
Abstract
Nicotinamide phosphoribosyltransferase (NAMPT) has been implicated in neuroprotection against ischemic brain injury, but the mechanism underlying its protective effect remains largely unknown. To further examine the protective effect of NAMPT against ischemic stroke and its potential mechanism of action, we generated a novel neuron-specific NAMPT transgenic mouse line. Transgenic mice and wild-type littermates were subjected to transient occlusion of the middle cerebral artery (MCAO) for 60 minutes. Neuron-specific NAMPT overexpression significantly reduced infarct volume by 65% (
INTRODUCTION
Cerebral ischemic injury leads to complex tissue pathology involving both gray matter and white matter (WM). White matter in the telencephalon encompasses the corpus callosum, internal capsule, anterior commissure, and fiber bundles in the striatum, and is mainly involved in sensorimotor signal transduction to and from the cerebral cortex (CTX). Blood flow within WM is relatively less than in gray matter and there is little collateral circulation, especially in deep WM. Oligodendrocytes (OLs) and OL precursor cells play an essential role in myelin formation in WM and are highly sensitive to ischemia. 1 Furthermore, WM becomes even more susceptible to ischemia with age. 2 Therefore, WM is highly vulnerable to ischemia and often more severely injured than gray matter. 3 Indeed, WM is affected in most cases of human stroke, accounting for half of the lesion volume. 4 Histologic changes of WM injury (WMI) range from demyelination, death of OLs, and axonal damage and loss, to mild reactive gliosis. Owing to the critical role of WM in neurotransmission, WMI may lead to sensorimotor disruption, neurobehavioral syndromes, and cognitive impairments.5–7 Despite the essential roles for WM in neural function and its susceptibility to ischemia, WMI has largely been overlooked in animal studies as well as in clinical treatments. 8 Thus, complete neuroprotection cannot be truly attained without WM protection, and complete analyses of WMI in models of cerebral ischemia and potential WM protectants are essential.
Nicotinamide phosphoribosyltransferase (NAMPT) converts nicotinamide to nicotinamide mononucleotide, the rate-limiting step in the salvage pathway of nicotinamide adenine dinucleotide (NAD) biosynthesis.9,10 Nicotinamide adenine dinucleotide is a major source of ATP production and also acts as the substrate for several important molecules, such as the DNA repair enzyme poly-[ADP-ribose]-polymerase 1 (PARP-1) and the transcription regulatory protein sirtuin 1. Nicotinamide phosphoribosyltransferase therefore plays a key role in the regulation of energy metabolism and stress responses. Previous studies indicated that lentivirus-mediated NAMPT overexpression is neuroprotective against ischemic brain injury, 11 while genetic deletion of NAMPT exacerbates ischemic brain injury. 12 Specifically, Wang and colleagues reported that NAMPT upregulation is part of a natural stress response to ischemic injury and that it protects gray matter through sirtuin 1-dependent modulation of the adenosine monophosphate-activated kinase pathway. These findings show that NAMPT is an endogenous neuroprotective molecule. However, beyond these observations, little is understood about the functions of the NAMPT molecule when overexpressed in the central nervous system (CNS) and the mechanisms underlying its protective effects, particularly in the context of ischemic injury.
In mammals, NAMPT protein can be found in both the intracellular (iNAMPT) and extracellular (eNAMPT) spaces. Whereas the function of iNAMPT as a NAD biosynthetic enzyme has been fully supported, the significance and function of eNAMPT remains unknown. Extracellular NAMPT does not exhibit classic secretory signaling. Instead, it can be secreted into the extracellular space or into the blood via a non-classical secretory mechanism 13 by a number of cell types, such as adipocytes, hepatocytes, macrophages, and leukocytes.14–16 Whether neuronal cells also secrete eNAMPT and its potential function in the CNS remain unknown. In the present study, we observed that transgenic mice overexpressing NAMPT in a neuron-specific manner were highly resistant to focal cerebral ischemic brain damage. Interestingly, WMI was significantly reduced in neuronal NAMPT transgenic (Tg-NAMPT) mice compared with WT littermates. Furthermore, we detected that ischemic neuronal cells secrete NAMPT into the extracellular space and that the conditioned medium from ischemic neurons overexpressing NAMPT is protective against ischemic injury in primary cultured OLs. Finally, recombinant NAMPT protein itself was also protective against ischemia-like insults when added to the medium of primary cultured OLs. These data support a novel role for eNAMPT in protection against ischemic WMI.
MATERIALS AND METHODS
Generation of Brain-Specific NAMPT Transgenic Mice
Human NAMPT cDNA tagged with hemagglutinin (HA) was PCR-amplified and inserted into pcDNA 3.1 vector (Invitrogen, Grand Island, NY, USA). Cytomegalovirus promoter was substituted with a 4.1 kb Thy-1 promoter (kindly provided by Dr Joshua Sanes, Harvard University). To enhance the expression of NAMPT, the human
Murine Model of Transient Focal Ischemia
All animal experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Focal cerebral ischemia was produced by intraluminal occlusion of the left middle cerebral artery (MCA) with a nylon monofilament suture, as previously described. 17 Briefly, 8- to 10-week-old Tg-NAMPT or WT male littermates were anesthetized with 1.5% isoflurane in a 30% O2/70% N2O mixture under spontaneous breathing conditions. Rectal temperature was controlled at 37.0°C ± 0.5°C with a temperature-regulated heating pad during surgery and MCA occlusion (MCAO). Mean arterial blood pressure was monitored during MCAO through a tail cuff, and arterial blood gas was analyzed 15 minutes after the onset of ischemia. The animals underwent MCAO for 60 minutes and then reperfusion for 72 hours for infarct volume measurement or for 2 weeks for behavioral tests. In all experiments, the experimenter was masked to genotype.
Regional Cerebral Blood Flow Measurement
Changes in regional cerebral blood flow at the surface of the left CTX were recorded using laser Doppler flowmetry (PeriFlux system 5000; Jarfalla, Sweden) with a flexible optical fiber probe. The tip of the probe was affixed with glue on the skull over the core area supplied by the MCA (1 mm posterior and 5 mm lateral from bregma). Regional cerebral blood flow was measured 5 minutes before MCAO (baseline), 5 and 55 minutes during MCAO, and 5 minutes after reperfusion and expressed as a percentage of baseline values.
Measurements of Infarct Volume
At 72 hours after MCAO, brains were removed and the forebrain was sliced into 1 mm thick coronal sections. Sections were stained with 2% 2,3,5-triphenyltetrazolium chloride. Infarct volume was determined using MCID image analysis (Imaging Research, St. Catherine's, Canada). The experimenter was masked to genotype during all measurements.
Neurologic Deficits and Behavioral Tests
Three neurobehavioral tests were performed by an observer masked to the experiments, as previously reported by us. 18 First, neurologic deficits were scored on a 0 to 5 scale as follows: no neurologic deficit (0); failure to extend the right forepaw fully (1); circling to the right (2); falling to the right (3); unable to walk spontaneously (4); or dead (5). Second, the corner test was performed on days 0, 1, 3, 5, 7, 10, and 14 after ischemia. A mouse was placed between two angled (30°) boards, facing the corner. Rearing and subsequent turning behavior to either side was recorded. The nonischemic mouse turns either left or right whereas the ischemic mouse turns preferentially toward the left side. The numbers of left turns were counted in 10 trials. For the third test, the rotarod, animals were placed on an accelerating rotating rod (from 4 to 40 r.p.m. over 300 seconds) and their latency to fall was recorded. Preoperative training was performed for three trials a day for 3 days and the last trial served as preoperative baseline. Postoperative testing was performed on day 1, 3, 5, 7, 10, and 14 postinjury. The data are expressed as the mean duration of time on the rotarod per day as a percentage of the presurgery control value.
Generation of Lentiviral Vectors
Based on our experience, the phosphoglycerate kinase promoter mediates better gene expression than the ubiquitin promoter in neurons. Therefore, we modified the lentiviral shuttle vector FUEW (kindly provided by Dr Carlos Lois from the Massachusetts Institute of Technology) in which green fluorescent protein (GFP) was driven by the ubiquitin promoter and generated two lentiviral vectors: lenti-GFP and lenti-NAMPT. Lenti-GFP was produced by substitution of the ubiquitin promoter in FUEW by the phosphoglycerate kinase promoter. Lenti-NAMPT was produced by substitution of GFP in lenti-GFP with NAMPT cDNA containing the HA tag. Lentiviral vectors were generated by CaPO4 precipitation transfection in 293 cells, concentrated by sucrose ultracentrifugation, and further purified by ion exchange chromatography using Mustang Q Acrodisc Units (PALL Corporation, Port Washington, NY, USA). The titer of the purified lentiviral vector was 1.2 × 10 9 /transfection units/ml.
Generation of NAMPT Protein
His6-tagged NAMPT cDNA was generated by PCR, inserted into pET-30a (Novagen, Darmstadt, Germany), and transformed into BL21
In Vitro Models of Ischemia-Like Insults
Primary cultures of cortical neurons were prepared from 17 day-old Sprague Dawley rat embryos (Charles River Laboratories, Horsham, PA, USA), as previously described.
17
Experiments were conducted at 8 to 10 days
Primary Oligodendrocyte Precursor Cells/Oligodendrocytes Culture and In Vitro Models of Ischemia-Like Insults
Primary OL precursor cell cultures were prepared as previously described.
19
Briefly, cerebral cortices from postnatal day 1 to 3 Sprague Dawley rats were dissected and minced. Dissociated cells from 2 to 3 rats were plated in a tissue culture flask coated with poly-
For the OGD model, OLs were subjected to OGD for 180 minutes, and returned to normal culture media and conditions. To induce alpha-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA) excitotoxicity, OLs were challenged with 50
In a parallel study, neurons were transduced with lenti-NAMPT or control lenti-GFP virus and subjected to OGD treatment for 60 minutes. Conditioned medium was collected 24 hours after OGD, coincubated with OLs, and then subjected to OGD treatment for 180 minutes. Cells were incubated for 24 hours with conditioned medium and cell death was analyzed as above. To deplete NAMPT released into culture medium, conditioned medium was incubated with NAMPT antibodies (10
Immunofluorescent Staining
After blocking with 5% bovine serum albumin in phosphate-buffered saline for 1 hour, sections were incubated with primary antibodies at 4°C overnight followed by the appropriate secondary antibodies for 1 hour at room temperature. The primary antibodies used in this study include: mouse anti-HA (1:100, Cell Signaling, Danvers, MA, USA), rabbit anti-NAMPT (1:500, Bethyl Laboratories, Montgomery, TX, USA), or mouse anti-NAMPT (1:300, Enzo Life Sciences, Farmingdale, NY, USA), mouse anti-NeuN (1:500, Millipore, Billerica, MA, USA), rabbit anti-GFAP (1:500, Dako, Carpinteria, CA, USA), rabbit anti-Iba1 (1:2000, Wako, Richmond, VA, USA), mouse anti-APC (1:400, Millipore), rat anti-CD31 (1:200, Abcam, Cambridge, MA, USA), and rabbit anti-myelin basic protein (MBP, 1:500, Abcam).
Luxol Fast Blue Stain
Brain sections were deparaffinized with xylene and dehydrated with alcohols, and then immersed in a 0.1% alcoholic solution containing Luxol fast blue (Sigma) at 56°C for 2 hours, followed by washing with distilled water. Sections were then incubated in 0.05% lithium carbonate, dehydrated through graded alcohols, mounted with Permount (Sigma), and examined by light microscopy. The quantitative analysis for remyelination in Luxol fast blue stained section was performed as described previously. 20 The area of reduced Luxol fast blue staining was measured and expressed as a percentage of the contralateral hemisphere.
Statistical Analysis
Results are reported as mean ± s.e.m. The difference between means was assessed by the Student's
RESULTS
Neuron-Specific Overexpression of NAMPT is Protective Against Cerebral Ischemic Injury in a Transgenic Mouse Line
Previous reports indicated that lentiviral-mediated NAMPT overexpression decreases cerebral ischemic infarct size
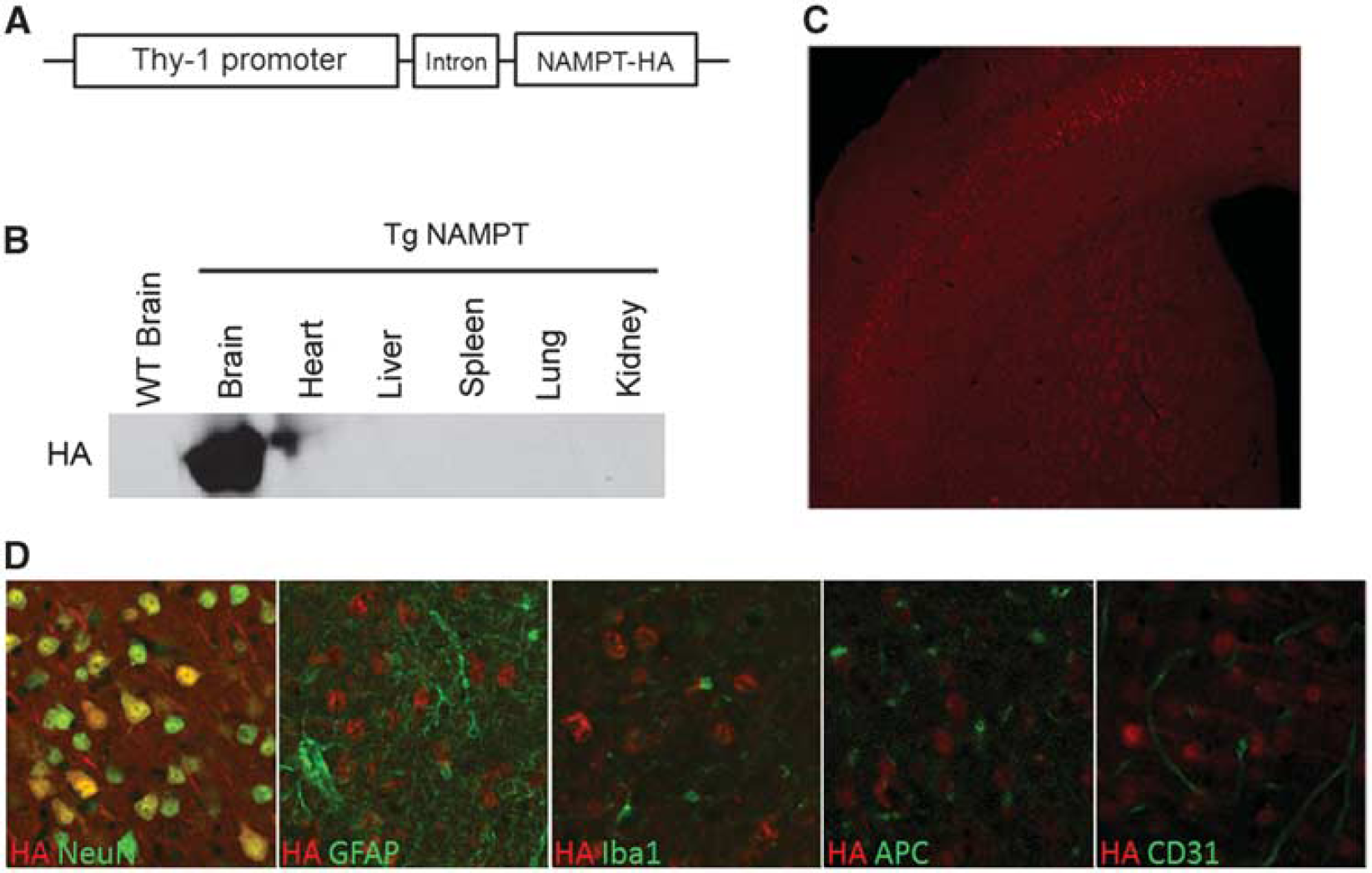
Generation of neuron-specific nicotinamide phosphoribosyltransferase (NAMPT) transgenic (Tg-NAMPT) mice. (
Using these novel neuron-specific Tg-NAMPT mice, we sought to determine if neuronal overexpression of NAMPT alone would be adequate to confer protection against focal ischemic injury. Neuronal Tg-NAMPT mice and WT littermates were subjected to 60 minutes MCAO and infarct volume and neurologic deficits were assessed 72 hours after reperfusion. Functional outcomes were evaluated by the corner and rotarod tests for 2 weeks. Neuronal NAMPT transgenic overexpression significantly reduced infarct volume by 65% compared with WT littermates (
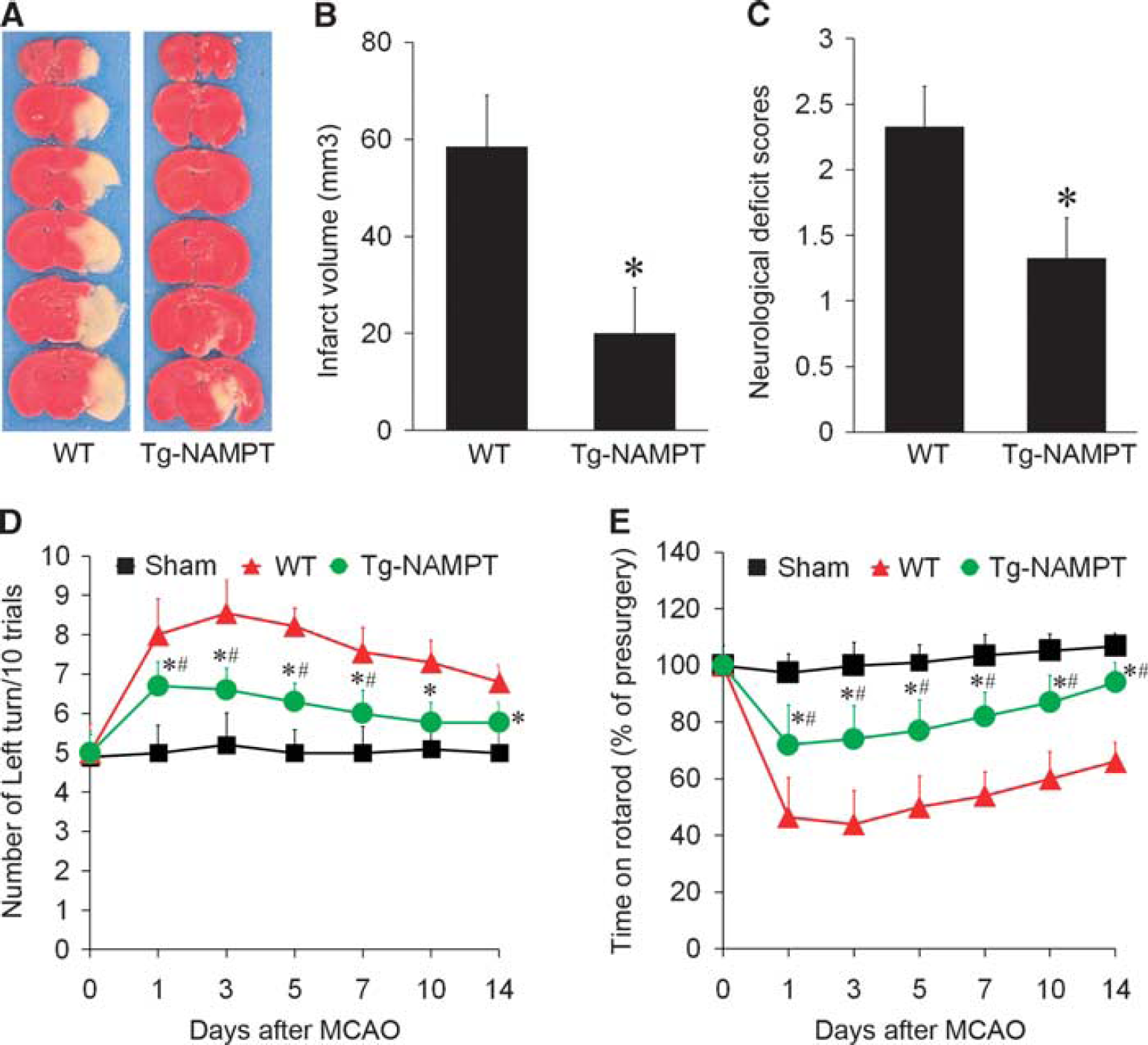
Nicotinamide phosphoribosyltransferase (NAMPT) overexpression protects against ischemic brain injury. (
Changes of rCBF before, during, and after MCAO in Tg-NAMPT mice and WT littermates
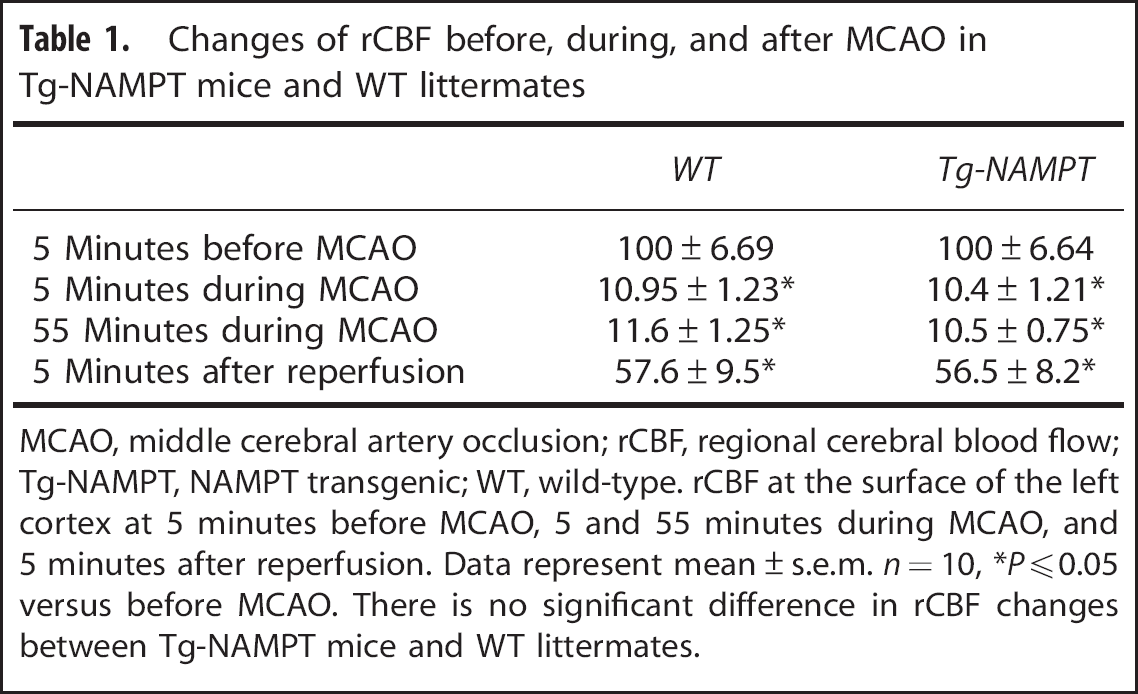
MCAO, middle cerebral artery occlusion; rCBF, regional cerebral blood flow; Tg-NAMPT, NAMPT transgenic; WT, wild-type. rCBF at the surface of the left cortex at 5 minutes before MCAO, 5 and 55 minutes during MCAO, and 5 minutes after reperfusion. Data represent mean ± s.e.m.
Nicotinamide Phosphoribosyltransferase Transgenic Overexpression Attenuates White Matter Injury after Ischemia
Previous studies using lentiviral vectors
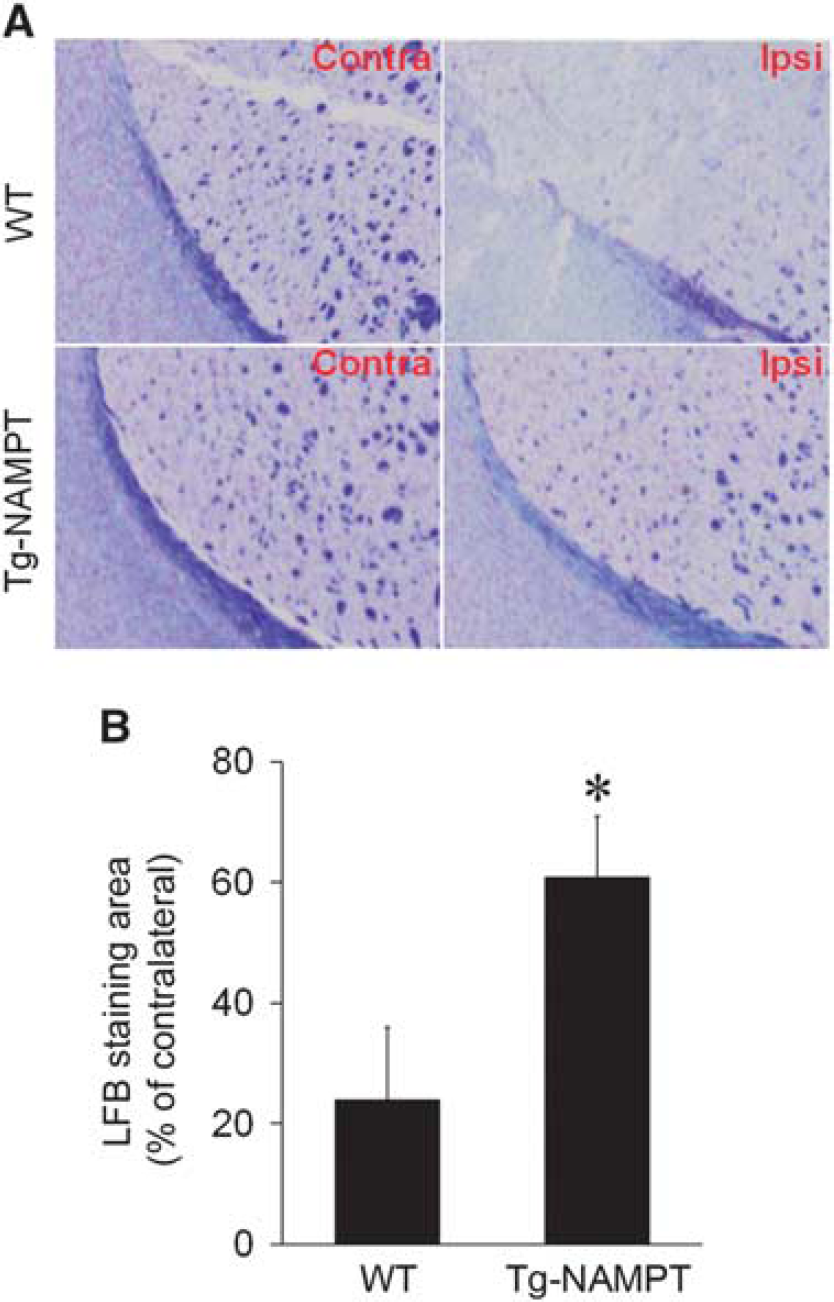
Nicotinamide phosphoribosyltransferase (NAMPT) overexpression attenuates white matter injury (WMI). (
Ischemic Injury Induces Nicotinamide Phosphoribosyltransferase Expression and Secretion from Neurons
Extracellular NAMPT has been found both locally in non-CNS systems and circulating systemically in the blood. Extracellular NAMPT can be distinguished from iNAMPT by its larger molecular weight arising from posttranslational modifications. 13 Whether the CNS also has eNAMPT and its function in the ischemic brain remains unknown. Both eNAMPT and iNAMPT protein expression was detectable with two different NAMPT antibodies in WT C57BL/6 mice. Although iNAMPT remained unchanged, eNAMPT was dramatically induced at 24 hours after ischemia, peaking at 72 hours but remaining at high levels even after 168 hours (Figure 4A). These findings suggest that eNAMPT is selectively induced after ischemia, perhaps as part of a long-term stress response pathway. Interestingly, NAMPT expression was induced in both neuronal cell bodies and WM regions, including striatal fiber bundles (Figure 4B, top panel) and the corpus callosum (Figure 4B, bottom panel) in WT mice after ischemia.
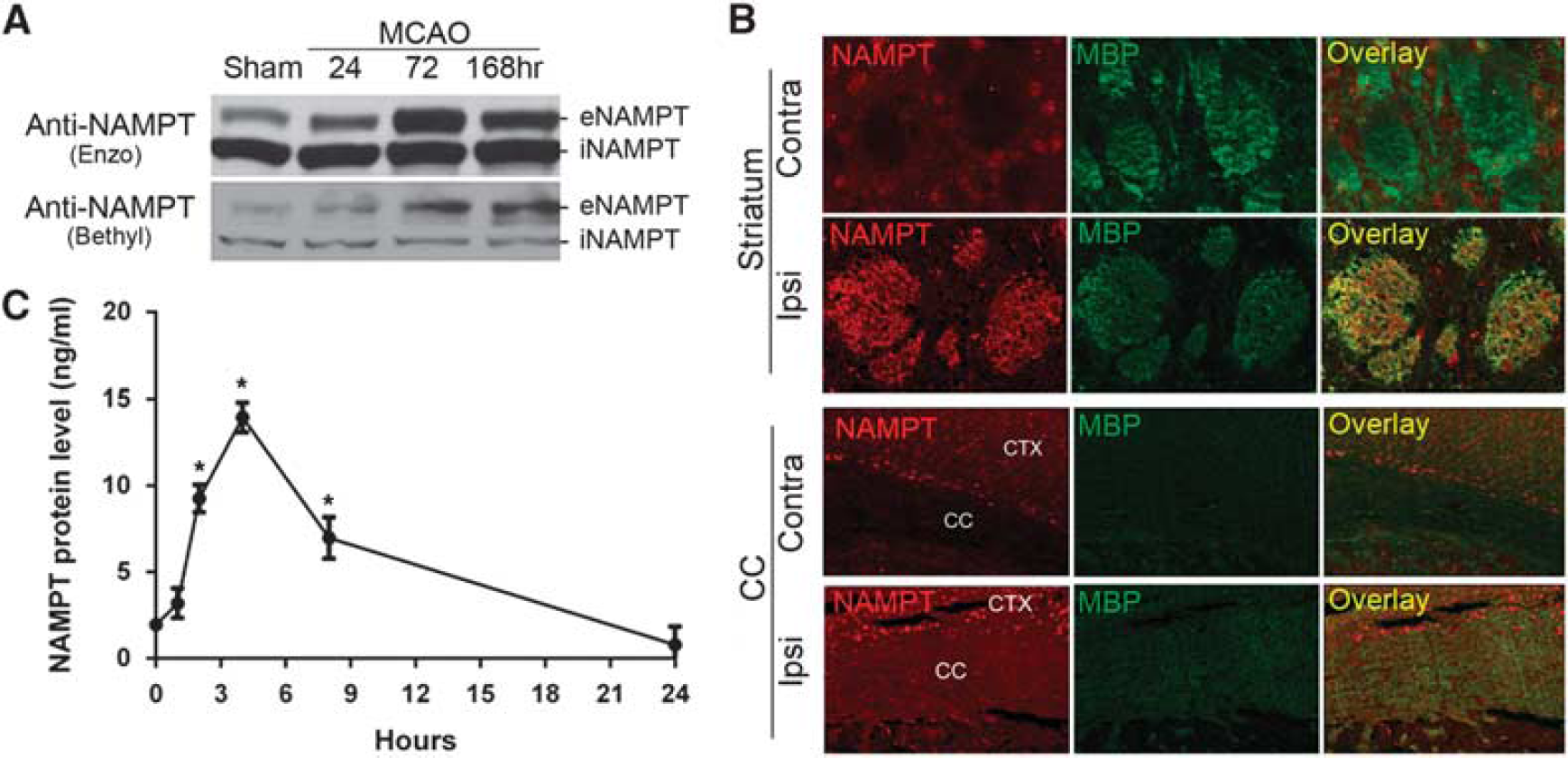
Extracellular nicotinamide phosphoribosyltransferase (eNAMPT) is expressed in the brain and induced after ischemia. (
To confirm that ischemic neurons are capable of secreting NAMPT protein into the extracellular space, we subjected neuronal cultures to OGD and measured NAMPT levels in the medium using ELISA. Consistent with the
Secretion of NAMPT can be Induced by Forced Overexpression in Neurons and Confers WM Protection
Given our
To directly address the extent of NAMPT secretion when overexpressed in neurons, we measured NAMPT levels in the culture medium of primary neurons transduced with either lenti-NAMPT–HA or a control lenti-GFP vector. Forced NAMPT overexpression in neurons significantly increased NAMPT secretion into the medium even without OGD, as detected by ELISA (Figure 5A), as well as by western blot (Figure 5B). Notably, the levels of protein found in the culture medium of transduced neurons exceeded the levels found from ischemic neurons by ~50-fold. As the endogenous levels of NAMPT secreted by ischemic neurons appear to be insufficient to confer full protection, exogenously delivered NAMPT protein may be necessary to effectively protect the brain against ischemic injury. We also observed that forced overexpression of NAMPT in Tg-NAMPT mice increased the levels of HA-tagged NAMPT within neuronal cell bodies as well as in the extracellular space under non-ischemic conditions (Figure 5C, upper panel). The expression of eNAMPT in the CTX and striatum of Tg-NAMPT mice was confirmed by western blot analysis. In contrast, no eNAMPT was detected in the brain of WT mice (Figure 5C, lower panel).
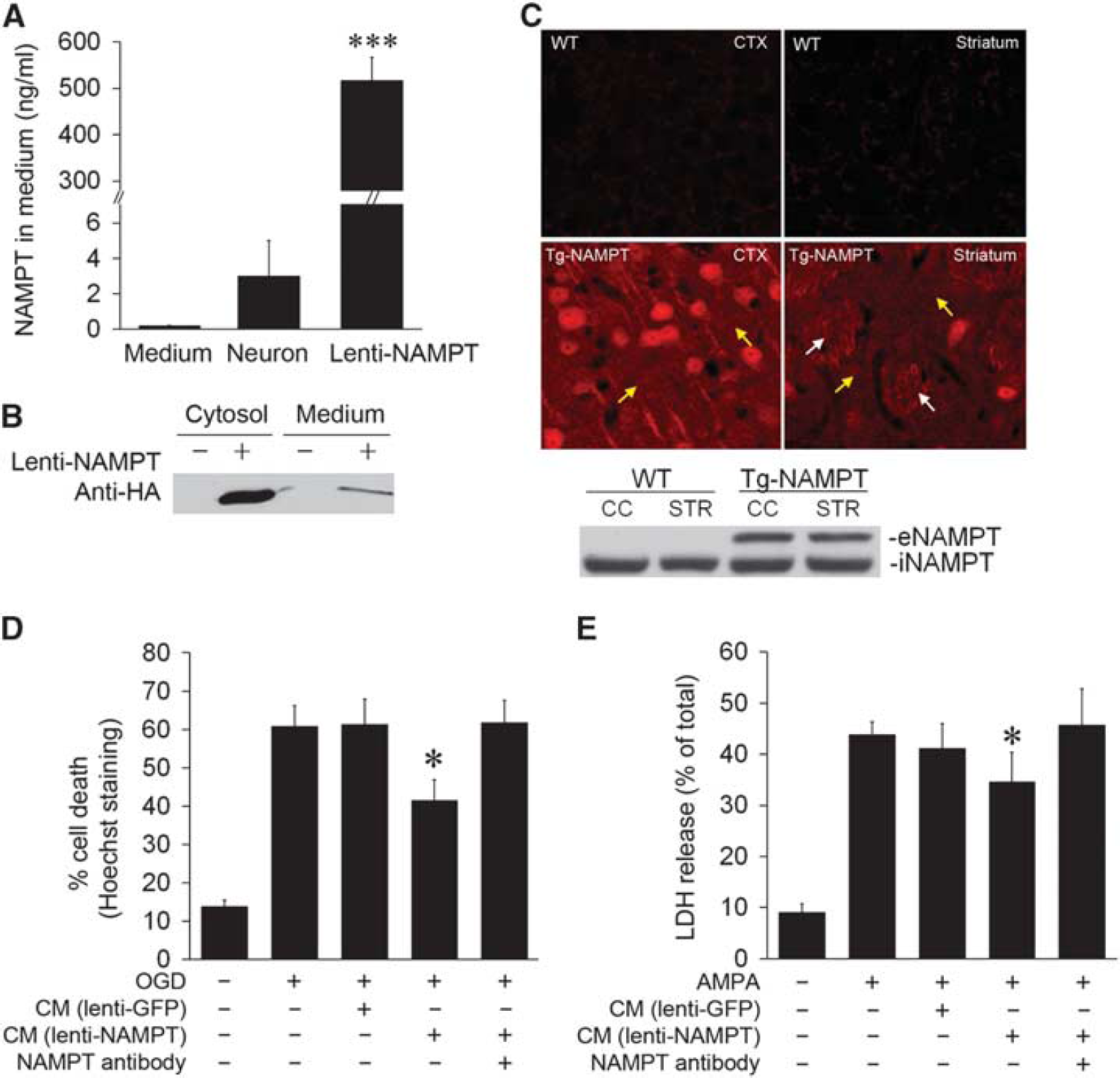
Secretion of nicotinamide phosphoribosyltransferase (NAMPT) from neurons can be induced by forced overexpression and confers white matter (WM) protection. (
Because the presence of eNAMPT in transgenic mice was correlated with WM protection
To more directly assess the role of eNAMPT on WM ischemic protection, we incubated primary oligodendrocytic cultures with recombinant NAMPT protein (5
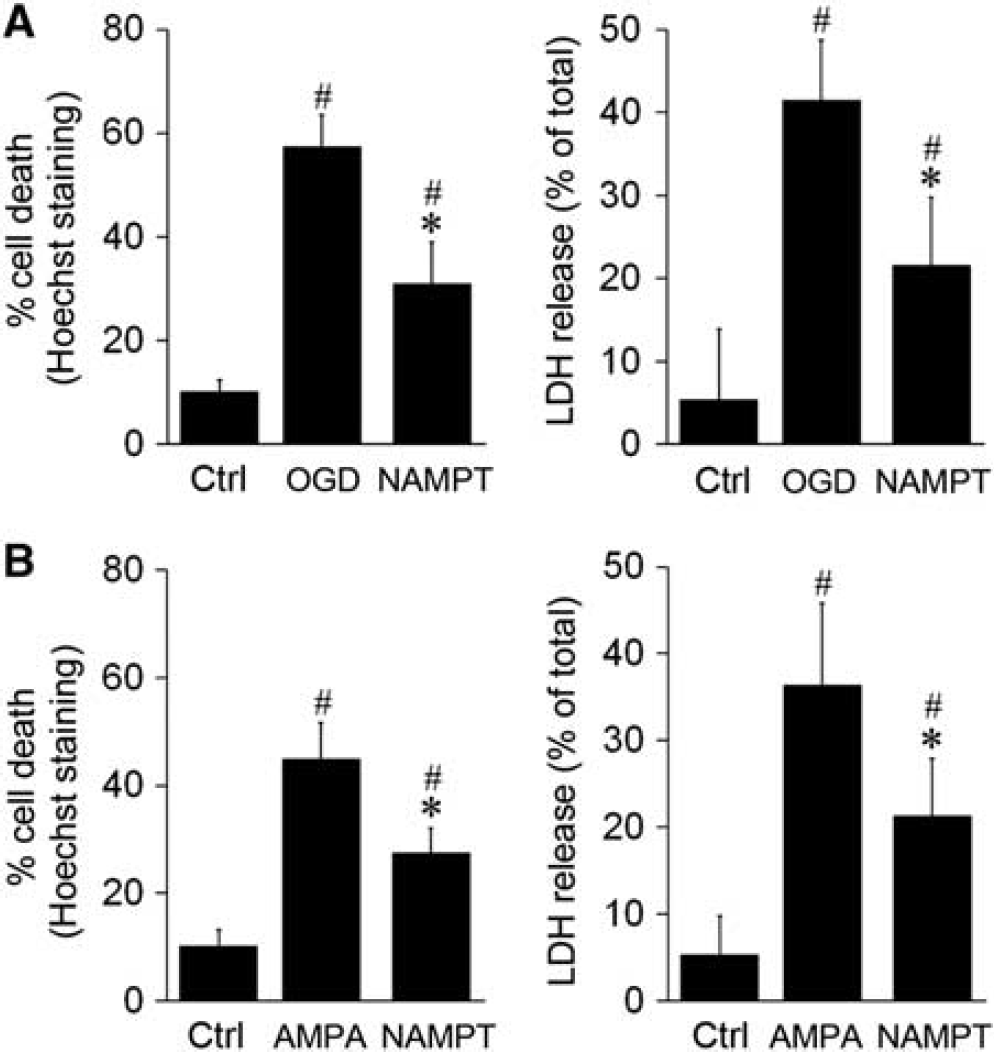
Exogenous nicotinamide phosphoribosyltransferase (NAMPT) protein reduces oxygen–glucose deprivation (OGD)- and alpha-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA)-induced oligodendrocytic death. Primary oligodendrocytes (OLs) were pretreated with NAMPT protein (5
Together, these observations indicate that neuronal overexpression of NAMPT leads to secretion of NAMPT into the extracellular space, which directly protects WM independent of the presence of neurons. As addition of NAMPT antibodies significantly reduced the protective effects of conditioned medium and the sole addition of recombinant NAMPT to oligodendrocytic cultures was sufficient to confer protection, we conclude that eNAMPT is responsible for WM protection against ischemic injury.
DISCUSSION
The role of NAMPT in both non-CNS and neural systems has focused primarily on its intracellular function in NAD biosynthesis. Although this function is extremely important in ischemic contexts, we have discovered a new role for NAMPT in the present report. We are the first group to show that NAMPT confers protection against ischemic WMI
In our transgenic mouse model, NAMPT was driven by the Thy-1 promoter, which has been widely used for neuronal overexpression of transgenes. We did not observe the presence of HA-tagged NAMPT within non-neuronal cells in the mouse brain (Figure 1D), although Thy-1 has been reported to be expressed in endothelial cells and in white blood cells such as T lymphocytes,21,22 two cell types that are intimately involved in the pathogenesis of ischemic stroke. However, T-cell infiltration into the brain usually occurs 3 days after cerebral ischemia, 23 whereas NAMPT already exerted WM protection effect within this timeframe in our studies. Therefore, it is unlikely that NAMPT functions through or is affected by NAMPT expression in T cells, at least not at the early time points observed in this study.
Nicotinamide phosphoribosyltransferase has been best characterized in terms of its function as the rate-limiting enzyme in the NAD biosynthetic pathway. As the availability of NAD is a critical determinant for neuronal ischemic sensitivity, the neuroprotective function of NAMPT has historically centered on the role of iNAMPT in NAD production. Consistent with the NAD biosynthetic function of NAMPT, inhibition of enzymatic activity of NAMPT by intraperitoneal administration of FK866 significantly reduced brain nicotinamide mononucleotide levels and substantially enlarged MCAO-induced infarction. 11 NAMPT has also been shown to protect against stroke by increasing the activity of sirtuin 1 through an increase in NAD. 11 However, our data suggest that a novel, extracellular function of NAMPT may also contribute to ischemic WM protection. On the other hand, NAMPT protection of WM could be distinct from NAD-dependent protection, especially given its extracellular localization. The function of NAMPT in the extracellular milieu could be either via NAD biosynthesis in the extracellular compartment, or via an unknown capacity, such as binding to a receptor and activating pro-survival signaling pathways. In line with this, we have found that incubation of neurons/OLs with recombinant NAMPT protein can activate several pro-survival signaling pathways, such as PI3K/Akt, ERK1/2, and Stat5a (unpublished data), indicating that these molecules might mediate the protective role of NAMPT. In other words, it seems likely that NAMPT binds to an unrecognized receptor on cell membranes and then exerts its protective effect via downstream pro-survival signaling pathways. We are currently attempting to identify the potential receptor for NAMPT on neurons/OLs. Further exploration into the mechanism of NAMPT and its protection of ischemic WMI may yield novel insights into both the function of NAMPT and the process of WMI under ischemic settings.
In addition to its function in NAD synthesis, NAMPT also has been characterized as a putative cytokine that can promote inflammation in peripheral systems. Nicotinamide phosphoribosyltransferase, also called pre-B-cell colony-enhancing factor 1, was first isolated from a human peripheral blood lymphocyte cDNA library and was shown to synergize with the pre-B-cell colony formation activity of stem cell factor and interleukin 7. 21 Nicotinamide phosphoribosyltransferase also regulates the activity of monocytes and neutrophils in response to inflammatory stimuli.22,23 The role of inflammation is well established in cerebral ischemic research and appears to exert a powerful influence over brain injury and recovery. As such, the acute overactivation of inflammatory processes likely contributes to the injured state, whereas a more subdued prolonged inflammatory response appears to assist in neural remodeling and repair, for which WM is critical. Our finding that NAMPT is secreted into the ischemic brain leads us to speculate that eNAMPT influences not only OLs, but also cells such as microglia, the resident inflammatory cells in the CNS, and contributes to the regulation of inflammatory processes after ischemia (data not shown). Further studies examining the role of exogenous NAMPT on inflammatory processes with longer survival periods may yield additional new insights into the effects of NAMPT on ischemic recovery. In addition to inflammation, oxidative stress has also been shown to propagate injury after ischemia. A recent study showed that NAMPT is essential for the benefits of calorie restriction against oxidative stress, 24 indicating that eNAMPT may also exert antioxidant effects. Thus, future studies to examine whether eNAMPT protects OLs by inhibiting oxidative stress are highly warranted.
As mentioned above, the significant contribution of WM to ischemic outcomes has been historically overlooked, and largely assumed to be secondary to neuronal or axonal injury. However, recent work underscores a unique role for WM health in determining ischemic severity. Several drugs that protect against ischemic injury are now being explored in their effects on WM, including the only FDA (Food and Drug Administration) approved therapeutic, tissue plasminogen activator 2 as well as delayed administration of the histone deacetylase inhibitor valproic acid. 25 Similar to the novel means (secretion) by which NAMPT appears to promote WM integrity, tissue plasminogen activator may also promote WM protection and recovery independent of its classic proteolytic activity, acting instead as a novel cytokine-like molecule. Although it is tempting to speculate that NAMPT may exert WM protection via its NAD biosynthetic activity, the mechanisms of WMI and recovery after cerebral ischemia are not well understood and may respond to NAMPT in a novel manner. Further exploration into both the process by which WM responds to ischemic injury as well as the mechanism of WM protection by NAMPT will serve to further the development of effective therapeutics for stroke recovery.
Several animal models, such as ET-1 injection and chronic hypoperfusion models,26,27 have been developed to specifically examine WMI after stroke and do not involve significant gray matter injury. However, our initial findings that neuronal NAMPT confers ischemic WM protection and that neurons readily release NAMPT
In conclusion, the present study shows that NAMPT may not function simply in a restricted intracellular manner during stroke recovery and that it protects against ischemic injury in both gray matter and WM. We have presented multiple lines of evidence supporting the extracellular secretion of NAMPT by neuronal cells either overexpressing transgenic NAMPT or subjected to ischemic insults. This secretion is correlated with the protection of OLs from ischemic injury. Furthermore, the addition of recombinant NAMPT itself is protective against ischemic WMI. These findings promote the concept that NAMPT is an endogenous protective molecule that may circulate throughout neural tissue and exert beneficial effects on a variety of cell types, likely via distinct mechanisms. The increase of NAMPT in ischemic tissue from non-transgenic mice also supports the notion that NAMPT is part of a natural stress response, consistent with previous reports. 11 Thus, the preservation and recovery of WM by molecules such as NAMPT may lead to improvements in axonal repair and function and promote long-term behavioral recovery after cerebral ischemia.
Footnotes
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The authors thank Carol Culver for editorial assistance, Pat Strickler for secretarial support, Dr Joshua Sanes from Harvard University for providing Thy-1 promoter, and Dr Carlos Lois from Massachusetts Institute of Technology for providing the lentivirus system.