE2 inverted fluorescence microscope equipped with an automated x, y stage. Cortical lesions were identified in 8 of 13 animals at E23 and 6 of 6 animals at E26 in both the parasagittal cortex and the lateral ventral/piriform cortex at the levels of the mid-septal nuclei and the anterior hippocampal formation. Only animals with cortical injury were used for subsequent analyses. Such lesions were found to comprise single continuous collections of neighboring AC-3-labeled cells. Lesion boundaries were readily defined because of the high density of degenerating AC-3-labeled cells that were well demarcated between cortical layers 2/3 and the subplate (e.g., Figure 6C) as visualized by fluorescent nuclear counterstain with Hoechst 33324. The boundary criteria excluded occasional AC-3-labeled cell outliers that exceeded ∼60 μm from the nearest labeled neighboring cells. Total AC-3-labeled cells within lesions were quantified in ImageJ (NIH, Bethesda, MD, USA) using an automated particle-counting algorithm. By this approach, 97 ± 2% of the total degenerating cells were contained within the lesion regions of interest. To obtain the density of AC-3-labeled cells within each region of interest, cell numbers were divided by the regions of interest area. For each level, the mean densities of AC-3-labeled cells in the parasagittal cortex and the lateral ventral/piriform cortex across both hemispheres were calculated using three near-adjacent sections. For comparison of lesion sizes between E23 and E26 (a period of active brain growth), lesion area was normalized to total section area. Total section area for control and lesion animals was quantified in ImageJ from low-resolution montages generated from digitized images acquired with a × 5 objective.
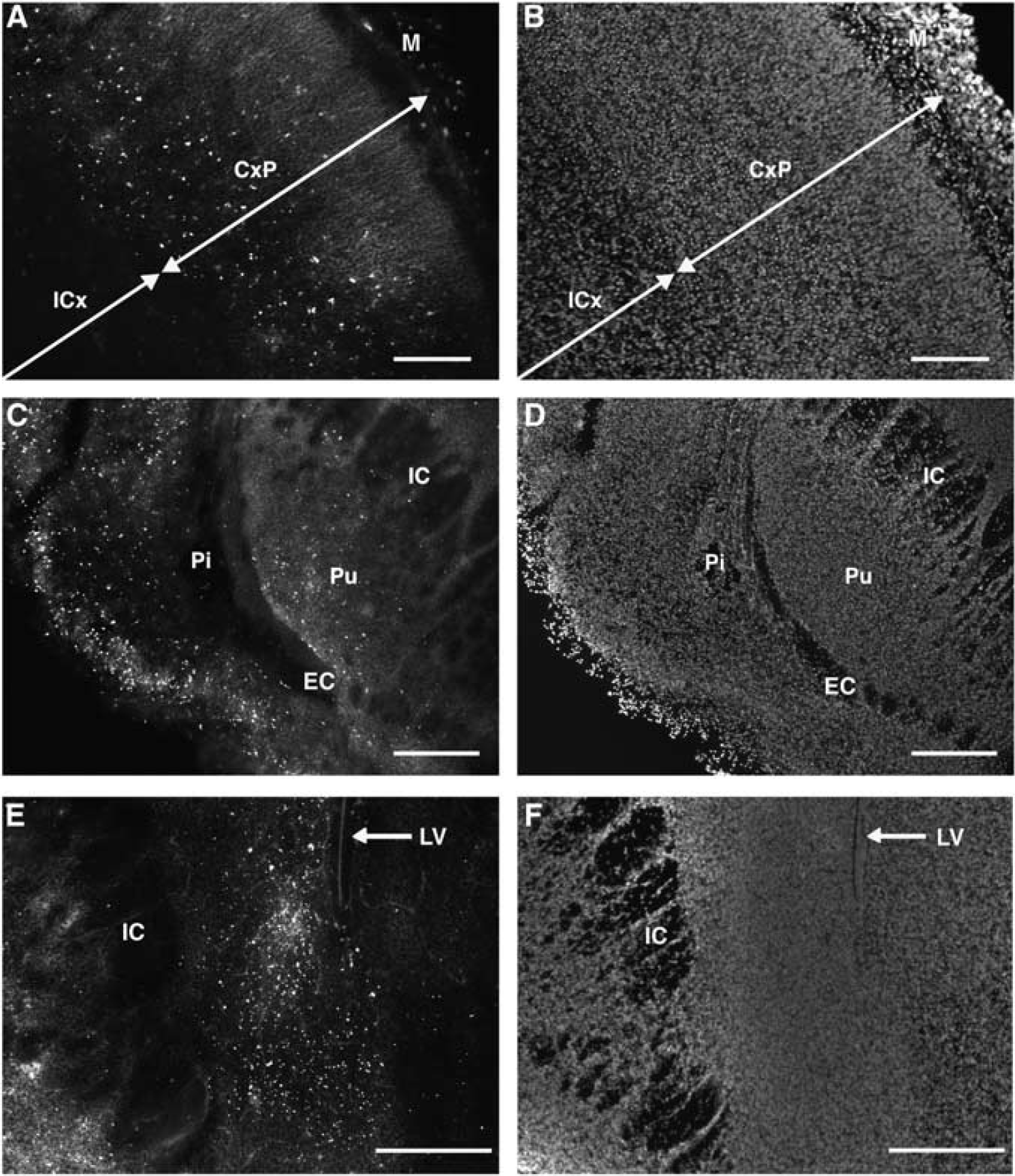
Cell degeneration in cerebral white matter of the embryonic day 22 (E22) rabbit was uniformly very low as determined by TUNEL staining (
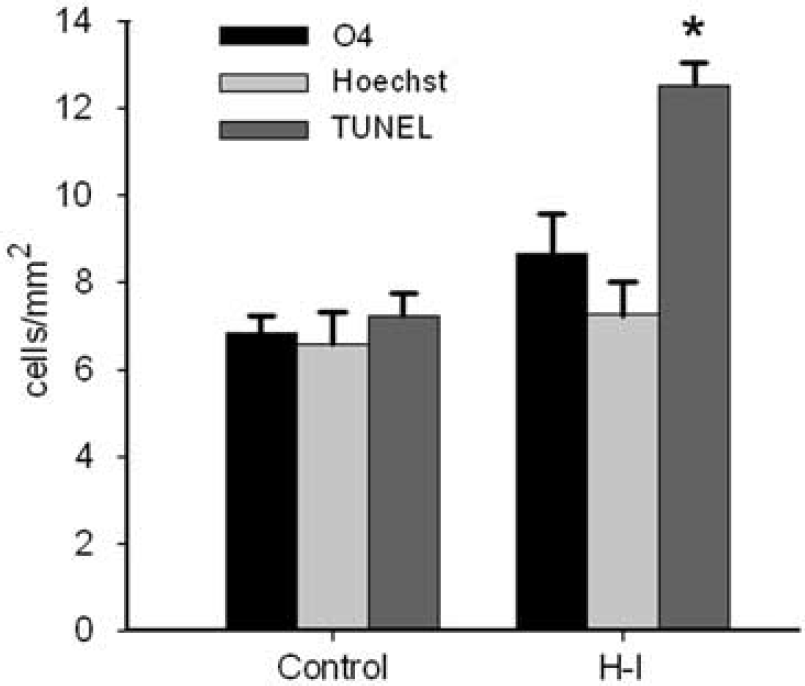
The overall density of degenerating cells, derived from the average from four major forebrain white matter tracts (corpus callosum, corona radiata, internal capsule, and external capsule), was very low at 24 h after hypoxia-ischemia (H-I) at E22, as determined by three complementary methods to assess cell death: O4-staining, Hoechst 33324-staining and TUNEL staining. *P < 0.001, independent samples t-tests. See ‘Materials and methods’ for details.
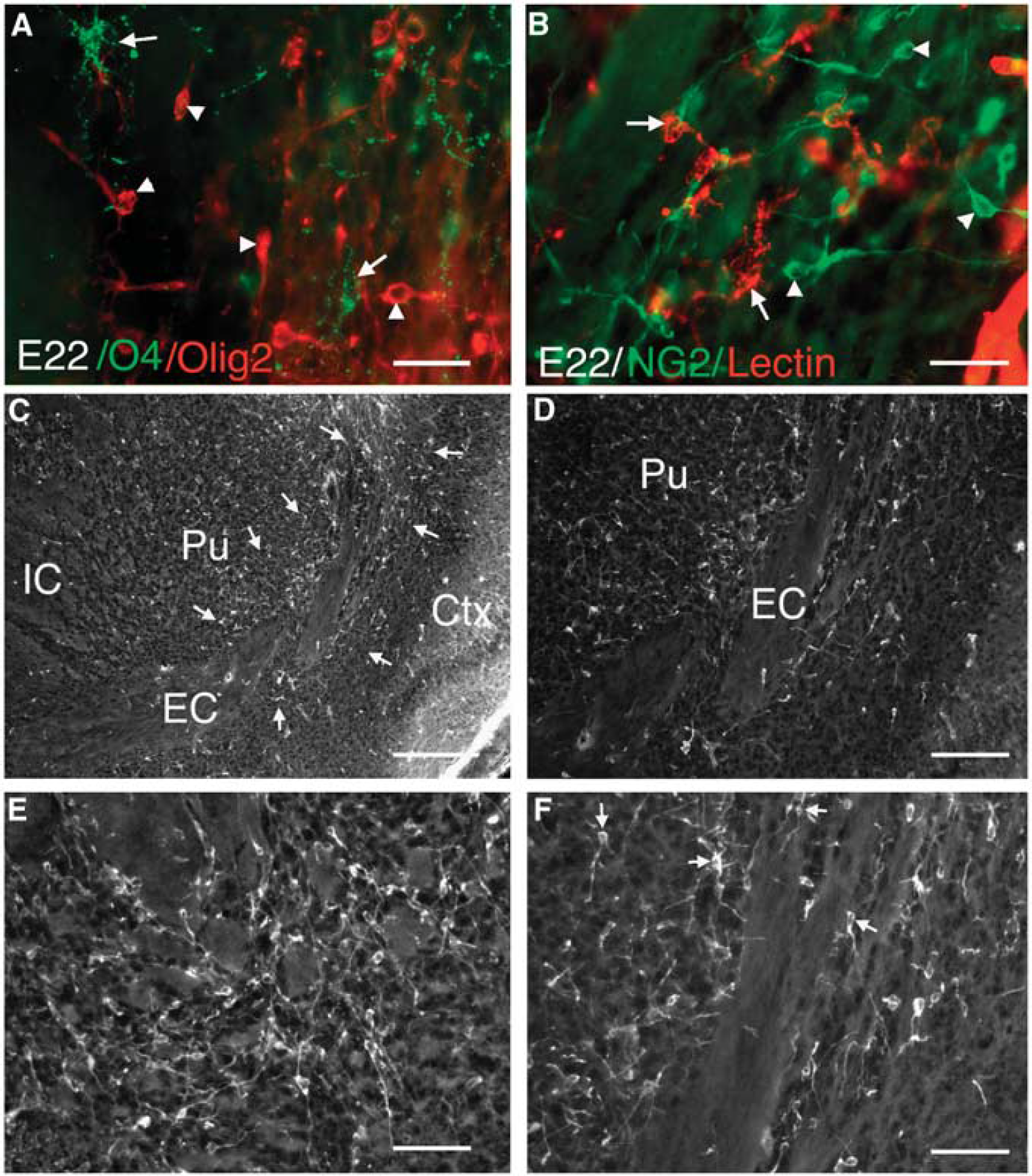
Numerous early oligodendrocyte (OL) progenitors are restricted to the basal ganglionic eminence at E22. (
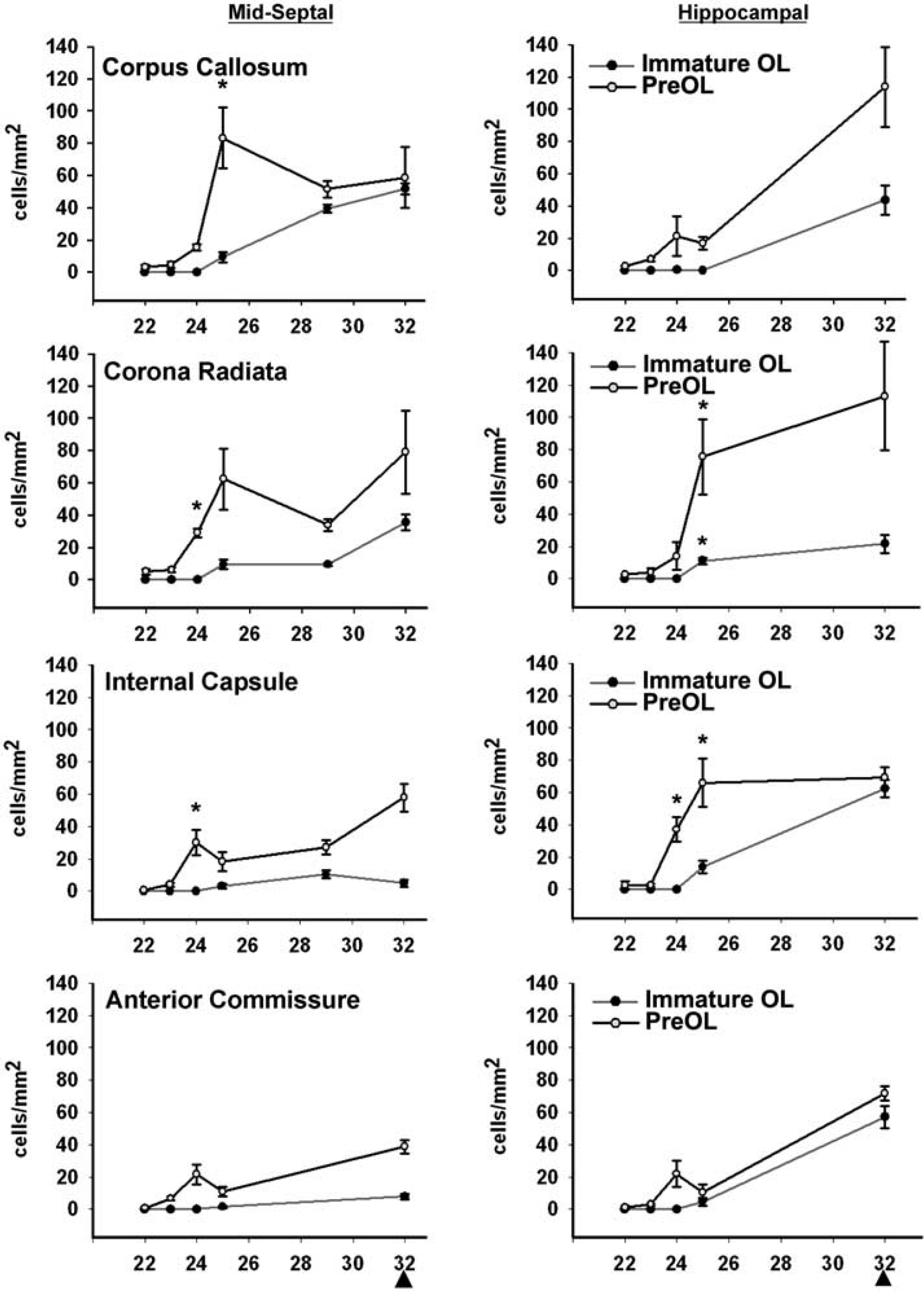
OL lineage progression in four major rabbit cerebral white matter tracts between E22 and E32 at the levels of the mid-septal nuclei and the rostral hippocampal formation. PreOL density (cells/mm2) was coordinately upregulated at E24 to E25; *P < 0.001, independent samples t-tests. Data presented as mean ± s.e.m. Arrowheads indicate the time of birth at E32.
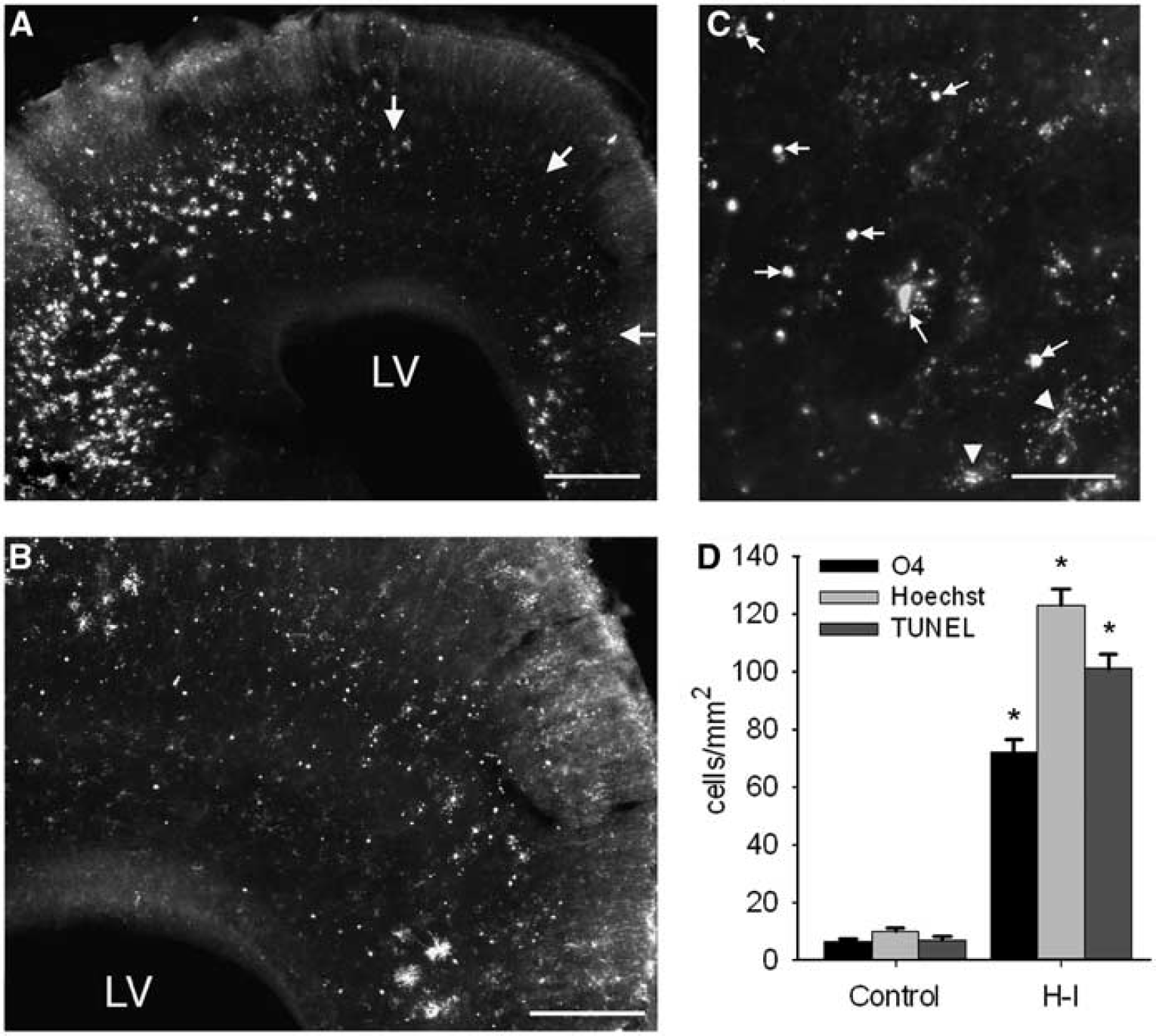
The preOL-enriched cerebral white matter is highly susceptible to injury 24 h after H-I at E25. (
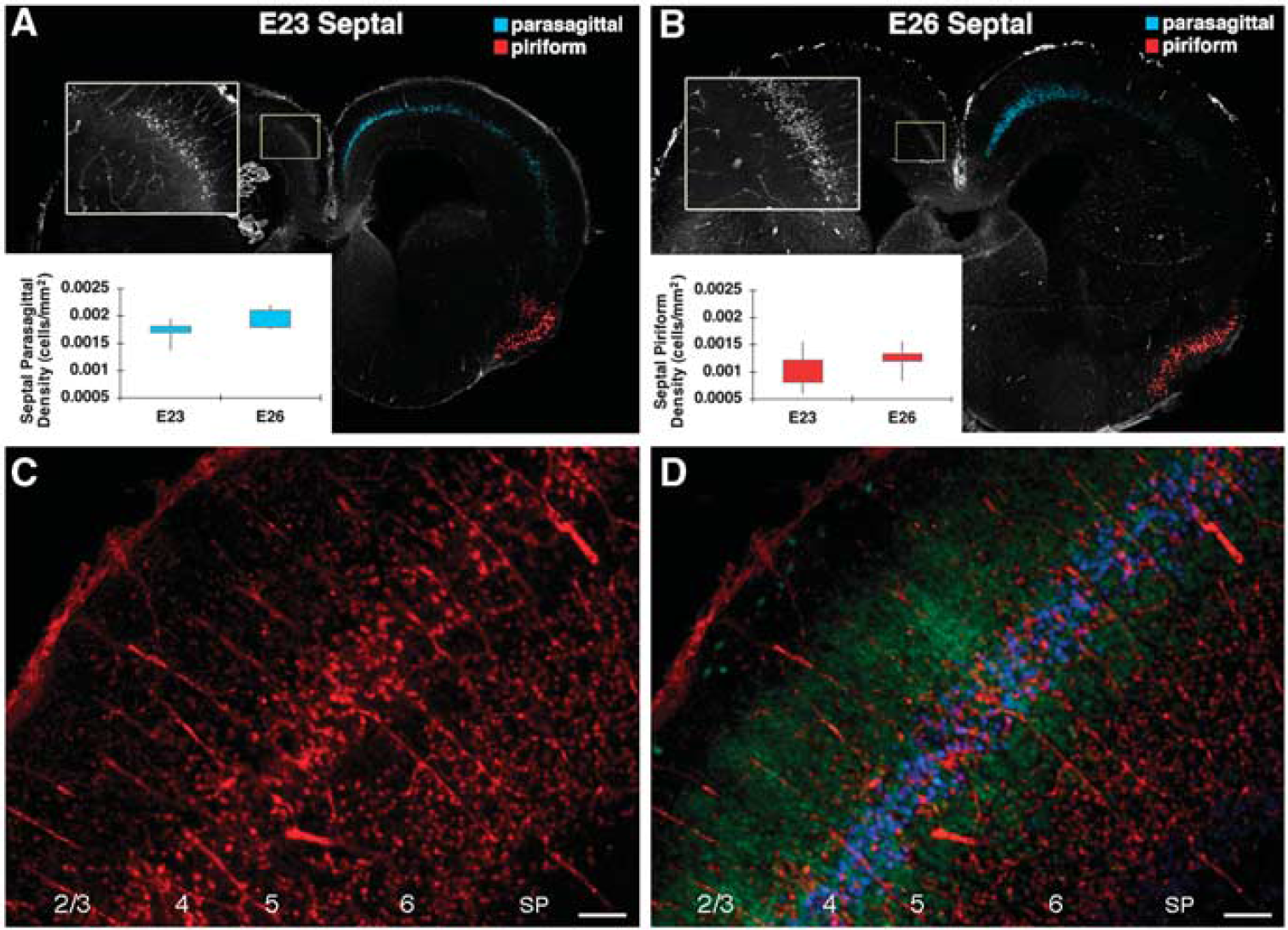
Cortical neuronal degeneration, as assessed by staining for activated caspase-3 (AC-3), was similar at E23 (
Estimation of Cerebral White Matter Atrophy and Statistical Analysis
Measurements of the area of white matter tracts were performed to determine the effect of cerebral atrophy on cell density measurements from animals that survived until E29 after H-I at E22 or E25 relative to E29 controls. After cell counts were completed, digitized images of white matter tracts, visualized with Hoechst fluorescent counterstain, were acquired with a × 2.5 objective. Areas of white matter tracts in three near-adjacent sections were acquired with Open Lab Morphometry software, version 4.0.3 (Improvision, Boston, MA, USA) from which a mean area measurement (mm2) was determined. Owing to reductions in the area of white matter tracts at E29 after H-I at E22 or E25, we incorporated the area and cell density measurements into a total cell count for O4- and O1-labeled cells for E22 and E25 treatment groups and controls.
An independent samples t-test was used to compare any two variables or groups in pre-hoc, planned comparisons. One-way ANOVA was used for comparisons of three groups, and significant differences were probed with a post hoc Tukey HSD test. Data are presented as mean ± s.e.m. P < 0.05 was considered statistically significant.
Results
Preterm Fetal Cerebral White Matter Is Markedly Resistant to Hypoxia-Ischemia at E22
We previously reported that the E22 rabbit fetus sustained multifocal gray matter injury to the cerebral cortex, basal ganglia, and anterior thalamus, as identified by TUNEL staining at 24 h after H-I (Derrick et al, 2004), which was confirmed in this study (Figure 1). However, this extensive gray matter injury was accompanied by a surprisingly low level of TUNEL staining in adjacent cerebral white matter tracts. Despite the presence of numerous TUNEL-labeled nuclei in the overlying cortical plate (CxP), there was a paucity of TUNEL-labeled nuclei in the intermediate zone (subcortical white matter; ICx) (Figures 1A and 1B). Similarly, the external capsule (EC) and the internal capsule (IC) rarely contained TUNEL-labeled nuclei, although the adjacent piriform cortex (Pi) and the putamen (Pu) contained markedly more degenerating nuclei (Figures 1C and 1D). The caudate neuroepithelium also contained numerous degenerating nuclei (Figures 1E and 1F), in contrast to the adjacent IC.
One explanation for the disparity between the extent of cell degeneration in E22 gray and white matter was the possibility that TUNEL was not a sensitive marker of cell death in the fetal rabbit white matter. We quantified the magnitude of white matter injury at 24 h survival with two additional markers (Figure 2). Each marker was quantified in four major forebrain white matter tracts (corpus callosum, corona radiata, IC and EC) that were adjacent to regions of gray matter injury (see Materials and methods). Collectively, these white matter tracts had a very low density of TUNEL-positive nuclei (13.0 ± 0.5 cells/mm2; e.g., Figure 1A) that was increased relative to control animals (7.0 ± 0.5 cells/mm2; P < 0.001). This was the case despite the high density of nuclei in these white matter tracts (e.g., Figure 1B). The density of Hoechst-labeled nuclei with morphologic features of degeneration was 7.0 ± 0.8 cells/mm2, similar to that of control animals (7.0 ± 0.7 cells/mm2). Adjacent sections were stained with the O4 monoclonal antibody to identify degenerating preOLs. The density of degenerating preOLs was 9.0 ± 0.9 cells/mm2, similar to that of control animals (7.0 ± 0.4 cells/mm2). Hence, three independent measures of cell death supported that the density of degenerating cells was low in cerebral white matter after H-I at E22, suggesting that white matter was markedly more resistant to injury than gray matter.
Early Oligodendrocyte Progenitors Are Restricted to the Basal Ganglionic Eminence at E22
We previously reported that OL progenitors are very resistant to H-I in lesions where preOLs markedly degenerate (Back et al, 2002). Hence, we determined whether the lack of injury from H-I at E22 might be related to a predominance of OL progenitors in the white matter. At E22, OL progenitors with a pseudo-unipolar or bipolar morphology were visualized with antibodies against the NG2 proteoglycan and the transcription factor Olig2 (Figure 3). Both Olig2-labeled (Figure 3A) and NG2-labeled cells (data not shown) were O4 antibody negative, supporting that they were OL progenitors rather than preOLs. As NG2 may label some microglia under pathological conditions, we confirmed that the NG2-positive cells were distinct from microglia and macrophages as visualized with isolectin-B4 (Figure 3B). Both NG2- and Olig2-positive cells were particularly concentrated in the ventral putamen between the adjacent IC and EC (Figures 3C and 3D) where they were intercalated among the white matter fasicles (Figures 3E and 3F). The pronounced enrichment of these OL progenitors in ventral forebrain at E22 is consistent with human developmental studies in which early OL progenitors originate from the basal ganglionic eminence and later migrate through subcortical white matter tracts to populate the dorsal cerebral hemisphere (Rakic and Zecevic, 2003). Hence, at E22 ventral white matter tracts contained mostly OL progenitors, with low numbers of preOLs.
The Olig 2 and NG2 expression in early OL progenitors is initially more diffusely localized in the cell and concentrates in the nucleus as these migrating cells reach their final target destinations in the white matter. At E26, some NG2- and Olig2-labeled cells persisted in the ventral putamen, but most were localized to the corona radiata and corpus callosum (Supplementary Figure 1). This distribution was consistent with a ventral-to-dorsal migration of progenitors between E22 and E26, through the EC and corona radiata to the corpus callosum. By E26, increasing numbers of cells showed a nuclear localization of Olig2 both in dorsal (Supplementary Figures 1A and 1B) and more ventral (Supplementary Figures 1C and 1D) white matter tracts. In the EC (Supplementary Figure 1E) and corpus callosum (Supplementary Figure 1F) these cells were O4-negative OL progenitors.
PreOL Maturation Is Coordinately Upregulated in Fetal Cerebral White Matter at E24 to E25
There is a well-defined window in cerebral development when white matter injury coincides with the selective vulnerability of preOLs to oxidative damage and H-I (Back et al, 2005). Hence, we defined the timing of preOL expansion and differentiation to immature OLs. Figure 4 shows the developmental changes in density of preOLs and immature OLs between E22 and E32 (term birth) in four white matter tracts at two distinct levels where gray matter injury was identified at E22 (i.e., mid-septal nuclei and hippocampal formation at the level of the anterior thalamic nuclei). At E22, the density of both OL stages was consistently low. However, at E24 to E25, a distinct increase in preOL density was observed in all white matter tracts. By contrast, immature OL density consistently showed little or no change from E22. Hence, at E25 the relative distribution of preOLs and immature OLs is similar to that in preterm human periventricular white matter during the high-risk period for injury (Back et al, 2001). The preOL expansion persisted between E25 and E32, and was accompanied by a progressive increase in immature OLs. Thus, the timing of expansion of the preOL population was tightly regulated in a coordinated manner, coinciding with the apparent dorsal migration of OL progenitors and preceding the differentiation and expansion of immature OLs.
Preterm Cerebral White Matter Tracts Show a Pronounced Increased in Susceptibility to H-I at E25
We hypothesized that the paucity of apparent white matter injury from 40 mins of H-I at E22 was related to the low density of vulnerable preOLs. Thus, we examined susceptibility of the white matter to H-I injury at E25, when preOL density is markedly higher. At 24 h after 40 mins of H-I at E25 (Figure 5), there was a pronounced increase in white matter damage compared with that at E22. The typical morphology and distribution of degenerating O4 antibody-labeled cells in white matter lesions is shown in Figures 5A to 5C. In many lesions, the majority of preOLs appeared to be degenerating.
White matter injury was quantified with three different markers to assess acute cell degeneration (Figure 5D). The density of TUNEL-positive nuclei in H-I animals was 101 ± 5 cells/mm2 (n = 7 from two separate litters) and was significantly increased relative to controls (7 ± 1 cells/mm2; P < 0.001; n = 4). Moreover, the density of TUNEL-positive nuclei in H-I animals was approximately an order of magnitude higher than that at E22 (Figure 5 versus 2). The density of Hoechst-labeled nuclei with features of degeneration was 123 ± 6 cells/mm2 and was significantly increased relative to controls (10 ± 1 cells/mm2; P < 0.001). The density of degenerating O4-labeled cells that contained a nucleus was 72 ± 4 cells/mm2 and was significantly increased relative to controls (7 ± 1 cells/mm2; P < 0.001). We also detected fragments of degenerating O4-labeled cells that lacked a nucleus; these cells were not counted. Hence, the density of degenerating preOLs was likely to have been underestimated by the rigorous criteria set for the cell counts. Overall, these data suggest that the timing of susceptibility of white matter injury to H-I coincided with the timing of expansion of preOLs in cerebral white matter tracts.
We determined whether other cell types in the white matter in E26 survivors were similar to preOLs in their susceptibility to injury from H-I (Supplementary Figure 2). In contrast to preOLs, features of acute degeneration were not observed in NG2 + or Olig2 + OL progenitors, microglia, astrocytes, or axons. Indeed, there was a distinct absence of GFAP-positive cells (Supplementary Figure 2A), a low number of activated microglia (Supplementary Figure 2B), and a paucity of degenerating axons (Supplementary Figure 2C) in the white matter at 24 h after H-I. Further, these cell types did not colocalize with AC-3.
Gray Matter Susceptibility to Injury Is Similar After H-I at E22 or E25
We examined whether the enhanced susceptibility to white matter injury at E25 was related to more severe global H-I at E25 than at E22. To quantify fetal cerebral blood flow in utero, we measured cortical microvascular flow with a laser Doppler perfusion monitor (Supplementary Figure 3). There were no significant differences in cortical microvascular blood flow during either H-I (−44 ± 1% versus −51 ± 6%) or during the initial 30 mins of reperfusion (16 ± 10% versus 16 ± 11%) between animals studied at E22 and E25 (n = 3 fetuses/group from three dams/group).
Consistent with these physiologic studies, there were no apparent differences in the magnitude or distribution of gray matter injury in the cerebral cortex at 24 h after H-I at E23 and E26. At both E23 (Figure 6A) and E25 (Figure 6B), the distribution of cell degeneration assessed by AC-3 staining was consistently localized to parasagittal and ventrolateral/piriform cortex at both the level of the mid-septal nuclei (Figure 6) and the hippocampal formation (not shown). Further, there were no differences in the density of AC-3-labeled cells between E23 and E26 (Figures 6A and B graph insets; Supplementary Figures 4A and 4B) or in the normalized cortical lesion area between E23 and E26 (Supplementary Figures 4C to 4F). Analysis of entire cortical lesions (see Materials and methods) showed a similar spectrum of bilateral, relatively symmetric injury at both ages that ranged from minimal to severe.
The neuron-specific marker NeuN and the cortical layer-enriched neuronal marker Ctip2 (Hevner, 2007) were used to define the distribution of AC-3 staining within cortical layers 1 to 6 (Figures 6C and 6D). With moderate injury at both ages, neurons in cortical layers 4 and 6 were particularly susceptible to degeneration, whereas severe injury resulted in more diffuse neuronal degeneration within layers 2 to 6. At both ages, there was also a similar predilection for neuronal injury within the head of the caudate nuclei, the putamen, the pyramidal cell layer of the hippocampal formation, and the anterior nuclear group of the thalamus (data not shown). Hence, despite more pronounced susceptibility to white matter injury at E26 than at E23, gray matter injury was similar in magnitude and distribution at both ages.
We also determined whether other cell types were damaged in the cerebral cortex in E23 and E26 survivors (Supplementary Figure 2). However, no acute degeneration of astrocytes or activated microglia was observed in the cortical lesions. Indeed, as for white matter regions, there was a distinct absence of GFAP-positive cells and low numbers of activated microglia in the cortical lesions after H-I at these ages.
Hypoxia-Ischemia at E25, but not at E22, Causes Delayed White Matter Injury
We hypothesized that the acute white matter injury after H-I at E25 would manifest as progressive white matter injury and disturbances in OL lineage progression. We further hypothesized that H-I at E22 would disrupt white matter maturation, despite the paucity of overt early white matter injury. When examined at E29 (Figure 7A), a significant reduction in the area of the corona radiata and IC was observed after H-I at E25 (P < 0.01), but not at E22, compared with control animals. To correct for the effect of area on cell counts, we determined the total number of cells by multiplying the area and cell density. After H-I at E25, but not after H-I at E22, there was a significant decrease in preOLs in the corpus callosum, corona radiata, and IC at E29 (Figure 7B). After H-I at E22 and E25, a significant decrease in immature OLs was only observed in the IC (Figure 7C). Hence, H-I at E22, when preOL density was low, did not cause significant chronic white matter injury. However, chronic white matter injury was significantly greater after H-I at E25, consistent with the pronounced acute degeneration of preOLs seen at E25.
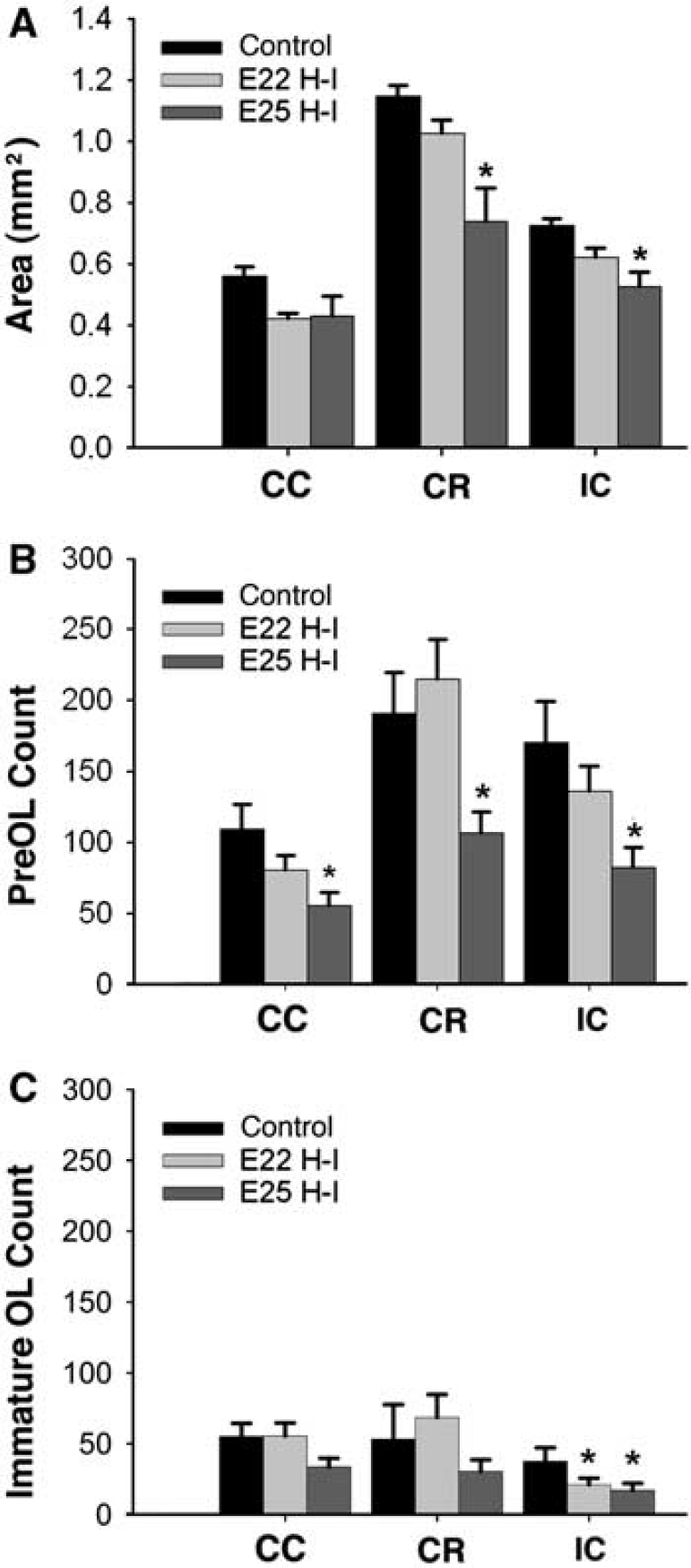
(
Discussion
This is the first study to define cellular mechanisms related to the susceptibility of fetal white matter to global H-I in an animal model of cerebral palsy. A distinct developmental window exists between ∼60% and 80% of term birth (∼26 to 32 weeks gestation) when preterm human cerebral white matter shows an enhanced predilection to injury. Thus, it was paradoxical that preterm rabbits at ∼70% of term birth (E22) showed minimal histologic evidence of acute white matter injury despite robust histologic injury to both cortical and subcortical gray matter (Derrick et al, 2004). Here, we identified a novel developmental window when fetal cerebral white matter transitions from a state that is mostly resistant to H-I to one that is highly susceptible. Our study yielded the following novel findings related to mechanisms that define the timing of enhanced susceptibility of fetal white matter to H-I: (1) the enhanced susceptibility to white matter injury at E25 was not related to more severe global H-I at E25 than at E22; (2) caspase-dependent cortical neuronal degeneration was extensive but similar at both E22 and E25, and was not sufficient to account for the markedly enhanced susceptibility of the white matter to injury at E25; (3) the enhanced susceptibility of E25 white matter to HI coincided with a tightly coordinated development window when OL maturation transitions from predominantly early OL progenitors to preOLs; (4) preOLs were the major population of cells susceptible to acute degeneration in white matter and this degeneration accounted for most of the cell loss sustained after H-I at E25; and (5) the magnitude of chronic white matter loss was related to the timing of H-I such that H-I at E22 did not produce significant delayed white matter injury in contrast to H-I at E25.
Recently, subcortical gray matter injury was reported to occur in association with human periventricular leukomalacia, particularly in the thalamus (Ligam et al, 2009). It currently remains unclear whether such neuronal degeneration contributes to the acute or delayed susceptibility of fetal white matter to H-I. Despite greater preOL death at E25, a similar spectrum of gray matter injury occurred after H-I at E22 and E25, which included a laminar pattern of cortical neuronal injury in parasagittal and piriform cortex and subcortical neuronal injury that involved the caudate nucleus, putamen, and anterior thalamus. Hence, the timing of onset of white matter injury appeared to occur independently of gray matter injury, which was substantial but similar at both developmental ages. Consistent with this observation, cortical microcirculatory flow was similar during H-I and early reperfusion at E22 and E25. In the preterm fetal sheep, the percentage changes in cerebral blood flow (CBF) during ischemia and reperfusion were also similar in cerebral gray and white matter structures (McClure et al, 2007). Hence, neither cortical neuronal injury nor global CBF showed developmental differences that provided an explanation for the timing of fetal white matter injury.
Glial-maturational factors appear to contribute substantially to the increasing predilection of fetal cerebral white matter to injury from H-I between E22 and E25. Among the cell types present in cerebral white matter lesions during this developmental window, only preOLs underwent significant degeneration, in contrast to astrocytes, microglia, and axons. Although we observed a disparity between the density of degenerating O4-labeled cells and nuclei identified by Hoechst or TUNEL staining, this disparity was consistent with our counting protocol, which excluded fragments of degenerating preOLs for which the association with the nucleus could not be definitively confirmed. Hence, preOL degeneration accounted for the majority of cell degeneration at both E22 and E25.
Several prior studies have defined OL lineage progression in the spinal cord or cerebrum where distinct dorsal-ventral gradients for the appearance and maturation of early OL progenitors have been defined (Richardson et al, 2006). To the best of our knowledge, this is the first study showing the onset of preOL maturation to be initiated in a coordinated manner across spatially distinct major cerebral white matter tracts during a narrow developmental window. It is unclear whether our observations are unique to the rabbit. In both humans and rabbits, myelination does not initiate uniformly across the entire forebrain, but rather progresses in sequential temporal and spatial maturational sequences (Drobyshevsky et al, 2005; Kinney et al, 1989). Hence, preOL maturation may be regulated by maturational mechanisms that are distinct both temporally and spatially from later stages in the OL lineage.
There are currently no ablative strategies to deplete white matter of preOLs. Nevertheless, several lines of evidence support that the timing of appearance of preOLs is a key factor that defined the susceptibility of fetal rabbit cerebral white matter to H-I: (1) OL progenitors, but not preOLs, were present in significant numbers at E22. In agreement with our prior findings (Back et al, 2002), rabbit OL progenitors also showed no apparent early susceptibility to H-I histologic injury; (2) a pronounced increase in the magnitude and distribution of injury at E25 coincided with a coordinated increase in preOL density in major forebrain white matter tracts; and (3) there was a paucity of preOLs at E22, a time when H-I produced no obvious acute or delayed injury, whereas at E25, a time of abundant preOL expansion in the white matter, H-I produced significantly greater white matter atrophy by E29.
Our studies to-date support that cerebral ischemia is necessary but not sufficient to generate white matter injury. We found that the spatial topography of injury coincides with the regional distribution of preOLs (Riddle et al, 2006). Contiguous regions of fetal ovine periventricular white matter showed markedly different susceptibility to injury that coincided with the density of susceptible preOLs. Together with the present findings, we propose that both the timing of appearance and the spatial distribution of preOLs contribute to the magnitude and extent of white matter injury. Consistent with this notion, maturation of preOLs to early myelinating OLs coincided with decreased H-I injury in fetal ovine white matter. The susceptibility for human perinatal white matter injury also appears to decline with the onset and progression of periventricular white matter myelination (Back et al, 2001).
Clinical Correlations
Several studies support that decline in incidence of PWMI is related to differentiation of preOLs to myelinating OLs (Back, 2006). In rabbits, the timing of reduction in preOL density and OL differentiation were coordinately regulated around the time of term birth. During human brain development, the decline in preOL density in parietal white matter occurs around 28 to 32 weeks gestation and coincides with the onset of OL differentiation and myelination in periventricular white matter (Back et al, 2001). However, even by term birth (37 to 40 weeks), the persistence of preOLs in parietal cerebral white matter tracts varies widely. In some cases, preOLs persist at high density at term birth, whereas in others, myelination is greatly advanced and preOLs are not present (Back and Luo, unpublished observations). Future studies are needed to define the spectrum of myelination in full-term infants as the persistence of preOLs may account for the increased predilection for PWMI seen in a subset of these infants. One such population is term infants with congenital heart disease who have a paradoxically increased risk for PWMI (Galli et al, 2004; Kinney et al, 2005; Miller et al, 2007).
That rabbit gray and white matter showed differential susceptibility to H-I at E22 supports that distinct cellular mechanisms mediate injury to developing gray and white matter. The enhanced susceptibility of preOLs to cell death from H-I and oxidative stress is related to maturation-dependent differences in the expression of ionotropic and metabotropic glutamate receptors, glutamate transporters, antioxidant enzymes, and cytotoxic cytokine receptors, as well as substantial differences in gene expression (Back, 2006; Blomgren and Hagberg, 2006; Volpe, 2009). PreOLs are selectively susceptible to degeneration from NMDA and AMPA/kainate receptor-mediated activation and reactive oxygen and nitrogen species-mediated toxicity (Baud et al, 2004; Deng et al, 2004; Fern and Moller, 2000; Fragoso et al, 2004; Manning et al, 2008; Sfaello et al, 2005). Antagonists of AMPA/kainate receptors reduce neuronal injury and myelin loss after H-I in the near-term rat (Choi and Kim, 2007; Follet et al, 2004; Noh et al, 2006). Antioxidants protect against fetal rabbit cerebral injury (Tan et al, 1998, 1999) and protect against rat preOL death from perinatal H-I (Lin et al, 2004). Thus, the selective susceptibility of the preOL to H-I is mediated through overlapping mechanisms, which supports the possibility that multiple classes of agents might provide additive protection against white matter injury in the preterm brain through reduction of preOL loss.
After H-I at E22, newborn rabbits at P1 show a spectrum of hypertonic motor deficits that persist at later postnatal age and resemble dystonic cerebral palsy (Derrick et al, 2007). Although we did not observe histologic evidence of early primary white matter injury at 24 h after H-I at E22, longer survival until P1 showed features consistent with secondary white matter injury (Drobyshevsky et al, 2007). By MRI analysis, P1 survivors with hypertonia showed a reduction in white matter area and in fractional anisotropy in the corpus callosum, corona radiata, and IC. These findings were explained by reduced axon staining for neurofilament protein that coincided with ventriculomegaly. The axon injury is consistent with the susceptibility of projection neurons in deeper cortical layers. Hence, H-I at E22 generates delayed cerebral white matter injury that arises from secondary neuro-axonal degeneration in the cerebral cortex, basal ganglia, and anterior thalamus. In the IC, there appears to be evidence of delayed OL injury at E29 after H-I at E22. Whether the delayed OL injury is a direct result of gray matter injury or a separate process requires further investigation.
The susceptibility to acute gray matter injury in the fetal rabbit differs from preterm fetal sheep where cortical injury is low after H-I (Riddle et al, 2006) and from preterm human where no significant oxidative damage was detected in either cerebral cortex or white matter axons (Back et al, 2005). The predilection for mixed gray and white matter injury at E25 in the rabbit provides a unique model system in which to evaluate the relative efficacy of potential therapeutic agents to prevent neuronal versus oligodendroglial degeneration. Future studies are needed to determine the pattern of motor deficits that occur in survivors of H-I at E25. It is possible that the combination of gray and white matter injury sustained after H-I at E25 would present with a mixed pattern of motor deficits involving both dystonia and spasticity, the latter related to increased susceptibility to white matter injury.
Footnotes
Acknowledgements
This study was supported by the NIH (National Institutes of Neurological Diseases and Stroke, KO2NS41343; 1RO1NS054044, R37NS045737, 1RO1NS043285, 1RO1NS051402) a Bugher Award from the American Heart Association and the March of Dimes Birth Defects Foundation. We are very grateful to Dr John Alberta for generously providing the Olig2 antisera and to Dr William Stallcup for generously providing the NG2 rabbit antisera used in these studies. We are grateful to Hongyan Du and Dr Larry Sherman for their helpful suggestions and advice.
The authors declare no conflict of interest.