
Abstract
Patients with aneurysmal subarachnoid hemorrhage (SAH) who experience delayed cerebral ischemia (DCI) have an increased risk of poor outcome. Delayed cerebral ischemia is considered to be caused by vasospasm. However, not all patients with DCI have vasospasm. Inversely, not all patients with vasospasm develop clinical symptoms and signs of DCI. In the past, treatments aiming at vasospasm were not successful in preventing ischemia. The purpose of this review is to give an overview of clinical data showing that DCI cannot always be attributed to vasospasm, and to present an in-depth analysis of clinical and autopsy studies on the role of microthrombosis in the pathogenesis of DCI. Clinical studies show that DCI is associated with an activation of the coagulation cascade within a few days after SAH, preceding the time window during which vasospasm occurs. Furthermore, impaired fibrinolytic activity, and inflammatory and endothelium-related processes, lead to the formation of microthrombi, which ultimately result in DCI. The presence of microthrombi is confirmed by autopsy studies. Insight in the pathophysiology of DCI is crucial for the development of effective therapies against this complication. Because multiple pathways are involved, future research should focus on drugs with pleiotropic effects.
Introduction
During aneurysmal subarachnoid hemorrhage (SAH), intracranial pressure increases, which results in a sharp decrease in cerebral perfusion pressure. This may lead to global (acute) cerebral ischemia and loss of consciousness. From day 4 to day 10 after SAH, patients are at increased risk of developing delayed cerebral ischemia (DCI), which may present as headache, confusion, focal neurologic deficits, and/or a deterioration of the level of consciousness (Hijdra et al, 1986; Roos et al, 2000). Delayed cerebral ischemia is sometimes reversible, but may also progress to infarction, which is associated with increased mortality and severe disability (Rabinstein et al, 2004).
The cause of DCI, also known as delayed ischemic neurologic deficit, is unknown. In the past decades, DCI and cerebral infarction after SAH have been associated with the presence of vasospasm in several studies (Fisher et al, 1977; Rabinstein et al, 2004; Fergusen and Macdonald, 2007). In fact, many clinicians use the term vasospasm to describe clinical signs and symptoms of DCI. Radiologic vasospasm should be distinguished from symptomatic vasospasm. Radiologic vasospasm is the narrowing of one or more of the major cerebral arteries observed at conventional, magnetic resonance, or computed tomographic angiography. Increased blood flow velocities on transcranial Doppler (TCD) examination are also indicative of vasospasm. Conventional angiography is still the gold standard to detect vasospasm, which is present in up to 70% of patients after SAH (Ohta and Ito, 1981; Vora et al, 1999). Symptomatic vasospasm is defined as clinical features of DCI combined with radiologic vasospasm in the same region, and occurs in a much lower percentage of patients (approximately 22%) (Dehdashti et al, 2004).
To date, nimodipine is the only drug that reduces the incidence of DCI and decreases the risk of poor outcome after SAH (Rinkel et al, 2005). Nimodipine, being a calcium antagonist, was first considered to decrease the incidence of DCI by preventing and diminishing vasospasm as a result of relaxation of the muscular vessel wall. However, a systematic review showed that nimodipine reduces the incidence of DCI without a clear effect on vasospasm (Feigin et al, 1998). A later study showed that nimodipine increases fibrinolytic activity in patients with SAH, and thereby may decrease the incidence of DCI (Roos et al, 2001). After it was observed that nimodipine reduces the incidence of DCI and decreases the risk of poor outcome after SAH without a clear effect on vasospasm, it is increasingly recognized that DCI cannot be fully explained by the development of vasospasm.
As a result, several additional hypotheses besides macrovascular vasospasm have been postulated to explain the occurrence of DCI. Some data suggest that DCI might be caused by constriction of cerebral microcirculation. This hypothesis of small artery spasm was mainly investigated in animal models that showed conflicting results (Herz et al, 1975; Ohkuma et al, 1997; Perkins et al, 2002). Studies that showed microcirculatory spasm after SAH in humans were not able to show a direct relation to the pathogenesis of DCI (Uhl et al, 2003). Thus, although small-artery spasm may occur after SAH, future studies are needed to elucidate its role in the pathophysiology of DCI. Another hypothesis is that of cortical spreading ischemia, which might be related to the microvascular spasm hypothesis (Dreier et al, 1998, 2006). This hypothesis is based on the association between DCI after SAH and repeated spreading depolarizations with prolonged electrocorticographic depression periods. The presence and absence of a delayed cluster of spreading depolarizations have high positive and negative predictive values (86% and 100%, respectively) for DCI. In SAH, spreading depolarizations might result from the breakdown products of erythrocytes in the subarachnoid space (Dreier et al, 2000). These local toxic effects of hemolysis products evoke spreading depolarizations, which induce transient but long-lasting microvascular spasm and in turn cause cortical spreading ischemia (Dreier et al, 2002; Shin et al, 2006). The beneficial effect of nimodipine in patients with SAH can also be explained by the observation that nimodipine transforms cortical spreading ischemia back to cortical spreading hyperemia (Dreier et al, 1998). Interestingly, cortical spreading depression is also assumed to be the pathophysiological correlate of the migraine aura. In a recent case-control study, it was suggested that migraine in women is a risk factor for DCI (Dreier et al, 2007).
In recent years, several studies showed that besides vasospasm microthrombosis might also play a role in the development of DCI. In this review, we will first present recent clinical evidence that DCI cannot always be attributed to vasospasm. There-after, we will present an in-depth analysis of clinical and autopsy studies on the role of microthrombosis in the pathogenesis of DCI. Data from experimental studies have not been included in the review. An insight into the mechanisms involved in DCI is crucial for the development of new and effective therapies against this much-dreaded complication.
Clinical evidence against vasospasm as the only explanation for delayed cerebral ischemia
Since the incidence of angiographic vasospasm is up to 70% and that of DCI around 30%, it is evident that not all patients with vasospasm develop clinical signs of ischemia (Ohta and Ito, 1981; Vora et al, 1999; Roos et al, 2000). The positive predictive value of angiography and TCD combined to predict cerebral infarction is 67% (Rabinstein et al, 2004). The location of angiographic vasospasm correlates with the location of cerebral infarction in 25% to 81% cases (Rabinstein et al, 2004, 2005; Weidauer et al, 2007). Although vasospasm remains one of the main predictors of cerebral infarction in patients with SAH, in one-fifth to one-third of patients with cerebral infarction, clinical or radiologic variables do not predict the location of infarction (Rabinstein et al, 2004; Weidauer et al, 2007). Furthermore, the temporal relation between angiographic vasospasm and symptoms of DCI is often poor, as symptoms often occur several days after peak vasospasm. In patients experiencing DCI, positron emission tomographic cerebral perfusion patterns do not correlate with increased blood flow velocities at TCD (Minhas et al, 2003). Finally, patients with SAH may also develop ischemia and cortical band-like infarctions without any evidence of vasospasm (Rabinstein et al, 2004; Naidech et al, 2006; Lee et al, 2006; Weidauer et al, 2007, 2008).
When treatment is considered, evidence is also accumulating that DCI cannot be fully attributed to vasospasm. Patients experiencing delayed ischemia are often treated with triple H (hemodilution, hypervolemia, hypertension) therapy. The rationale for this treatment is that triple H reduces the clinical signs and symptoms of cerebral ischemia by a beneficial effect on vasospasm. A systematic review investigating the effect of the triple H therapy indeed showed a significant reduction of angiographic vasospasm, but without a reduction of DCI (Treggiari et al, 2003).
Modern treatment strategies often suggest endovascular treatment of vasospasm as a treatment option. A nonrandomized study investigating the effect of balloon angioplasty on the evolution of vasospasm-related cerebral infarction showed a significantly lower incidence of infarctions in the distribution of vessels undergoing angioplasty (Jestaedt et al, 2008). However, a recent large randomized trial investigating the effect of prophylactic endovascular treatment showed a nonsignificant reduction of DCI without a beneficial effect on outcome (Zwienenberg-Lee et al, 2008). Thus, although transluminal angioplasty and intraarterial drug administration reduce vasospasm, improvement in clinical outcome is less obvious (Polin et al, 2000). Some patients show direct clinical improvement after angioplasty, but this effect is often not sustained. This can result from a subsequent reduction in vessel caliber, but might theoretically also result from other pathophysiological pathways leading to DCI.
As endothelin has been suggested to play a role in the pathophysiology of vasospasm, it was hypothesized that drug-induced antagonism of endothelin receptors might decrease the incidence of vasospasm. The drug TAK-044 was investigated in a double-blind, randomized trial, which showed no effect on the occurrence of DCI, the incidence of cerebral infarction on computed tomographic scan, or on clinical outcome (Shaw et al, 2000). The lack of clinical efficacy was attributed to unselective inhibition of both the ETA and ETB receptors, which respectively mediate the vasoconstrictive and vasodilative effects of endothelin-1. Thereafter, it was suggested that the ETA receptor antagonist clazosentan could decrease vasospasm after SAH. Indeed, in a phase IIa randomized, controlled trial, clazosentan reduced the frequency and severity of vasospasm (Vajkoczy et al, 2005). Clazosentan might also reduce vasospasm-associated impairment of cerebral perfusion (Barth et al, 2007). However, no statistically significant reduction in the incidence of cerebral infarction was observed (Vajkoczy et al, 2005). A phase III study is presently being conducted.
Finally, studies investigating the effect of the antifibrinolytic agent tranexamic acid showed a significantly decreased incidence of rebleeds after SAH. However, there was no effect on the overall outcome as a result of an increased incidence of DCI, and impeded recovery from DCI, if this occurred (Vermeulen et al, 1984; Roos, 2000; Roos et al, 2003). As tranexamic acid does not influence the rate of cerebral vasospasm, direct hemostatic effects are a more plausible explanation for the increased incidence of DCI (Tsementzis et al, 1990). Therefore, it seems that the incidence and recovery from DCI can be influenced by drugs without a clear effect on vasospasm. Other trials showed that intracisternal thrombolysis with thrombolytic drugs decreases the incidence of DCI and improves outcome after SAH (Kinouchi et al, 2004; Kawamoto et al, 2004; Amin-Hanjani et al, 2004). The beneficial effect of intracisternal thrombolysis is less likely to be intravascular and probably results from lower degrees of vasospasm in recombinant tissue plasminogen activator (tPA)-treated patients (Findlay et al, 1995; Suzuki et al, 1998; Kawada et al, 1999). It could be possible that this effect is directly exerted on the blood vessel tone or related to increased subarachnoid blood clearance (Nassar et al, 2004; Reilly et al, 2004). Since an autopsy study showed that intravascular microclot burden is associated with the volume of subarachnoid blood, it could be that increased subarachnoid blood clearance as a result of intracisternal thrombolysis leads to a lower microclot burden and therefore a lower incidence of DCI (Stein et al, 2006).
In summary, clinical deterioration after SAH cannot always be attributed to radiologic vasospasm. Therapies aiming at radiologic vasospasm, such as endothelin receptor antagonists, were not successful, as no clear effect on DCI was observed. In contrast, drugs not aiming at vasospasm, such as antifibrinolytic agents, influenced the incidence of DCI. Apparently, other factors besides vasospasm also play an important role.
Supporting evidence from serological and cerebrospinal fluid examinations
In the following paragraphs we will present the results of various laboratory investigations in SAH patients, supporting the evidence that DCI cannot be fully attributed to vasospasm and that microthrombosis might be an additional explanation. A schematic overview of several processes involved in the pathogenesis of DCI is presented in Figure 1.
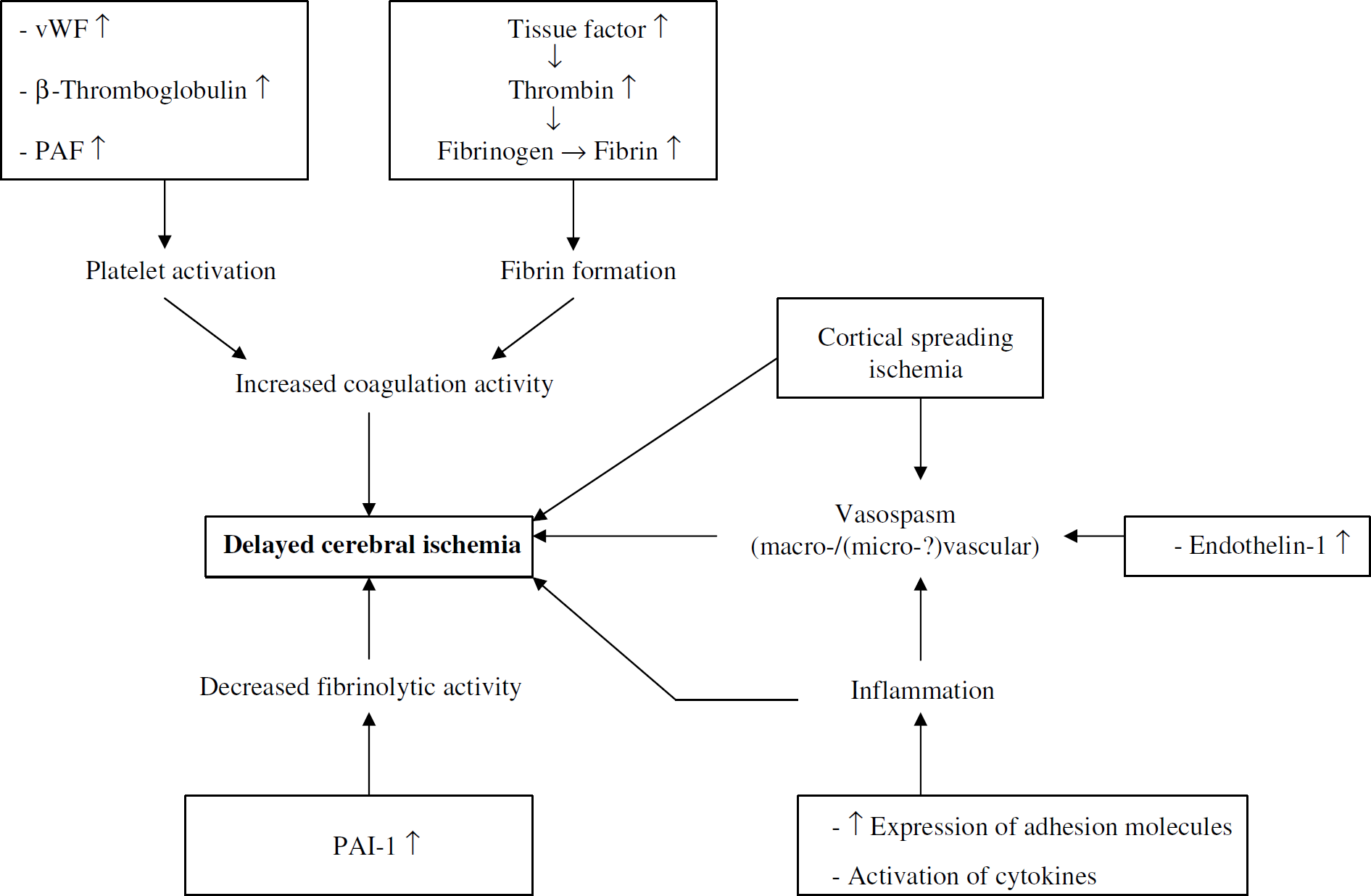
Schematic overview of pathways leading to delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. PAF, platelet-activating factor; PAI-1, plasminogen activator inhibitor-1; vWF, von Willebrand factor.
Activation of the Coagulation Cascade
In general, after vessel injury, platelet adhesion to the endothelium is one of the earliest steps in the coagulation pathway. Platelets are activated by several factors, such as collagen, von Willebrand factor (vWF), and thrombin. On activation of platelets, exocytosis of platelet granules results in the release of a wide array of procoagulant substances, which ultimately results in the formation of a stable hemostatic clot of both platelets and fibrin complexes.
Several studies in patients with aneurysmal SAH show that levels of serological coagulation markers correlate with the development of DCI and cerebral infarction (Ohkuma et al, 1991; Hirashima et al, 1997a; Peltonen et al, 1997; Suzuki et al, 1999;
The above-mentioned studies show that the coagulation cascade is already activated within a few days after SAH, preceding the time window during which vasospasm occurs. Although this early activation of the coagulation pathway may result from acute ischemia during the ictus of the hemorrhage, there is evidence from several studies that it is an early predictor of the occurrence of DCI and infarction after SAH.
Finally, evidence of coagulation activation is provided by the detection of micro-emboli with TCD examination (Giller et al, 1998). In the first study that described the presence of micro-emboli in SAH patients, micro-emboli were observed in only 4% of patients during routine vasospasm monitoring (Giller et al, 1998). The possibility that micro-emboli play a role in the pathophysiology of DCI was highlighted by the presence of low-density areas on computed tomographic scans from 82% of 11 SAH patients with micro-emboli compared with low-density areas in 24% of 123 patients without micro-emboli. Later, in a well-performed prospective study in which SAH patients were systematically monitored with TCD ultrasonography, micro-embolic signals were present in up to 70% patients, with a trend toward an increased incidence of micro-emboli in patients with symptomatic vasospasm (Romano et al, 2002). In contrast, the incidence of micro-embolic signals was lower in vessels with radiologic vasospasm compared with those without vasospasm, although not significantly different, which suggests that micro-emboli are probably not a direct result of vasospasm. As the incidence of micro-emboli was similar in vessels with and without proximal aneurysm, it is also unlikely that micro-emboli are generated within the aneurysmal sac. A trend was observed toward an increased incidence of emboli in patients with coiled aneurysms compared with clipped aneurysms; however, because of the small number of patients available for subgroup analyses, no preliminary conclusion can be drawn. In the absence of possible cardiac and carotid artery sources of emboli, it was concluded that micro-emboli in SAH patients are probably generated in large intracranial vessels as a result of a generalized hyperaggregable condition.
The micro-emboli studies are of special interest as they give a possible explanation for the observation that not all patients with vasospasm have neurologic deterioration. Probably, vasospasm is at higher risk to become symptomatic when the coagulation system is simultaneously strongly activated. Furthermore, the micro-emboli studies give an explanation why patients can experience DCI in the absence of vasospasm, as the coagulation cascade can also be activated in the absence of vasospasm. However, because not all patients with micro-emboli have symptoms of DCI, the relation of micro-emboli and DCI is not bidirectional. An inconsistent relation between micro-emboli and cerebral infarction was also observed in patients undergoing carotid endarterectomy or carotid stent placement (Wolf et al, 2004; Pinero et al, 2006). Thus, vasospasm and micro-emboli should not be considered as two separate entities in the pathogenesis of DCI, but rather as complimentary entities.
Impairment of the Fibrinolytic Cascade
As described before, clinical trials investigating the effect of antifibrinolytic drugs illustrate that the incidence of DCI can be influenced by drugs without any effect on radiologic vasospasm. Other studies support the observation that the fibrinolytic system is involved in the pathogenesis of DCI.
The natural ‘antagonist’ of thrombin generation is obviously fibrinolysis, resulting in the degradation of fibrin by the enzyme plasminogen. The activity of plasminogen is regulated by an activator, tPA, balanced by an inhibitor of this activator, plasminogen activator inhibitor-1 (PAI-1). Patients with DCI after SAH have significantly higher levels of PAI-1 antigen in the CSF compared with patients without DCI, suggesting that overactive inhibition of fibrinolysis is associated with DCI (Ikeda et al, 1997). In another study, nimodipine was associated with increased tPA activity and decreased PAI levels after SAH, whereas patients not receiving nimodipine did not show any change in the tPA/PAI balance (Roos et al, 2001). Interestingly, discontinuation of nimodipine resulted in normalization of both tPA and PAI-1 toward baseline levels. The results of this study imply that nimodipine, like other dihydropyridine calcium antagonists, increases fibrinolytic activity and, thereby, possibly decreases the incidence of DCI after SAH (Roos et al, 2001; Vergouwen et al, 2007). As a systematic review showed that nimodipine reduces the incidence of DCI without a clear effect on vasospasm, the study on the relation between nimodipine and fibrinolytic activity in SAH patients provides an alternative explanation for the beneficial effect of nimodipine (Feigin et al, 1998). A genetic study showed that PAI-1 polymorphism is also involved in the occurrence of DCI after SAH (Vergouwen et al, 2004). Patients with the 4G allele, who have impaired fibrinolytic activity, compared with patients homozygous for the 5G allele, are at increased risk to develop DCI. A trend was noted toward increased poor outcome in patients with the 4G allele. A study investigating the level of plasmin/alpha-2-antiplasmin complex, another indicator of fibrinolytic activity, in the acute phase of SAH, found no association with the occurrence of DCI (Itoyama et al, 1994).
Elevated levels of fibrin degradation products in the CSF, and of D-dimer, are also associated with the occurrence of delayed ischemia, cerebral infarction, and poor outcome after SAH (Peltonen et al, 1997; Vermeulen et al, 1985; Nina et al, 2001). Although increased CSF concentrations of fibrin degradation products suggest locally increased fibrinolytic activity, it can also reflect a damaged blood-CSF barrier as a result of ischemia (Ikeda et al, 1997; Vermeulen et al, 1985).
Activation of Inflammation
Besides the coagulation and fibrinolytic cascade, inflammatory processes are likely to be involved in the development of DCI after SAH. As discussed previously, patients with DCI after SAH also have increased concentrations of sP-selectin, an adhesion molecule involved in leukocyte adherence to the endothelium. During an inflammatory reaction, stimuli, such as thrombin, lead to the mobilization of P-selectin from the endothelium. Cytokines such as tumor necrosis factor-α stimulate the expression of P-selectin. Adhesion molecules bind to leukocytes, which results in slower leukocyte rolling and subsequent leukocyte adhesion. Consequently, leukocytes can penetrate the vessel wall. Several proinflammatory cytokines, such as interleukin (IL)-1, IL-6, and IL-8, are associated with vasospasm and DCI after SAH (Osuka et al, 1998; Hendryk et al, 2004). It is not completely understood how cytokines lead to ischemia. Interleukin-6 has vasoconstrictive properties
Endothelium Related Processes
Many parameters described above, such as vWF and P-selectin, are released by the endothelium or regulate processes within the endothelium. Another peptide released by the endothelium is endothelin, which is presently the peptide with the most potent vasoconstrictive properties. Endothelin-mediated vasoconstriction has been implicated in the pathophysiology of SAH-related vasospasm. However, data on the relation between endothelin-1 concentrations and DCI are conflicting (Juvela, 2000; Kessler et al, 2005). Interestingly, a phase IIa study investigating the effect of the selective ETA receptor antagonist clazosentan found a significantly decreased incidence and severity of vasospasm, without clear effect on cerebral infarction (Vajkoczy et al, 2005). Obviously, therapies aiming at preventing DCI after SAH should not selectively focus on vasospasm, but also on other processes involved in the pathogenesis of DCI.
Evidence from autopsy studies
Although pathology studies investigating SAH patients are rare, the available autopsy studies show that vasospasm is not the only cause of DCI. A landmark autopsy study from 1964, investigating 159 SAH patients who died more than 24 h after hospital admission, showed that angiographic vasospasm was present only in 37% of 119 patients with cerebral infarction and in 12% of patients without infarction (Crompton, 1964). Thus, a bidirectional relation between vasospasm and cerebral infarction is unlikely. Although pathologic changes are found in all layers of vasospastic vessels, ranging from concentric intimal thickening by subendothelial fibrosis to necrosis in the tunica media, pathologic findings are not restricted to the vessel wall alone (Hughes and Schianchi, 1978). Micro-thrombi were detected for the first time in 1983 upon autopsy in a patient with cerebral infarction after SAH, and later confirmed in other studies (Suzuki et al, 1983, 1990; Neil-Dwyer et al, 1994; Stein et al, 2006). In an autopsy study investigating six SAH patients, including four patients who supposedly died from DCI, it was observed that patients who die from delayed ischemia have significantly more microthrombi in clinically ischemic regions and in areas showing cerebral infarction on computed tomographic scan, when compared with patients who die from rebleeding or acute hydrocephalus (Suzuki et al, 1990). Micro-thrombi mostly consisted of a combination of fibrin thrombi and aggregated platelets, eventually mixed with multinuclear leukocytes. No distinction was made between the presence of microthrombi in symptomatic and asymptomatic vasospastic vessels. However, because microclots are not only present in regions with clinical signs of delayed ischemia or infarction but are widespread in the brain, an additional factor is apparently necessary for the development of clinical signs of focal cerebral ischemia (Suzuki et al, 1990; Neil-Dwyer et al, 1994; Stein et al, 2006). This is supported by a recent autopsy study that investigated 29 SAH patients and showed pathologic evidence of ischemia in 93% patients, but clinical signs of DCI were present in only 48% of the studied patients (Stein et al, 2006). These data suggest that microclot formation and vasospasm are probably complementary mechanisms in the pathogenesis of DCI. Interestingly, not only vasospasm but also microclot burden is influenced by the volume of subarachnoid blood (Stein et al, 2006). In a large autopsy study investigating microthrombosis in SAH patients, which showed that cortical ischemic lesions were present in 41 of the 53 patients studied, no significant association was observed between the presence of cortical ischemic lesions and angiographic vasospasm or location of the aneurysm (Neil-Dwyer et al, 1994). Interestingly, 80% patients with moderate/severe cortical ischemic lesions had fluctuating blood pressures, implying a profound disturbance of cerebral autoregulation, which particularly affects the cortical microcirculation.
Studies investigating drugs to prevent delayed cerebral ischemia: future directions
As activation of several cascades leading to the formation of microthrombi might be involved in the pathogenesis of DCI, treatments that aim at coagulation or fibrinolysis presently attract much attention. One studied agent was aspirin, which might reduce the incidence of DCI by antagonizing increased platelet activity. However, a randomized, controlled trial investigating the effect of aspirin was stopped prematurely, as an interim analysis showed that the probability of a beneficial effect was negligible (van den Bergh and MASH Study Group, 2006). Explanations provided were that a dosage of 100 mg once daily might not be powerful enough, or that aspirin might not be the right antiplatelet drug for preventing DCI. Another explanation can be that the effect of aspirin is too selective by only inhibiting platelet function and not affecting vasospasm. Enoxaparin, a low-molecular-weight antithrombin III agonist, was studied in two randomized, controlled trials with contradictory results (Wurm et al, 2004; Siironen et al, 2003). A large, well-powered, randomized, controlled trial is needed to investigate whether enoxaparin is beneficial to prevent DCI.
Other studies investigated the effect of intrathecal fibrinolysis. A systematic review found a significant 14% absolute risk reduction of delayed ischemia, and a significant 10% absolute risk reduction of poor outcome (Amin-Hanjani et al, 2004). However, of the nine included studies, only one study was randomized. Thus, a well-powered, randomized trial is also needed to investigate the effect of intrathecal fibrinolysis. Besides, if beneficial, it remains to be investigated whether this effect results only from a decreased incidence of vasospasm or from increased intravascular fibrinolytic activity as well.
Presently, magnesium sulfate is under intensive investigation as a potential agent to reduce the incidence of DCI after SAH and improve outcome. Magnesium sulfate is a noncompetitive calcium channel antagonist, which probably does not reduce the incidence of vasospasm after SAH, but possibly reduces its duration (Macdonald et al, 2004; Wong et al, 2006). No effect has been shown on the fibrinolytic system or coagulation cascade. In patients with preeclampsia, magnesium sulfate decreases endothelin-1 levels (Mastrogiannis et al, 1992). The pathophysiology of preeclampsia has much in common with that of DCI after SAH. Although magnesium sulfate has been shown to be beneficial in patients with preeclampsia by reducing neurologic complications and improving outcome, the mechanism of improvement by this treatment has not yet been elucidated in patients with preeclampsia.
Currently, the role of statin treatment as an additional strategy to prevent DCI is also being investigated. The possible beneficial effect of statins attracted attention when two studies showed that patients who use statins during aneurysmal hemorrhage less often have vasospasm and cerebral infarctions than patients not using statins (Parra et al, 2005; McGirt et al, 2006). However, in another study, the incidence of vasospasm, but not of DCI, was increased in patients using statins prior to SAH (Singhal et al, 2005). Because of the interesting observation in two of these studies, the effect of statin treatment in SAH patients was investigated in two small randomized, placebo-controlled clinical trials. The results of these trials were impressive, as acute statin treatment decreased the incidence of not only vasospasm, but also of DCI and mortality (Lynch et al, 2005; Tseng et al, 2005, 2007). Statins may be an interesting treatment option in patients with SAH as statins possibly not only decrease the incidence of vasospasm, but also reduce plasma levels of vWF, indicating decreased endothelial activation (Lynch et al, 2005; Tseng et al, 2005). Furthermore, statins have been shown to enhance fibrinolysis and inhibit blood coagulation (Krysiak et al, 2003). However, a recent retrospective cohort study showed that after implementation of the routine use of statin treatment in SAH patients, the incidence of vasospasm, DCI, cerebral infarction, and poor outcome did not change (Kramer et al, 2008). Therefore, more randomized studies are needed to investigate the effect of acute statin treatment in patients with SAH, and whether statins exert cholesterol-dependent or independent effects in this group of patients.
We conclude that further insight into the pathogenesis of DCI is instrumental in optimizing future strategies to decrease DCI. Because multiple pathways are involved, future research to prevent and treat this much-dreaded complication should focus on drugs with strong pleiotropic effects that do not only affect vasospasm but also microthrombosis.
Conclusions
Although vasospasm is associated with clinical deterioration and poor outcome after SAH, several studies indicate that the development of DCI cannot be fully attributed to the occurrence of radiologic vasospasm. Previously, alternative hypotheses, such as microvascular spasm and cortical spreading ischemia, were postulated. This review shows that an additional cause of DCI after SAH might be microthrombosis, as a result of various other processes, such as activation of the coagulation cascade and impaired fibrinolytic activity. It remains to be investigated whether microthrombosis after SAH is associated with the occurrence of microvascular spasm and cortical spreading ischemia. It could be that they all are rather complementary than competing hypotheses. The possible relation between these hypotheses is illustrated by the observation that microthrombosis provokes cortical spreading depression (Dietrich et al, 1994).
Footnotes
The authors state no conflict of interest.