
Abstract
Despite extensive effort to elucidate the cellular and molecular bases for delayed cerebral injury after aneurysmal subarachnoid hemorrhage (aSAH), the pathophysiology of these events remains poorly understood. Recently, much work has focused on evaluating the genetic underpinnings of various diseases in an effort to delineate the contribution of specific molecular pathways as well as to uncover novel mechanisms. The majority of subarachnoid hemorrhage genetic research has focused on gene expression and linkage studies of these markers as they relate to the development of intracranial aneurysms and their subsequent rupture. Far less work has centered on the genetic determinants of cerebral vasospasm, the predisposition to delayed cerebral injury, and the determinants of ensuing functional outcome after aSAH. The suspected genes are diverse and encompass multiple functional systems including fibrinolysis, inflammation, vascular reactivity, and neuronal repair. To this end, we present a systematic review of 21 studies suggesting a genetic basis for clinical outcome after aSAH, with a special emphasis on the pathogenesis of cerebral vasospasm and delayed cerebral ischemia. In addition, we highlight potential pitfalls in the interpretation of genetic association studies, and call for uniformity of design of larger multicenter studies in the future.
Keywords
Introduction
Pathogenesis of Delayed Cerebral Ischemia and Cerebral Vasospasm after aSAH
Delayed cerebral ischemia (DCI) has been implicated as major, potentially preventable contributor to poor outcome after aneurysmal subarachnoid hemorrhage (aSAH) (Hijdra et al, 1987). It is a reversible failure of cerebral perfusion that can progress to infarction and has been shown to increase the risk of mortality and severe disability in patients with aSAH (Hijdra et al, 1988). Though DCI can result from cerebral vasospasm, not all patients with DCI have reported radiographic vasospasm, suggesting a more complex set of contributors (Dankbaar et al, 2009; Naidech et al, 2006; Stein et al, 2006a).
Pathogenetic mechanisms leading to DCI are multifactorial and are thought to include cortical spreading ischemia (Dorsch, 1995; Dreier et al, 1998, 2006
Vasospasm after aSAH is a complex entity composed of reversible vasoconstriction, coupled with dysfunction of normal cerebral vascular autoregulation (Khurana and Besser, 1997), leading to a reduction of cerebral blood flow and the development of regional cerebral ischemia (Keyrouz and Diringer, 2007). Proximal vessels at the base of the brain are preferentially affected by cerebral vasospasm, however distal arteries may also develop impaired autoregulation and lead to further alterations of blood flow (Soehle et al, 2004). The mechanisms underlying the development of cerebral vasospasm after aSAH have been the subject of extensive study, both in animal models and in human observational cohorts (Fisher et al, 1980; Horowitz et al, 1996; Zabramski et al, 1986). The process begins with the presence of breakdown products of blood in the subarachnoid cisterns, which incite a reactive process that ultimately results in spasm of the cerebral vasculature. On a molecular level, initiation of cerebral vasospasm by the putative spasmogens is manifested by both Ca2+-dependent and -independent processes, resulting in abnormal and prolonged constriction of vascular smooth muscle (Laher and Zhang, 2001; Tani, 2002; Tani and Matsumoto, 2004). A considerable body of experimental evidence implicates oxyhemoglobin as the primary spasmogenic agent in the development of vasospasm (Macdonald and Weir, 1991; Nishizawa and Laher, 2005). This is supported by observations of high concentrations of oxyhemoglobin in the cerebrospinal fluid of patients during the vasospasm period after aSAH, and that changes in oxyhemoglobin concentration show vasospasm evolution (Nishizawa and Laher, 2005). Furthermore, erythrocytes have been identified as a necessary component of blood for the induction of experimental cerebral vasospasm (Macdonald and Weir, 1991). However, evidence suggests that vasospasm after aSAH is dependent on multiple factors, as hemoglobin does not produce as severe spasm as whole blood, and hemolysates of erythrocytes are more potent spasmogens than Hb alone (Macdonald, 2001). In addition, arachidonic acid metabolites, endothelins, free radicals, serotonin, adenosine, and bilirubin oxidation products have all been implicated as having causative functions in the development of vasospasm (Asano et al, 1984; Cook and Vollrath, 1995; Sano et al, 1980; Suzuki et al, 1999).
Inflammation has also been implicated in the pathogenesis of cerebral vasospasm, as well as in the mechanisms of ensuing ischemic injury. For example, rapid upregulation of inflammation-related genes has been observed in spastic arteries (Dumont et al, 2003). Elevation of adhesion molecule levels and complement cascade proteins have also been noted in the cerebrospinal fluid and serum of patients after aSAH (Dumont et al, 2003; Fassbender et al, 2001; Mack et al, 2007). Furthermore, monoclonal antibodies directed against intercellular adhesion molecule 1 and the common β-chain of the integrin superfamily reduces vasospasm in a rabbit model (Bavbek et al, 1998). Further support for an inflammatory basis underlying SAH pathophysiology comes from several other reports describing an association between cytokines in the cerebrospinal fluid of patients with SAH and increased transcranial Doppler levels, and the attenuation of experimental vasospasm after complement inhibition (German et al, 1996).
Delayed Cerebral Ischemia and Vasospasm as Clinical Outcomes after aSAH
The understanding of these multiple pathogenic processes and recent clinical evidence highlight that a critical distinction must be made between end points such as radiographic vasospasm, symptomatic vasospasm, cerebral infarction, DCI, and other functional outcome measures. A number of studies have shown that not all patients with DCI have angiographic vasospasm, and that DCI is associated with poor outcome whereas angiographic vasospasm alone is not (Frontera et al, 2009; Schmidt et al, 2008). Furthermore, recent results from the CONSCIOUS-1 trial, a phase IIb, randomized, placebo-controlled trial of clazosentan administration (Macdonald et al, 2008), provide further evidence of a disconnect between angiographic vasospasm and poor outcome. Clazosentan, a nonpeptide endothelin receptor-A antagonist, was shown in high dose to be associated with a 65% relative risk reduction of moderate and severe vasospasm (Macdonald et al, 2008). Despite such a significant reduction in vasospasm, clazosentan-treated patients had no significant reduction in morbidity and mortality within 6 weeks of hemorrhage; secondary end points included cerebral infarct, delayed ischemic neurologic deficit due to vasospasm, and rescue therapy for radiographic vasospasm (Macdonald et al, 2008). Additional evidence regarding a disconnect between vasospasm and clinical outcome derives from study of the calcium channel blocker, nimodipine. This agent is the only approved drug that reduces the incidence of poor outcome after aSAH, and a systematic review shows that nimodipine reduces the risk of poor outcome without showing any clear effect on vasospasm (Feigin et al, 1998). Subsequent work suggested that nimodipine increases fibrinolytic activity, lending support for additional mechanisms of ischemic injury other than vascular contraction in patients with aSAH (Roos et al, 2001).
The disjunction between the rates of angiographic and clinical vasospasm after aSAH, as well as failure of Clazosentan to improve outcome despite effectively ameliorating vasospasm, has prompted efforts to better elucidate alternative contributors to DCI after aSAH (Hansen-Schwartz et al, 2007; Macdonald et al, 2008). In theory, genotypic variability may serve to define in part an individual's susceptibility to both the development of vasospasm and the injury incurred secondary to ischemia throughout both the early and chronic course of this disease. Understanding the genetic determinants of vasospasm and DCI may become invaluable in defining the pathophysiology of vasospasm, and may well aid in developing targeted therapies.
Genetic Association Studies of aSAH
Association studies constitute a common approach to investigating the genetic basis of aSAH. In this type of analysis, the frequency of a specific gene allele is determined and compared between aSAH patients and controls. Using this approach, it was found that a large number of genes have been implicated in the development and subsequent rupture of intracranial aneurysm (IAs), many related to structural proteins of the extracellular matrix that are suspected to be deficient in a majority of aneurysms (Nahed et al, 2007). These findings are supported by the fact that these matrix proteins are altered in various genetic diseases associated with high rates of aneurysm formation, such as adult polycystic kidney disease (Chapman et al, 1992), Marfan syndrome (ter Berg et al, 1986), and Ehlers–Danlos syndrome Type IV (de Paepe et al, 1988). Although the majority of genetic research has focused on gene expression and linkage studies of these markers as they relate to the development of IA and their subsequent rupture, far less work has centered on the genetic determinants of cerebral vasospasm, the predisposition to DCI, and the determinants of ensuing functional outcome after aSAH. The candidate gene suspects are diverse, and encompass multiple functional systems including fibrinolysis, inflammation, vascular reactivity, and neuronal repair.
The literature implicating a genetic basis for vasospasm, DCI, and other clinical outcomes has to date provided a preliminary framework in the form of small- to medium-sized genetic association studies. Authors have begun to define relevant genetic polymorphisms and their value in predicting clinical end points. However, the existing literature shows significant variability in the both the above-mentioned end points and their definitions, which highlights at once the wide influence of genetic variability in aSAH and the need for uniformity to define the genetic determinants of outcome more conclusively. To this end, we present a review of the current evidence implicating a genetic basis for outcome after aSAH and discuss potential pitfalls in the interpretation of often conflicting data resulting from genetic association studies.
Materials and methods
All combinations of the following keywords were used to query the PubMed database for potential studies: subarachnoid hemorrhage, outcome, genetics, vasospasm, polymorphism, aneurysm, delayed cerebral ischemia, and delayed ischemic neurological deficit (DIND). The searches were restricted to the time period January 1980 to November 2009. Studies of all design types were included in the initial survey. Reference lists of selected studies and tables of contents from selected journal websites were hand-searched for additional relevant publications. From the 5,310 articles initially retrieved, 640 were selected for more detailed analysis. Studies were only retained if they analyzed one or more genetic polymorphisms in a patient population after aSAH, with vasospasm, DCI, and/or a clinical outcome as outcome measures. Of 640 studies analyzed, 21 original studies met criteria and were included in the present review.
Genetic Outcome Studies of aSAH
Endothelial nitric oxide synthase:
Much of the work specifically investigating the genetic underpinning of vasospasm, both angiographic and clinical, has focused on the gene encoding the endothelial isoform of nitric oxide synthase (eNOS) (Pluta, 2005; Table 1). Nitric oxide (NO) normally inhibits inflammation and smooth muscle proliferation, both of which are pathologic changes that occur during cerebral vasospasm. Impairment of NO signaling is evident after SAH in experimental animals (Khurana and Besser, 1997; Weir and MacDonald, 1993), and abnormal cerebrospinal fluid NO levels have been reported in human patients after SAH (Sadamitsu et al, 2001). In gene-transfer experiments, eNOS overexpression in animal and human intracranial arteries is vasoprotective after aSAH (Khurana et al, 2000, 2002). Multiple investigations have established the functional relevance of human eNOS gene polymorphisms by showing associations of these with increased susceptibility to various pathologies, including coronary vasospasm and the formation of IAs (Khurana et al, 2004a; Yoshimura et al, 2000b).
Genetics studies of vasospasm and delayed cerebral ischemia after aSAH
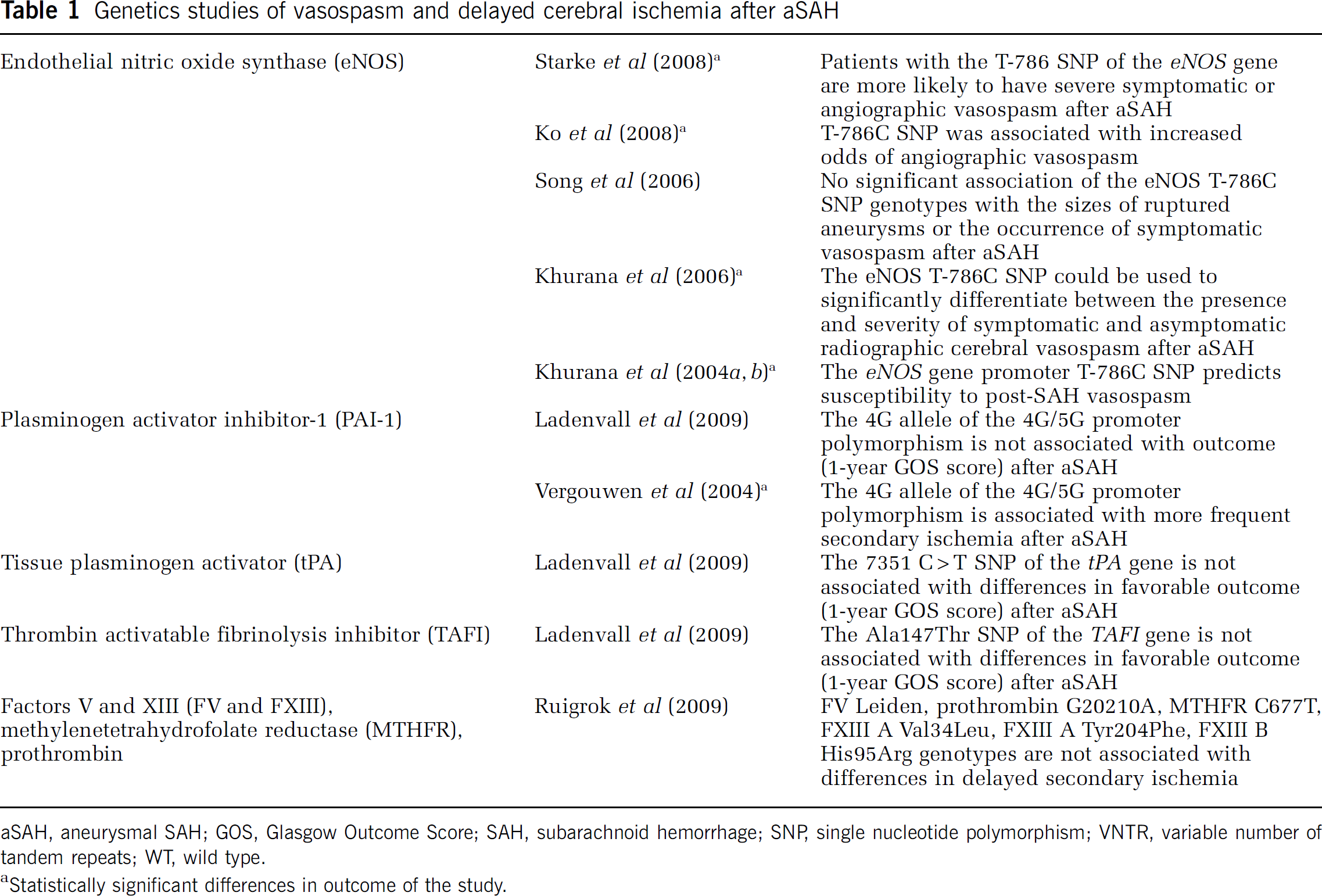
aSAH, aneurysmal SAH; GOS, Glasgow Outcome Score; SAH, subarachnoid hemorrhage; SNP, single nucleotide polymorphism; VNTR, variable number of tandem repeats; WT, wild type.
Statistically significant differences in outcome of the study.
Of the known eNOS gene polymorphisms, which include a variable number tandem repeat in intron 4 (Wang et al, 1996), a promoter region single nucleotide polymorphism (SNP, T-786C) (Yoshimura et al, 2000a), and a coding SNP in exon 7 (G894T) (Hingorani et al, 1999), the eNOS T-786C SNP has been most extensively investigated for associations with outcome after aSAH. The initial study used gene microarray technology and samples from 141 subjects; 90 controls were randomly selected from a nationwide adult registry and 51 persons were admitted with aSAH (Khurana et al, 2004b). Using multiple logistic regression analysis and adjustments for age, gender, and smoking history, the authors found a significant difference in the frequency of the eNOS variable number tandem repeat polymorphism between patients with aSAH compared with controls (
In another well-designed study, Ko et al (2008) prospectively enrolled a large cohort of patients admitted with aSAH (
The latest investigation into associations between eNOS and outcome after aSAH was performed by Starke et al (2008). In this study, 77 patients with aSAH were genotyped, prospectively enrolled and followed up for the occurrence of symptomatic or angiographic cerebral vasospasm. In a multivariate logistic regression, T-786C genotype was the only factor predictive of vasospasm, with an OR for symptomatic vasospasm of 3.3 in patients possessing one T allele (95% CI 1.1 to 10.0,
These interstudy differences also highlight the lack of understanding of the contributions of the various eNOS haplotype combinations to the pathophysiology of vasospasm after aSAH. One attempt to explain this discrepancy is that endothelial cell damage as a result of aSAH leads to a decrease in expression of the
Fibrinolysis-related genes:
In keeping with the integral involvement of microvascular thrombosis in the pathogenesis of cerebral ischemia (Siesjo, 1992), and the involvement of microthrombi in DCI after aSAH (Suzuki et al, 1990; Vergouwen et al, 2008) several investigators have attempted to determine associations between fibrinolytic genes and outcome (Table 1). In one study, Vergouwen et al (2004) investigated whether the plasminogen activator inhibitor-1 (PAI-1) 4G allele of the 4G/5G promoter polymorphism influences outcome after 126 patients with aSAH. They found that secondary ischemia occurred more often in patients harboring a 4G allele than in patients homozygous for the 5G allele (RR 3.3, 95% CI 1.1 to 10.0). Despite this, they noted no increased rate of rebleeding between the cohorts, but patients with the 4G genotype did tend to have higher risk of poor outcome (determined by 3-month Glasgow Outcome Scale (GOS) scores) relative to the 5G/5G genotype (RR 1.2, 95% CI 0.7 to 2.2). The authors concluded that the 4G allele of the
Another recent study investigated associations between several different genes involved in fibrinolysis and functional outcome after aSAH (Ladenvall et al, 2009). In this study, 183 patients presenting with aSAH were consecutively recruited and compared with healthy, age, sex, and region-matched controls. Single nucleotide polymorphisms of the tissue plasminogen activator, PAI-1, thrombin-activatable fibrinolysis inhibitor, and factor XIII
Similarly, a study by Ruigrok et al (2009) investigated polymorphisms in the Factor V (
Inflammatory genes:
As discussed previously, inflammation likely has a pivotal role in both the pathogenesis of cerebral vasospasm and as a downstream mediator of DCI. As such, polymorphisms of inflammatory genes are logical choices for investigation in genetic association studies (Table 2). Proinflammatory cytokines, however, have been shown to provide both detrimental and beneficial effects after brain injury depending on the spatial and temporal aspects of their expression (Stoll et al, 2002). Furthermore, proinflammatory cytokines result in increased expression of growth factors, particularly in the subacute and chronic phases of cerebral injury, and thus the role of inflammation after aSAH is likely extremely complex.
Genetics studies of clinical outcome after aSAH
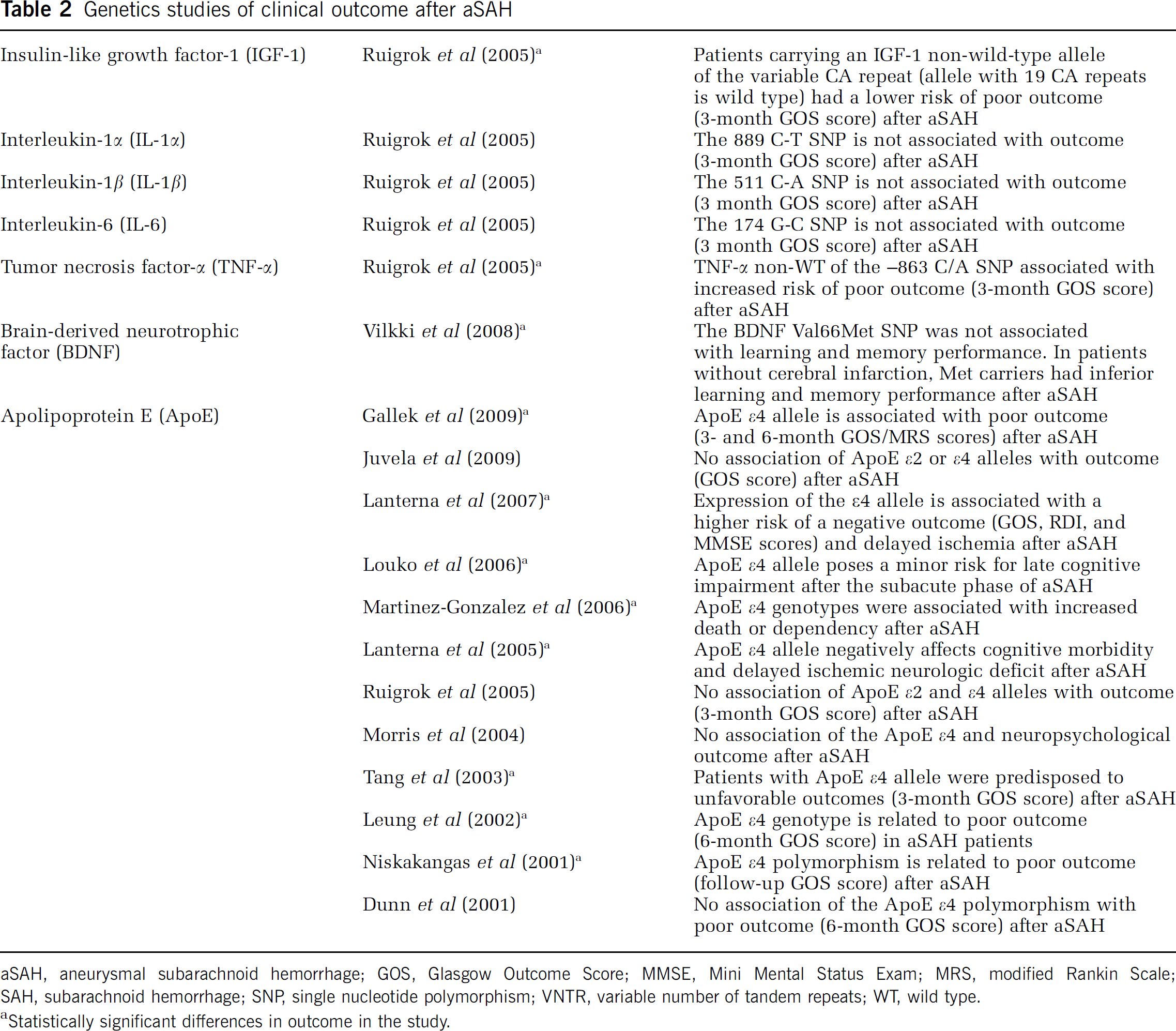
aSAH, aneurysmal subarachnoid hemorrhage; GOS, Glasgow Outcome Score; MMSE, Mini Mental Status Exam; MRS, modified Rankin Scale; SAH, subarachnoid hemorrhage; SNP, single nucleotide polymorphism; VNTR, variable number of tandem repeats; WT, wild type.
Statistically significant differences in outcome in the study.
Tumor necrosis factor-α (TNF-α) is one of the principal proinflammatory cytokines in the body, and is thought to have a role in the formation and rupture of IAs based on evidence showing significantly increased levels of TNF-α mRNA in IA samples (Jayaraman et al, 2005). The
Investigators have also examined whether functional polymorphisms of a number of ischemia-related genes including insulin-like growth factor-1 (
Although the previously discussed studies were mainly focused on identifying injurious gene polymorphisms and mechanisms, another approach is to investigate genes thought to be important for the process of functional recovery after aSAH. These genes include those coding for various growth factors, such as brain-derived neurotrophic factor (
Apolipoprotein E:
A myriad of studies have been performed over the past decade seeking to identify associations between apolipoprotein E (ApoE) and outcome after aSAH (Table 2). ApoE is a polymorphic protein that is associated with plasma lipoproteins and is involved in the metabolism and transport of lipids in the central nervous system. Three common alleles of the
The first genetic association study examining the association of ApoE alleles with aSAH included 126 patients and was performed in 2001 (Niskakangas et al, 2001). Apolipoprotein E genotype and outcome data (GOS) on follow-up examination (the specific time point was undefined) were examined in 86 patients for whom the data were available. In this study, ApoE ε4 genotype was strongly associated with unfavorable outcome (GOS score) after adjustment for age, rebleeding, clinical status on admission, and Fisher grade (OR 7.1, 95% CI 1.9 to 26.3;
A more subtle effect of the ApoE ε4 allele on outcome after aSAH may be revealed through the use of more sensitive outcome markers, such as neuropsychometric tests. Morris et al (2004) investigated whether the ApoE ε4 genotype was associated with cognitive and emotional outcome at a mean of 16 months after aSAH. There was no consistent association between the presence of the ε4 allele and performance on neuropsychometric tests, GOS, or SF-36. Lanterna et al (2005) also studied 101 noncomatose patients admitted to their institution between 1996 and 2003, with 26 patients found to have the ε4 allele. The presence of the ε4 allele negatively affected overall outcome, as assessed using the Rankin Disability Index and the Mini-Mental Status Exam at 6 months after SAH (P = 0.0087). Patients with the ε4 allele also experienced more frequent DIND (
In contrast, other studies suggest that the
Two studies focusing specifically on Asians were conducted and reached similar conclusions. Leung (2002) showed a significant association between the ApoE ε4 allele and poor outcome (GOS score) at 6 months in a multivariate model (OR 11.3, 95% CI 2.2 to 57.0;
Meta-analyses of ApoE genotype:
Given the large number of studies with conflicting results, meta-analysis of the relationship between ApoE genotype and functional outcome after SAH has been performed (Lanterna et al, 2007). The authors included eight observational studies, with 696 patients for the analysis of clinical outcome, and 600 patients for the analysis of delayed ischemia. They were able to obtain raw data from the original studies, allowing for analysis of the distribution of baseline variables, quantification of patients lost to follow-up, inclusion of pertinent patients, and better insight into population-stratification bias. In addition, effect sizes were pooled with a random effects model to account for underlying heterogeneity. The authors showed that the risk of a poor outcome (OR 2.558, 95% CI 1.61 to 4.065) as well as delayed ischemia (OR 2.044, 95% CI 1.269 to 3.291) was increased in carriers of the ε4 allele. Poor outcome was defined as death, functional dependency (GOS 2 to 3 or mRS 3 to 5), or severe cognitive impairment (Mini Mental Status Exam score < 24). The secondary end point was the development of DCI. Taking into account existing experimental evidence, the authors proposed that ApoE exerts it effects to scavenge free radicals, modulate the inflammatory response, and helps with membrane repair and synaptic plasticity. In addition, individuals with the ε4 allele likely have deficits in these critical functions, predisposing to poor outcome after cerebral injury. As such, it is likely that ApoE mediates susceptibility to, and recovery from, secondary ischemic injury after aSAH rather than having an integral part in the initial pathogenesis of this disease. The specific mechanism by which ApoE is associated with outcome after aSAH, as well as the functional effect of its various polymorphisms, remains unclear. Further studies are necessary to answer these questions.
Pitfalls of Genetic Association Studies in aSAH
Initial findings of genetic association studies of aSAH have often been difficult to replicate in later studies. Several factors can be identified as reasons why genetic studies in general, and those focusing on aSAH in particular, have encountered these difficulties. The single most common limitation is the small sample size included in these investigations. Aneurysmal SAH is a relatively low-incidence disease, thus the numbers of patients available for enrollment into genetic research protocols is limited, particularly for single-center studies. The negative effect of small sample sizes is compounded by the heterogeneity of the aSAH population in general (i.e., variations in Hunt–Hess grade, Fisher grade, aneurysm location/size, etc.), which further reduces the power of genetic studies to detect clinically significant associations. In addition, when investigations focus on a process that affects only a subset of aSAH patients, such as development of cerebral vasospasm, available subject numbers are reduced even further. The net result is that the majority of small, single-center genetic studies in aSAH are underpowered, increasing the likelihood of type II errors. Negative studies should therefore be viewed critically, particularly when the number of subjects is small or the end point is a relatively low-frequency event (i.e., infarction from vasospasm). For this reason, publication bias is a significant issue in this field. Nevertheless, negative studies, even if underpowered, have the potential to be useful, particularly in providing pilot data that can then be used to design an adequately powered investigation. Dissemination of these results to the scientific community should therefore be encouraged.
Also thought to affect the power of association studies is the type of genetic marker used in the analysis. The majority of aSAH association studies have focused on SNP analysis rather than haplotype analysis, which serves as another possible explanation for inconsistent, sometimes contradictory results seen across multiple studies. A haplotype is a set of genetic markers, as in a set of SNPs, transmitted together on the same chromosome. Haplotype analyses, in some instances, are thought to provide greater statistical power than association studies based on their underlying SNPs (Bader, 2001). An alternative source of error is rooted in the design of studies requiring selection of a control group. Control group selection in these populations is crucial, as any unknown allele variation between cases and controls may appear as disease association. This becomes particularly relevant in eNOS association studies, for example, the majority of which are not matched for ethnicity. Failure to properly match for ethnicity is problematic in light of recent evidence showing striking interethnic disparities in the distribution of eNOS polymorphisms among African Americans and Caucasian Americans (Tanus-Santos et al, 2001), as well as black, white, and Amerindian Brazilians (Luizon et al, 2009).
Much of the inconsistency in genetic association studies across multiple disease processes may also result from methodological differences between studies (Dichgans and Markus, 2005). Perhaps the greatest impairment to interpreting data from genetic studies is their lack of consensus regarding the definition of clinical events and the use of varying outcomes measures. Widely disparate end points (Table 3) limit the ability to synthesize data from multiple smaller genetic studies of aSAH and perform adequate meta-analyses. For example, the definition of vasospasm may alternatively be defined as angiographic vasospasm, vessel narrowing as detected by transcranial Doppler ultrasound, or on a clinical basis only (DIND) depending on the individual study. In fact, genetic association studies frequently define vasospasm using measures other than conventional angiography (Tables 1 and 3), the gold standard for radiographic confirmation of vasospasm. Aside from lack of uniformity across studies, these alternative definitions preclude a conclusive determination regarding genetic susceptibility to radiographic vasospasm because of their relative lack of sensitivity. Transcranial Doppler ultrasound, for example, was determined in a meta-analysis as approximately 67% sensitive for middle cerebral artery spasm and 42% sensitive for anterior cerebral artery spasm (Lysakowski et al, 2001).
Comparison of outcomes evaluated in genetic studies of aSAH
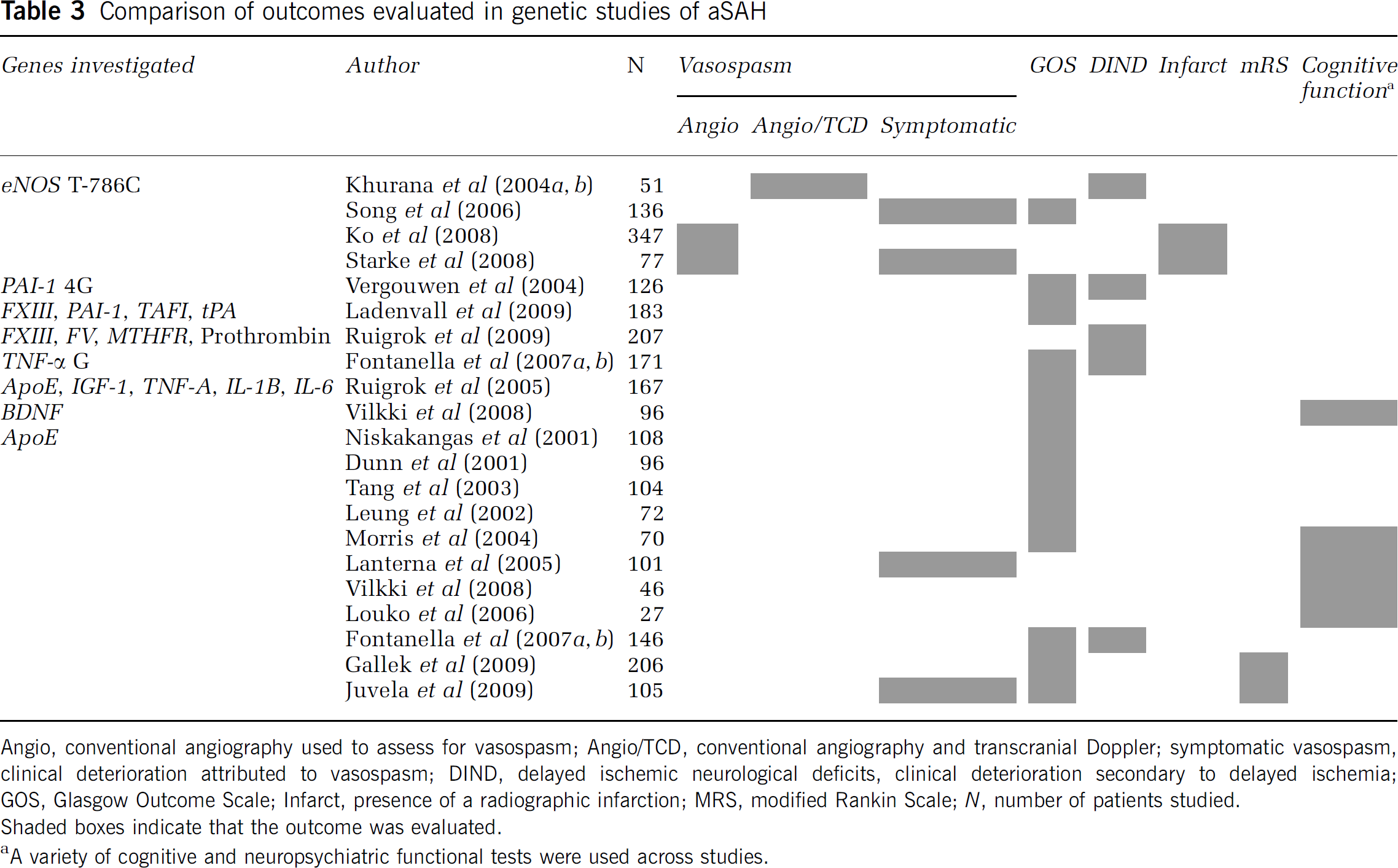
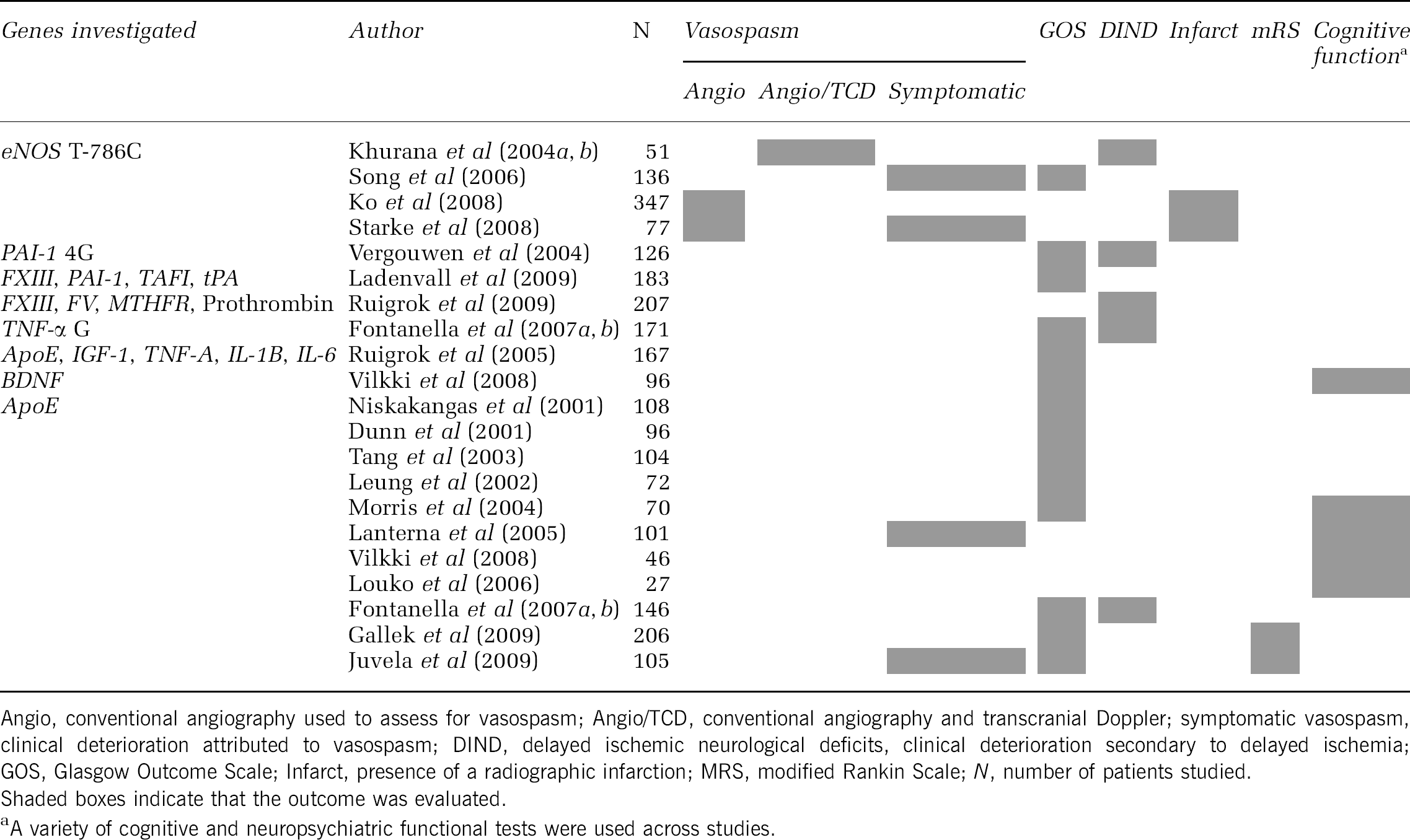
Functional outcome has been assessed using an assortment of grading scales at variable time points, including the mRS, GOS, extended Glasgow Outcome Scale (GOS-E), and neuropsychometric scales. This is compounded by the fact that neurologic outcome is complex, making definition of target phenotypes essential. For instance, many genetic studies have appropriately distinguished between stroke subtypes in large studies (i.e., aSAH versus ICH), however this also significantly decreases the number of available subjects for study. Studies that use more sophisticated outcome measures targeting specific domains of neurologic recovery may more successfully uncover genetic associations.
Although type II error is arguably the greatest limitation of genetic studies of aSAH, positive associations should likewise be viewed critically, as contributions to the observed effect by other factors, both clinical and genetic, may remain uncharacterized. In addition, multiple testing increases the possibility of false-positive associations if appropriate corrective statistics are not used, such as the Bonferroni procedure (Zou and Zuo, 2006). Moving forward, large, multicenter, prospective studies that are able to enroll greater numbers of subjects and investigate the effect of multiple genes/polymorphisms simultaneously and combine these results in a multivariate model are likely to provide the most robust data. As such, collaboration and sharing of data between investigators and research centers is paramount and should be fostered.
Conclusion
Increasing evidence points to a genetic role in the pathophysiology of the ensuing vascular response with development of cerebral vasospasm, occurrence of DCI, and impairment of functional outcome after aSAH. These studies suggest that the mechanisms of injury after aSAH may result from an association of varied gene products throughout the progression of the disease process. However, given the inconsistencies observed between many of these emerging studies, researchers must interpret data and conclusions with caution, and it is essential to reproduce previous results, particularly for smaller genetic studies. Large, multicenter genetic studies involving collaborations between several tertiary care centers will be required to provide the most rigorous results. Moving forward, investigators designing both small- and large-scale genetic association studies should determine consensus outcome definitions and relevant outcome selection early in the course of disease study. Uniformity in study design will allow for more informative results, and will enable investigators to pool data in the event of conflicting results and inadequate power. Although further research is needed, insight into the genetic determinates of outcome after aSAH will help to unravel the complex pathophysiology of this disease and ultimately translate into improved care for these patients.
Footnotes
Acknowledgements
We thank Dr Emilia Bagiella (the Mailman School of Public Health, Columbia University) for her thoughtful appraisal of the paper.
The authors declare no conflicts of interest.