
Abstract
Oxidative and nitrosative stress are targets for intervention after ischemia/reperfusion. The aim of this study was to explore the effect of CR-6, a vitamin-E analogue that is antioxidant and scavenger of nitrogen-reactive species. Sprague–Dawley rats had the middle cerebral artery (MCA) occluded either for 90 mins or permanently. Cortical perfusion was continuously monitored by laser–Doppler flowmetry. CR-6 (100 mg/kg) was administered orally either at 2 and 8 h after MCA occlusion, or at 2 h only. Infarct volume, neurological deficit, and signs of reperfusion injury were evaluated. CR-6 was detected in plasma and brain by HPLC. CR-6 reduced glutathione consumption in the ischemic brain and superoxide generation in the isolated MCA. CR-6 decreased infarct volume and attenuated the neurological deficit at 1 and 7 days after ischemia/reperfusion, but not after permanent ischemia. Immediately after reperfusion, cortical blood flow values returned to their baseline (±20%) in several animals, whereas others showed hyper-perfusion (>20% of baseline). Reactive hyperemia was associated with adverse events such as increased cortical BBB leakage, edema, protein nitrotyrosination, COX-2 expression, and neutrophil accumulation; and with a poorer outcome, and CR-6 attenuated these effects. In conclusion, oral CR-6 administration after transient ischemia protects the brain from reperfusion injury.
Introduction
Reperfusion is currently the best way to protect the brain against ischemic damage, and thrombolysis with recombinant tissue plasminogen activator (rtPA) is the only approved stroke treatment in Europe and North America. Also, preclinical studies have shown that transient middle cerebral artery occlusion (MCA) occlusion plus reperfusion causes less damage than permanent MCA occlusion in rats (Memezawa et al, 1992; Rogers et al, 1997). However, reperfusion carries some risks, such as hemorrhagic transformation and fatal edema, which could be attributed, at least in part, to reperfusion injury (Pan et al, 2007). Post-ischemic damage associated to reperfusion injury is well-documented in the heart (Kutala et al, 2007), but demonstration of reperfusion injury in human brain has become more elusive. Experimental data obtained from certain rat models of intraluminal MCA occlusion (Dietrich, 1994; Yang & Betz, 1994; Aronowski et al, 1997) provide evidence for reperfusion injury in the preclinical setting. For instance, Yang and Betz (1994) showed that 6-h (h) permanent MCA occlusion causes less damage than 3-h transient MCA occlusion plus 3 h of reperfusion in the rat. Aronowski et al (1997) reported that a period of ischemia ranging between 2 and 5 h followed by reperfusion up to 24 h causes more damage than 24-h permanent occlusion in Lewis rats, but not in spontaneously hypertensive rats. These studies evidence that the manifestation of reperfusion injury might depend on several features, such as the degree and duration of MCA occlusion, and genetic factors, among others. This suggests that reperfusion injury in humans, if any, might be variable, as genetic and comorbidity factors could be involved and the length, degree, and location of flow reduction might differ in each patient.
Reperfusion injury is attributed to several effects of reperfusion, such as oxidative stress (Chan, 1996), blood–brain barrier (BBB) damage (del Zoppo and Mabuchi 2003), leukocyte accumulation in blood vessels (del Zoppo et al, 1991) and infiltration into the brain parenchyma (Zhang et al, 1994), platelet (Chong et al, 2001) and complement (D'Ambrosio et al, 2001) activation, and hemorrhagic transformation (Pan et al, 2007). Also, reperfusion might induce increases in blood flow above basal levels, as it has been reported in animals (Tamura et al, 1980; Traupe et al, 1982; Gourley and Heistad, 1984; Todd et al, 1986; Tsuchidate et al, 1997) and in humans (Kidwell et al, 2001).
Antioxidants exert beneficial effects in preclinical studies of ischemia/reperfusion, and they might protect against brain damage by inhibiting reperfusion injury (Tasdemiroglu et al, 1994). Here we investigated whether the antioxidant CR-6 (3,4-dihydro-6-hydroxy-7-methoxy-2,2-dimethyl-1(2H)-benzopyran) exerted beneficial effects in transient and permanent cerebral ischemia. CR-6 is a synthetic, structurally simpler, analogue of vitamin-E, with the additional capacity to scavenge nitrogen-reactive species (Montoliu et al, 1999). Previous reports showed beneficial effects of CR-6 against oxidative stress injury in vitro (Montoliu et al, 1999; Sanvicens et al, 2006) and in vivo (Miranda et al, 2007). We searched for signs of reperfusion injury by examining the development of early reactive hyperemia, BBB leakage, edema, signs of oxidative and nitrosative stress, and markers of inflammation and leukocyte accumulation.
Materials and methods
Animals
Adult, male Sprague–Dawley rats (Charles River, Lyon, France) (300 to 350 g body weight) were used in this study. Animal work was performed according to the local legislation (Decret 214/1997 of 30th of July by the ‘Departament d'Agricultura, Ramaderia i Pesca de la Generalitat de Catalunya’) under the approval of the Ethical Committee (CEEA) of the University of Barcelona, and in compliance with the European legislation. Animals were kept under a 12-h day/night light cycle, controlled environmental conditions of temperature and humidity, and were given food and water ad libitum. Surgical interventions were performed under isofluorane anesthesia.
Induction of Focal Ischemia
Focal brain ischemia was induced by intraluminal occlusion of the right middle cerebral artery (MCA) with reperfusion, as reported (Justicia et al, 2006). A group of rats was subjected to 90-min ischemia followed by reperfusion, whereas permanent ischemia was induced in another group of rats. Briefly, rats were anesthetized with isoflurane in a mixture of O2 and N2O (30:70) and intubated through the trachea for controlled ventilation. Blood samples were taken to determine blood gases (EasybloodGas Analyzer; MedicaCorp., Bedford, MA, USA) and glucose concentration (Accu-chek Sensor Comfort; Roche Diagnostics Spain, St Cugat del Valles, Barcelona) at various time points during the surgical procedures. Mean arterial blood pressure was monitored and body temperature was maintained between 36.5°C and 37.5°C during surgery. Briefly, the common and external carotid arteries were exposed and the pterigopalatine artery was ligated. A filament (nylon monofilament 4/0, Look-sutures; Harvard-Apparatus, Holliston, MA, USA) heat-rounded at the tip was introduced (21 mm) through the external carotid artery to the level where the MCA branches out and a ligature was placed on the internal carotid artery. In the reperfusion group, the ligature of the internal carotid artery was taken off and the filament was cautiously removed 90 mins after occlusion to allow reperfusion. In the permanent ischemia group, the intraluminal filament was not removed. Sham-operated rats were subjected to all the surgical procedures and the filament was introduced in the MCA and immediately removed. Rats were allowed to recover from the anesthesia, and at 8 h, 24 h, or 7 days after MCA occlusion; they were deeply anesthetized with isofluorane and killed. Rats of the same gender and body weight that were not subjected to surgery were used as controls for biochemical determinations.
Measurement of Cerebral Blood Flow
Cortical cerebral blood flow (CBF) was assessed with a laser–Doppler system (Perimed AB, Järfalla, Sweden). The day before induction of ischemia, rats were anesthetized and a guide was implanted at the surface of the skull after reducing the thickness of the bone with a 1-mm-thick microdrill. The guide was fixed to the skull with dental cement and two miniature screws at the following stereotaxic coordinates: 2 mm posterior and 3.5 mm lateral to bregma. This location corresponds to the margin of the MCA territory. On the following day, the laser–Doppler probe was introduced through the guide and registration was started. After achieving a stable registry, ischemia was induced as described above. Perfusion was continuously monitored before ischemia, through the 90-min ischemia period, and during the first 15 mins after reperfusion. CBF data were used as criteria to exclude animals from the study, as follows: (1) rats, which after introducing the MCA occlusion filament, did not show a perfusion rate lower than 65% of basal were excluded, as it was considered that ischemia was not successfully induced; (2) rats, which at reperfusion did not show recovery higher than 80% of the basal value, were excluded; and (3) rats with surgical complications, such as subarachnoid hemorrhage, were discarded.
Treatment
CR-6 was chemically synthesized as reported (Casas et al, 1992). CR-6 is a rather lipophilic molecule and for this reason it was dissolved in commercial edible olive oil (Carbonell, SOS Corporación Alimentaria S.A., Madrid, Spain) and it was orally administered to rats at a dose of 100 mg/kg in 1mL using a feeding gauge. This dose was previously shown to protect rats against oxidative-stress damage (Miranda et al, 2007). The same oil was used as vehicle. Rats received CR-6 or the vehicle either twice, at 2h and 8 h after the onset of ischemia, or at 2h only. Rats received a code that did not reveal the allocated treatment, for the purpose of performing all further studies in a masked manner as experimenters analyzing the samples were not aware of the treatment assigned to each rat.
Evaluation of Infarct Volume
Rats were anesthetized with isofluorane and killed by decapitation. The brain was removed and sliced in 2-mm-thick coronal sections, which were stained with a 1% solution of 2,3,5-triphenyltetrazolium chloride for 10 mins at 37°C. Sections were then immersed overnight in a 4% paraformaldehyde solution in 0.1 mol/L phosphate buffer (pH 7.4) and then washed and stored in the same buffer. Images of the sections were scanned and analyzed with an image analysis system (Scion Image; Scion Corporation, Frederick, MD, USA). Correction for brain edema was made by multiplying the infarct area by the ratio of the contralateral area to the ipsilateral area. Areas were integrated to calculate infarct volume. The extent of edema was assessed by calculating the percent increase of the ipsilateral/contralateral region volume.
Neurological Score
A simple neurological test on a nine-point scale (0 = no deficit to 9 = highest handicap) was performed before killing the animals, as reported previously (Justicia et al, 2006). Four tests were followed: (1) spontaneous activity (moving/exploring= 0, moving without exploration= 1, no moving or only when pulled by the tail = 2); (2) circling to the left (none = 0, when elevated by the tail and pushed or pulled = 1, spontaneously = 2, circling without displacement (spinning top) = 3); (3) resistance to left forepaw stretching (not stretching allowed = 0, stretching allowed = 1, no resistance = 2); and (4) parachute reflex (symmetrical = 0, asymmetrical = 1, contralateral forelimb retracted = 2). Animals, which died are reported separately for each treatment group.
RNA Isolation, cDNA Synthesis, and Real-Time Detection of iNOS, TNF-α, and IL-1β mRNA
Total RNA from ipsilateral cortex was isolated using Trizol® reagent (Invitrogen Carlsbad, CA, USA) according to the manufacturer's protocol. RNA quantity and purity were determined using an ND-1000 micro-spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). A 2-μg weight of RNA was used for cDNA synthesis using the AMV First-Strand cDNA Synthesis kit (no.-12328 to 040; Invitrogen) and the cDNA was stored at −20°C until use.
Real-time quantitative reverse transcription-PCR analysis was performed by SYBR green-I dye detection using the iCycler iQTM Multicolor Real-Time Detection System (Bio Rad Laboratories Inc., Hercules, CA, USA). Inducible nitric oxide synthase (iNOS), tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and β-actin were amplified in duplicate in a PCR optical 96-well reaction plate (Bio Rad Laboratories Inc., Hercules, CA, USA). A 15-μl volume of the PCR mixture per well contained 3 μl of 1:4 diluted cDNA, 0.75 μl of each sequence-specific primer (10 μmol/L), 7.5 μl of 1 × iQ SYBR Green Supermix (Bio-Rad), and 3.75 μl of nuclease-free water. Primers were synthesized (F. Hoffmann - La Roche AG, Basel, Switzerland) and reconstituted in nuclease-free water before use. The following primers were used for iNOS (forward): 5′-TGGTGCAGAAGC ACAAAGTC-3′ and (reverse) 5′-GAACTGGGGGAAACCATT TT-3′; for TNF-α: (forward)5′-GGGGCCACCACGCTCTTCTG TC-3′ and (reverse) 5′-TGGGCTACGGGCTTGTCACTCG-3′; for IL-1β: (forward) 5′-TGAAGCAGCTATGGCAACTG-3′ and (reverse) 5′-TGCCTTCCTGAAGCTCTTGT-3′; and for β-actin: (forward) 5′-CAACCTTCTTGCAGCTCCTC-3′ and (reverse) 5′-TTCTGACCCATACCCACCAT-3′. Optimized thermal cycling conditions were as follows: 5 mins at 95°C followed by 45 cycles of 15 secs at 95°C, and 45 secs at 59°C and 1:30 mins at 72°C. Data were collected after each cycle and presented graphically (iCycler iQTM Real-time Detection System Software, version 3.1; Bio-Rad). Specificity was confirmed by melting-curve analysis of the PCR products. Quantification was performed by analyzing the cycle threshold (Ct) values by the 2–ΔΔCt method (Livak and Schmittgen, 2001).
BBB Breakdown
A 4-mL/kg (body weight) amount of a 2% (w/v in saline) solution of Evans Blue (Sigma-Aldrich, St Louis, MO, USA) was injected through the femoral vein 4 h after the induction of reperfusion, and 2 h later, rats were anesthetized and perfused through the heart with saline. The brain was cut in 2-mm-thick coronal sections, which were used for Evans Blue extraction. The ipsilateral and contralateral cortex and striatum were dissected out, weighted, and immersed in a solution containing 50% trichloroacetic acid (1.3 mL/g of tissue). The tissue was homogenized with a polytron device and centrifuged at 12,000 g for 20 mins. The supernatant was mixed with ethanol (1:3). Fluorescence intensity was measured with a fluorimeter at 680 nm using a 110-μl sample aliquot. The concentration of Evans Blue (ng/mL) was calculated by using a standard curve generated with known concentrations of the dye.
Determination of Glutathione Concentration
Twenty-four hours after ischemia rats were anesthetized and killed. Brain tissue was dissected out to separate the ipsilateral and contralateral cortex and striatum, and it was rapidly frozen and kept at −80°C until further analysis. The contralateral hemisphere was used as control. Total (GSx) and oxidized (GSSG) glutathione concentrations (nmol/mg of protein) were measured as described by Baker et al (1990), with minor modifications. Tissue was sonicated in 3.3% 5-sulfosalicylic acid (1 mL per 200 mg of tissue) and centrifuged at 12,000 g for 30 mins at 4°C. The supernatant was used to determine GSx and GSSG, whereas the pellet was used to determine protein concentration (Bradford Method; Bio-Rad). The reaction is based on the oxidation of DTNB (5,5′-dithio-bis-(2-nitrobenzoic acid)) followed by measurement at 405 to 412 nm in a spectrophotometer. Formation of GSSG was determined in 100 μl of the supernatant after addition of 2 μl of 2-vinylpyridine. The content of reduced glutathione (GSH) was estimated as follows: GSH = GSx–(2 * GSSG).
Western Blotting
Western blotting was performed using monoclonal antibodies against myeloperoxidase (MPO) (PeliCluster; A Menarini Diagnostics, Badalona, Barcelona, Spain), diluted 1:500, and against nitrotyrosine (Abcam Limited, Cambridge, UK), diluted 1:500. A rabbit antibody against cyclooxygenase-2 (COX-2) (Cayman Chemical Co., Ann Arbor, MI, USA) was used diluted 1:1,000. Mouse monoclonal antibodies against β-tubulin (Sigma-Aldrich, St Louis, MO, USA), diluted 1:50,000; anti-glyceraldehyde 3-phosphate dehydrogenase (Assays Designs, Ann Arbor, MI, USA), diluted 1:5,000; or a rabbit polyclonal antibody against actin (Stressgen, Victoria, Canada), diluted 1:100,000, was used as loading controls. The optical density of the bands was measured by densitometric analysis (GS-800 Densitometer; Bio-Rad). The ratio between band intensity of specific proteins and the corresponding loading control was calculated to correct for differences in protein gel loading. For each gel, values are expressed as the percentage of control.
Inmunohistochemistry
Protein expression was studied at 24 h by immunohistochemistry. Two-millimeter-thick coronal brain sections were immersed in 4% paraformaldehyde in phosphate buffer (pH 7.4) overnight and then washed in the same buffer. Sections measuring 50mm were obtained with a vibratome and were used free-floating for immunostaining using a rabbit polyclonal antibody against COX-2 (Cayman Chemical Co., Ann Arbor, MI, USA) diluted 1:200, as previously reported (Planas et al, 1999). Immunohistochemistry against MPO was performed with a rabbit polyclonal antibody (Dako, Glostrup, Denmark), diluted 1:800, in 5-mm-thick paraffin sections. After the primary antibody, sections were incubated with biotinylated secondary antibodies followed by the ABC procedure (Serotec AbD, MorphoSys UK Ltd, Kidlington, Oxfordshire, UK). The reaction was developed with diaminobenzidine. MPO + cells were examined with a light microscope in four different areas per brain section and in two different brain sections. For each area, MPO + cells were counted in a systematic manner in four different zones by an investigator who was not aware of the treatment, and the count average was calculated for each rat.
Isolation of the MCA and Evaluation of Oxidative Stress
The ipsilateral MCA was isolated (n = 17) at 24 h after ischemia, as previously reported (Jiménez-Altayó et al, 2007). This study was conducted with ischemic rats that received either the vehicle (n = 7) or CR-6 (n = 6). Shamoperated animals (n = 4) were also studied to underscore any mechanical effect of the filament on the MCA. Segments of the MCA were placed in Krebs–HEPES buffer (130mmol/L NaCl, 5.6mmol/L KCl, 2mmol/L CaCl2, 0.24 mmol/L MgCl2, 8.3 mmol/L HEPES, 11 mmol/L glucose, pH 7.4) containing 30% sucrose overnight and transferred to a cryomold (Bayer Química Farmacéutica, Barcelona, Spain) containing Tissue-Tek OCT embedding medium (Sakura Finetek Europe B.V., Zoeterwoude, The Netherlands) for 20 mins, and then they were immediately frozen in liquid nitrogen for storage at −70°C until further measurements. The oxidative fluorescence dye dihydroethidium (DHE) was used to evaluate in situ production of superoxide radicals, as described by Jiménez-Altayó et al (2006). The frozen MCA was cut into 14-μm-thick sections and placed on a glass slide. Serial slices were equilibrated under identical conditions for 30 mins at 37°C in Krebs–HEPES buffer. Fresh buffer containing DHE (2 μmol/L) (Ex 546 nm and Em 610 nm) was applied topically on each tissue section, coverslipped, and incubated for 2 h in a light-protected humidified chamber at 37°C, and then viewed by fluorescence laser-scanning confocal microscopy (Leica TCS SP2, Heidelberg, Germany; × 63), using the same imaging settings in all samples. Parallel sections were incubated with polyethylene glycol superoxide dismutase (PEG–SOD; 500 U/ml) for 2 h at 37°C. Fluorescence was detected with a 568-nm long-pass filter. For quantification, integrated optical densities were calculated from four sampled areas per ring for each experimental condition using the MetaMorph image analysis software (Universal Imaging; Molecular Devices, Downingtown, PA, USA). The integrated optical densities in the target region were calculated.
Determination of CR-6 Concentration in Plasma and Brain
The concentration of CR-6 in plasma and brain tissue was evaluated by high-performance liquid chromatography (HPLC) with fluorimetric detection. CR-6 (100 mg/kg) was orally administered to ischemic rats after 30 mins of reperfusion or to control non-ischemic animals. Blood samples were taken at different time points (ranging from 15 mins to 4 h) under isoflurane anesthesia. At either 15 mins or 4 h after CR-6 administration, animals were anesthetized and perfused through the heart with saline to remove blood from brain vessels. The method used for CR-6 extraction from biological samples and detection was an adaptation of a method for detection of vitamins by direct liquid chromatography (Ruperez et al, 2004). Briefly, plasma (50 μL) and brain tissue (200 mg) were deproteinized in acetone (150 μL for plasma and 750 μL for brain tissue) and the samples were sonicated for 10 secs (plasma) or 20secs (brain tissue) with an ultrasound microprobe (Labsonic M; Sartorius AG, Goettingen, Germany), followed by centrifugation for 4mins at 12,000 g (Centrifuge 5415 D; Eppendorf, Hamburg, Germany). Aliquots (10 to 50 μL) of the supernatant were injected into the HPLC system (Water Associates, Milford, MA, USA) made of a 600E Pump, a 717 plus Auto sampler, and a 2475 fluorimeter. A reverse-phase Nucleosil C18 5-μm column (10 × 0.4 cm) was used (Teknochroma, Sant Cugat del Valles, Spain). The mobile phase was methanol/H2O (80/20 v/v), which was run with an isocratic regular low flow rate of 0.8 mL/min. For CR-6 detection, the fluorimeter was adjusted to 296 nm Ex/334nm Em according to the fluorescence spectrum of the compound. CR-6 eluted from the column at 2 mins of retention time. Quantification was performed by external calibration. The CR-6 detection limit in plasma samples was 10 ng/mL. In good agreement with data of Ruperez et al (2004), the recovery from plasma was close to 100%.
Statistical Analyses
Comparisons between two groups were made using t-test or Mann–Whitney non-parametric test, depending on whether or not the data passed the normality test (D'Agostino & Pearson omnibus normality test). Comparisons between more than two groups were made by oneway analysis of variance followed by Bonferroni's post hoc analysis for data conforming normality. Otherwise, data were analyzed using Kruskal–Wallis test followed by Dunn's multiple comparison test. Correlation analysis was performed using Pearson's test. Statistical analyses were carried out with the GraphPad Prism software.
Results
CR-6 Reduces Infarct Volume and Ameliorates the Neurological Score after Transient but not Permanent Ischemia
In the 90-min ischemia group, administration of the antioxidant agent CR-6 twice, at 2 h and 8 h after the onset of MCA occlusion, reduced infarct volume (P < 0. 05) (Figure 1A) and ameliorated (P < 0.05) the neurological score at 24 h (Figure 1B) as compared with that in rats receiving the vehicle. Infarct volume (mean ± s.d.) was 75.3 ± 40.8mm3 (n = 15) in the CR-6-treated group versus 141.1 ± 64.2 mm3 (n = 23) in the vehicle-treated group. Neurological score (mean ± s.d.) was 3.4 ± 1.7 points (n = 15) in the CR-6-treated group versus 4.8 ± 1.8 (n = 23) in the vehicle-treated group. CR-6 protected the cortex and the striatum (Figures 1C and 1D). There were no statistical differences between the groups regarding the physiological parameters measured during surgery (Supplementary Table 1). In this experiment, mortality was 14% in the vehicle-treated group and 8 % in the CR-6-treated group.
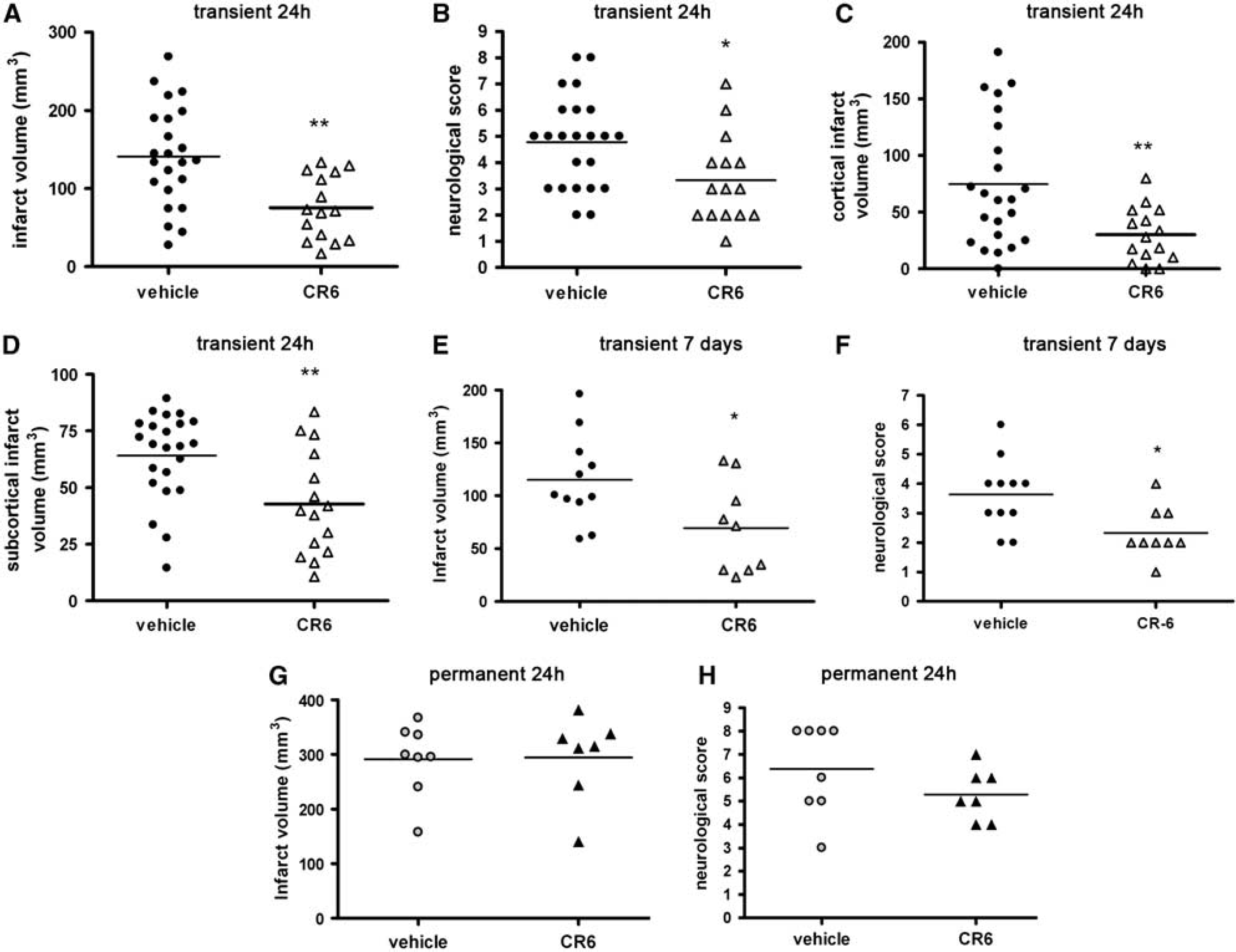
Effect of CR-6 after transient and permanent ischemia. Rats received the vehicle or CR-6 twice, that is, at 2 and at 6 h after the onset of ischemia. Animals were examined at 24 h (
Ninety-minute transient ischemia was induced in an additional group of rats to examine whether the beneficial effect of CR-6 persisted for a longer time period. Rats received CR-6 or vehicle, as above, and were killed at 7 days. Only one rat in this experiment died and it belonged to the CR-6-treated group. There were no differences between the groups regarding the physiological parameters measured during surgery (Supplementary Table 2). Infarct volume (mean ± s.d.) at 7 days in the CR-6-treated group (69.6 ± 43.3 mm3, n = 9) was smaller (P < 0.03) than that in the vehicle-treated group (115.1 ± 41.9 mm3, n = 11) (Figure 1E). Likewise, CR-6 attenuated the neurological deficit (2.3 ± 0.8 points, n = 9) in relation to the vehicle (3.6 ± 1.2 points, n = 11) at 7 days (Figure 1F). Therefore, the benefit of CR-6 in transient ischemia that was found at 24 h was maintained at 7 days.
However, this CR-6 treatment regime was not protective in a model of permanent ischemia, as it neither reduced infarct volume (P = 0.94) (Figure 1G) nor ameliorated the neurological score (P = 0.21) (Figure 1H), at 24 h. Mortality after permanent ischemia was 11% in the vehicle-treated group, whereas none of the rats of the CR-6-treated group died within the first 24 h. Comparison of the extent of brain damage in the vehicle-treated group depending on whether the MCA was transiently (90 mins) or permanently occluded, showed that infarct volume (mean ± s.d.) was smaller (P < 0.001) after transient (141.1 ± 64.2 mm3) than after permanent (291.5 ± 66.3 mm3) ischemia at 24 h. Also, neurological function was less affected (P < 0.05) after transient (4.8 ± 1.8 points) than after permanent (6.3 ± 1.9 points) ischemia. Likewise, in rats treated with CR-6, infarct volume (mean ± s.d.) was significantly (P < 0.001) larger after permanent (294.4 ± 79.4 mm3) than after transient (75.3 ± 40.8 mm3) ischemia, and neurological score (mean ± s.d.) was worst (P < 0.05) after permanent (5.3 ± 1.1 points) than after transient (3.4 ± 1.7 points) ischemia.
One single administration of CR-6 at 30 mins after reperfusion was not sufficient to induce protection in the transient ischemia group, as it did not significantly (P = 0.70) reduce infarct volume (141 ± 82 mm3 (n = 9) in controls versus 123 ± 72 mm3 (n = 8) in the CR-6-treated group) and did not ameliorate (P = 0.70) the neurological score either (4.8 ± 2.2 (n = 9) points in controls versus 4.4 ± 1.3 points (n = 8) in the CR-6-treated group) at 24 h. This suggests that a prolonged antioxidant action might be necessary to neutralize the effects of reperfusion.
Consumption of Endogenous Glutathione is Attenuated in the Animals Treated with CR-6
The concentration of total (GSx) and oxidized (GSSG) glutathione was measured in the ipsilateral and contralateral cortex and striatum 24 h after ischemia in rats treated with either vehicle or CR-6 (two doses), and reduced glutathione (GSH) content was calculated from the previous measurements (see section Materials and methods). At 24 h after transient ischemia, the concentration of GSH significantly decreased in the ipsilateral cortex (P < 0.05) (Figure 2A) and striatum (P < 0.001) (Figure 2B) of rats receiving the vehicle. This effect was attenuated by CR-6 treatment, as the GSH content in this group was not significantly different from that of the controls (Figures 2A and 2B). A negative relationship was found between cortical GSH content and infarct volume (Figure 2C), suggesting that the extent of GSH consumption was indicative of the magnitude of tissue damage after ischemia/reperfusion.
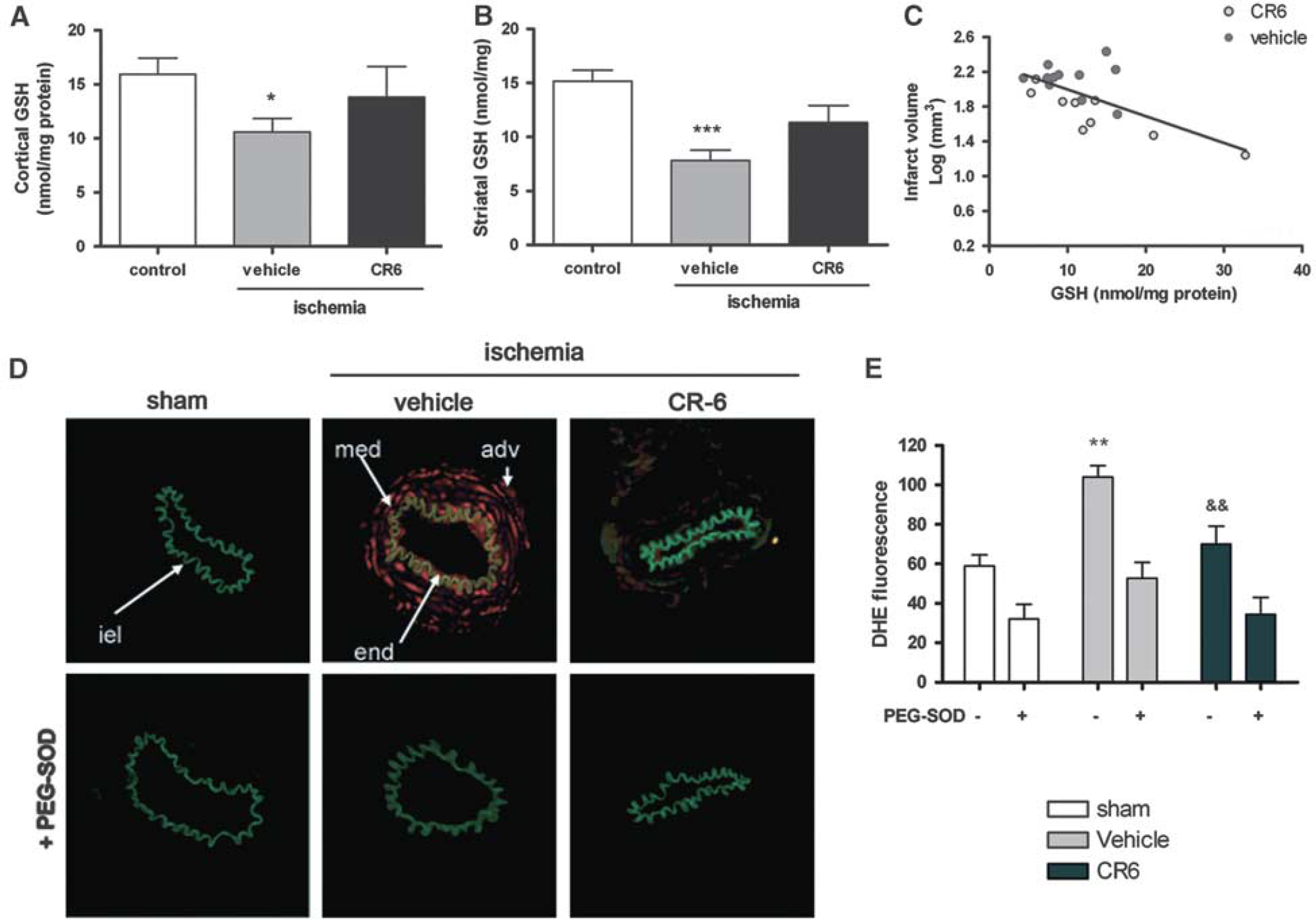
CR-6 attenuates oxidative stress in brain tissue and in the isolated MCA. (
CR-6 Attenuates Oxidative Stress in the Isolated MCA
To test whether CR-6 might have an effect on blood vessels, we isolated the MCA of sham-operated and ischemic rats treated with either CR-6 (two doses) or the vehicle. Compared with sham-operated rats, transient ischemia induced a positive DHE reaction (P < 0.01) that was detected in the three layers of the MCA vascular wall of the vehicle group. CR-6 strongly attenuated this DHE reaction (P < 0.01) (Figures 2C and 2D). To verify the involvement of superoxide anions in this effect, we used the permeable superoxide scavenger, PEG–SOD, which strongly diminished DHE fluorescence in all the groups (Figures 2C and 2D).
Relation of Reactive Hyperemia after Transient Ischemia and Infarct Volume
In exploratory analyses of cortical CBF, we observed that immediately after reperfusion (before any treatment) blood flow values returned to baseline levels in several rats, whereas other rats showed values well above basal (see example in Supplementary Figure 1). To find out whether this effect was related to the outcome of ischemia, we first analyzed data from rats that received the vehicle. Reactive hyperemia at the time of reperfusion tended to correspond with larger infarct volumes, suggesting that increases of blood flow above basal levels (hyperemia) at the time of reperfusion were associated to larger infarct volumes. A significant positive correlation was found between flow at reperfusion and infarct volume (P < 0.05) (Figure 3A), suggesting that, at reperfusion, high increases of blood flow were associated to larger infarct volumes. This phenomenon was not attributable to the degree of blood flow reduction during ischemia, as blood flow at occlusion was not significantly correlated to blood flow at reperfusion or to infarct volumes (Supplementary Figure 2). Therefore, under these experimental conditions, the occurrence and magnitude of hyperemia was not strictly attributable to the measured CBF reduction during ischemia. However, we cannot discard that it might be related to higher reductions of CBF in the core during ischemia, as our CBF measurement was taken in a peripheral location.
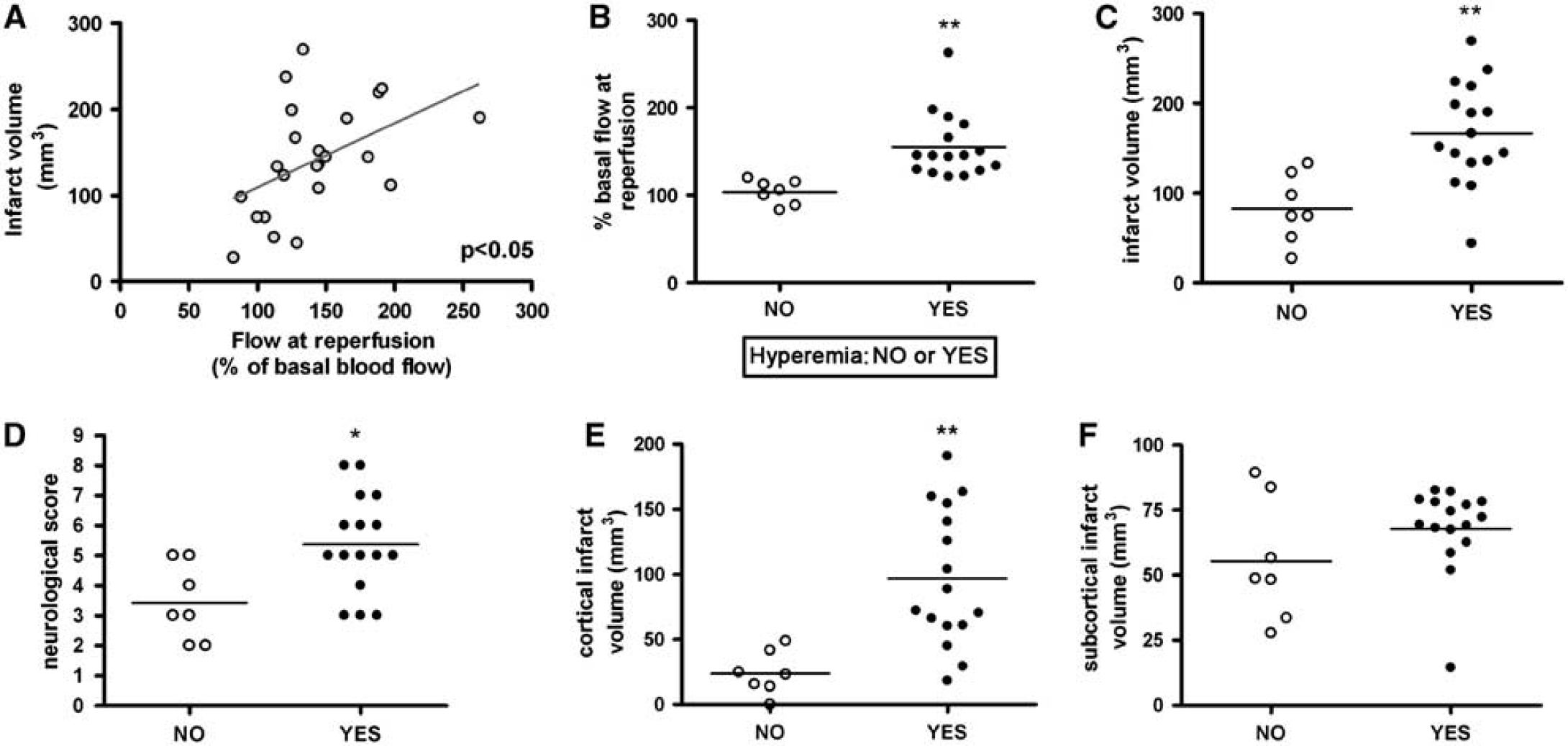
CBF at reperfusion influences tissue outcome. Only rats subjected to ischemia/reperfusion and receiving the vehicle are shown in this figure (n = 23). (
Values of cortical blood flow immediately after reperfusion (first 15 mins) that recovered to their corresponding baseline pre-ischemia values (± 20%) were considered as similar to basal, whereas increases of cortical blood flow above 20% of basal values were considered as hyper-perfusion and were considered a sign of reactive hyperemia. According to this criterion rats were divided in two groups, which were termed hyperemic (Yes) and non-hyperemic (No) (Figure 3B). The mean ± s.d. percent blood flow value during ischemia in the group of rats with hyperemia (44.1 ± 15.7 % of basal flow) was not significantly different from that in non-hyperemic rats (48.4 ± 12.9 % of basal flow). Then, infarct volumes were compared as a function of the presence or absence of reactive hyperemia. Infarct volume (mean ± s.d.) was larger (P < 0.01) in the group of rats showing hyperemia (166.6 ± 56.6 mm3) than in the group without hyperemia (82.9 ± 37.8 mm3) (Figure 3C). Likewise, neurological score (mean ± s.d.) was worst (P < 0.05) in hyperemic (5.4 ± 1.6 points) than in non-hyperemic (3.4 ± 1.3 points) rats (Figure 3D).
Examination of the size of infarction in cortical and subcortical regions showed that cortical infarction (mean ± s.d.) was larger (P < 0.01) in the hyperemic (97.0 ± 52.8 mm3) that in the non-hyperemic (24.0 ± 16.7 mm3) group (Figure 3E). In contrast, no significant differences in infarct volume (mean ± s.d.) were found in the striatum between the group with cortical hyperemia (67.8 ± 16.7 mm3) and that without cortical hyperemia (55.4 ± 23.4 mm3) (Figure 3F). This might be expected since hyperemia here refers to cortical hyper-perfusion, that is, the laser–Doppler probe measured cortical blood flow, but we had no information on subcortical flow. These results suggest that cortical hyperemia at reperfusion was a predictor of exacerbated cortical damage, and therefore it might be a marker of reperfusion injury affecting the cerebral cortex.
CR-6 is Protective in Animals Showing Reactive Hyperemia after Reperfusion
Treatment (two doses of CR-6 or vehicle) was initiated 30 mins after reperfusion, whereas blood flow was studied prior to administration of treatment. Animals were randomly allocated to a given treatment regardless of whether they developed hyperemia or not, as hyperemia was only identified a posteriori after calculation of the blood flow registry. In the group of animals used to study infarct volume, the incidence of reactive hyperemia was 60%. However, hyperemia was not distributed evenly between the two treatment groups, as hyperemia was retrospectively found in 70% of the rats allocated to the vehicle-treated group (16 out of 23 rats), whereas only 47% of rats in the CR-6-treated group (7 out of 15) developed hyperemia. This might be a confounding factor in neuroprotection studies, as hyperemic rats showed larger infarct volumes than non-hyperemic rats (see the last section). Therefore, we took into account the factor hyperemia Yes or No in the comparisons between animals receiving the vehicle or CR-6.
The magnitude of increase in cortical perfusion in hyperemic rats was similar in the vehicle-treated and CR-6-treated groups (Figure 4A), and the extent of blood flow reduction during ischemia was similar in all the groups (vehicle and CR-6, either hyperemic or non-hyperemic) (Figure 4B). As shown above, hyperemic animals had larger infarct volume than non-hyperemic animals in the vehicle-treated group (Figure 4C). However in CR-6-treated rats, infarct volume (mean ± s.d.) in hyperemic rats (85.7 ± 39.2 mm3) was similar to that in non-hyperemic rats (73.8 ± 40.6 mm3) (Figure 4C). Furthermore, hyperemic rats treated with CR-6 showed reduced infarct volume (P < 0.01) as compared with hyperemic rats receiving the vehicle (Figure 4C), whereas no statistical differences were found for non-hyperemic rats (Figure 4C). Similar results were found for the neurological score, as hyperemic rats treated with CR-6 performed better that those receiving the vehicle, whereas there were no significant differences in the score between hyperemic and non-hyperemic rats treated with CR-6, or between non-hyperemic rats receiving either vehicle or CR-6 (Figure 4D). CR-6 protected more the cortex than the striatum (Figures 4E and 4F). These results show that CR-6 was more beneficial in hyperemic than in non-hyperemic rats, suggesting that this antioxidant was more effective under conditions with signs of reperfusion injury as seen in the cortex in this experimental model.
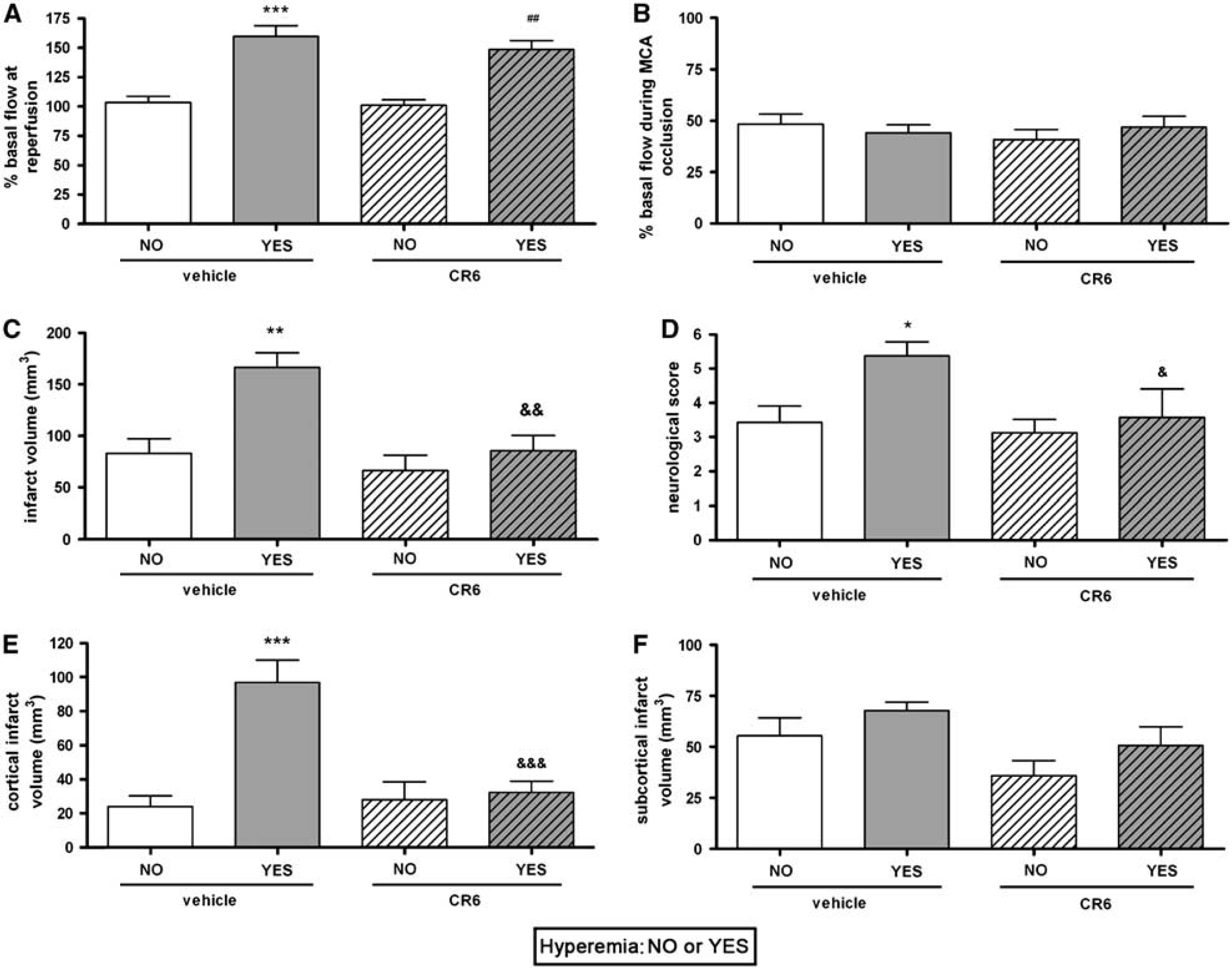
CR-6 protects hyperemic rats. In each treatment group, rats are separated according to whether they developed cortical hyperemia at reperfusion (Yes) or they did not (No). In the vehicle group, 7 rats did not develop hyperemia, whereas 16 rats did. In the CR-6 group, 8 rats did not develop hyperemia, whereas 7 rats did. (
Effect of Hyperemia and CR-6 on the Expression of mRNA of Proinflammatory Molecules
Expression of iNOS, TNF-α, and IL-1β mRNA at 6 h after reperfusion was significantly increased (P < 0.05) in rats showing the hyperemic response as compared to the controls, but not so in the non-hyperemic groups (Figures 5A–5C). CR-6 did not reduce the expression of iNOS and TNF-α mRNA, but showed a tendency (P = 0.087) to reduce the expression of IL-1β mRNA in hyperemic rats (Figures 5A–5C).
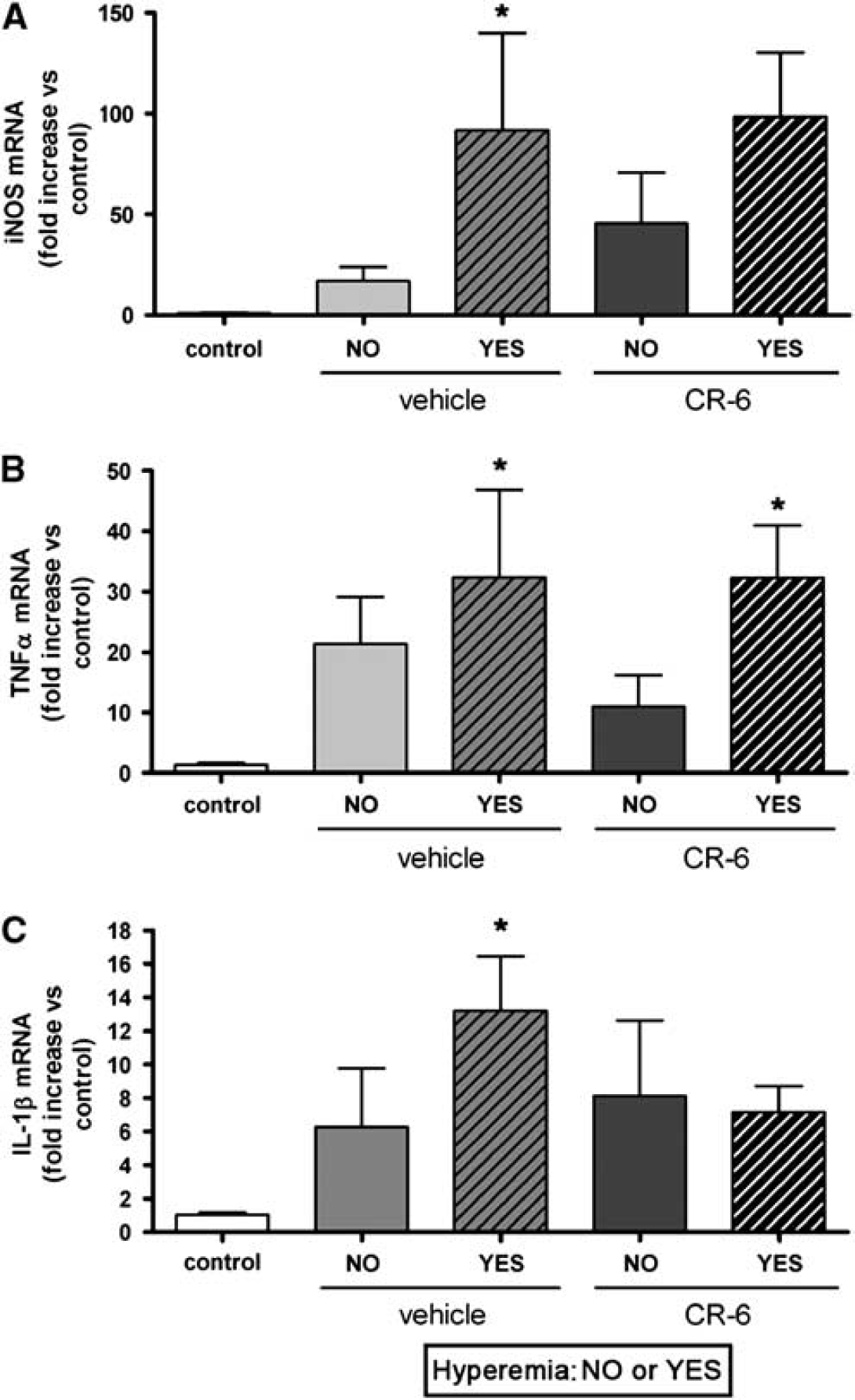
Expression of iNOS, TNF-α, and IL-1β mRNA. Expression of the mRNA of proinflammatory molecules was studied at 6 h after reperfusion. Hyperemic rats showed higher (P < 0.05) expression of iNOS (
CR-6 Attenuates Alterations in BBB Permeability
Alterations in BBB permeability were assessed by Evans Blue technique at 6 h after the onset of reperfusion. In this group of animals we measured the physiological parameters of the rats during surgery and immediately after surgery, and the results for both treatment groups were similar (Supplementary Table 1). Rats with cortical hyperemia at reperfusion had more (P < 0.01) Evans Blue extravasation in the cortex than non-hyperemic animals, which did not show signs of BBB alteration (Figure 6A). In the former animals, treatment with CR-6 attenuated alterations of BBB permeability in the cortex. In subcortical regions, no significant differences were observed between rats showing cortical hyperemia and rats that did not develop hyperemia, and CR-6 did not significantly reduce the tissue Evans Blue content (not shown).
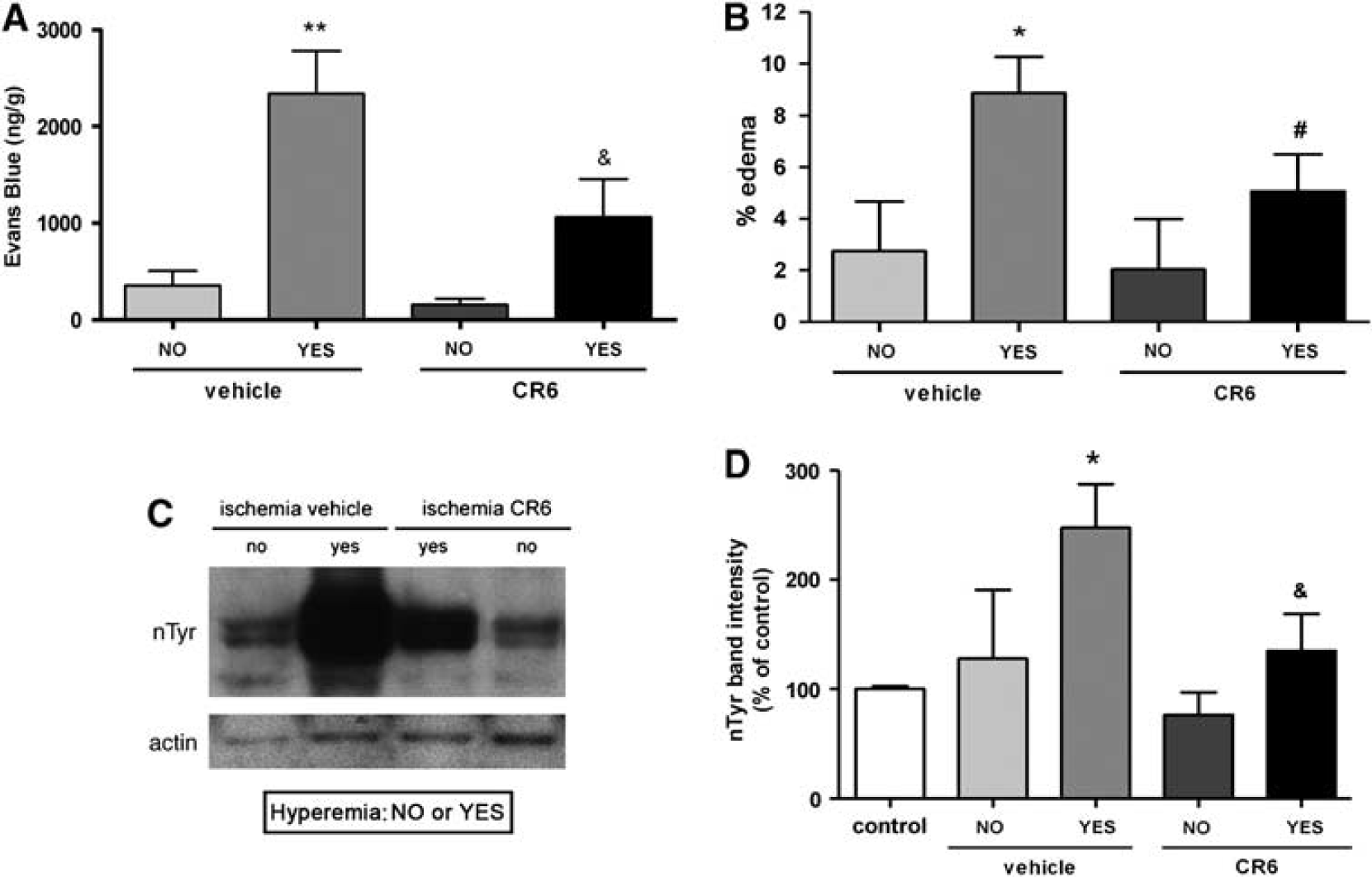
BBB breakdown, edema, and protein nitrotyrosination are exacerbated in the cortex of hyperemic animals and these effects are attenuated by CR-6. (
In agreement with the previous finding, cortical brain edema was highly increased (P < 0.05) at 24 h in hyperemic rats as compared with non-hyperemic rats of the vehicle-treated group (Figure 6B), and CR-6 tended to attenuate (P = 0.06) this effect (Figure 6B).
Ischemia Induces Protein Nitrotyrosination in Hyperemic Rats and CR-6 Reduces it
Certain reactive oxygen species (superoxide) can react with NO to produce peroxynitrite, a highly reactive anion that causes protein nitrotyrosination (Beckman et al, 1990). This is an irreversible process that can alter the function of proteins. We examined protein nitrotyrosination after ischemia as an indicator of peroxynitrite production and the action of CR-6 on this process, as CR-6 has the capacity to scavenge reactive nitrogen species (Montoliu et al, 1999). A significant increase in protein nitrotyrosination was detected 24 h after ischemia/reperfusion as compared with the controls (P < 0.05) in hyperemic animals only, and CR-6 attenuated this effect (P < 0.05) (Figures 6C and 6D).
CR-6 Decreases the Expression of COX-2 Induced by Ischemia
Ischemia induced the expression of COX-2 protein at 24 h (Figure 7A), and this effect was strongly attenuated by CR-6 (two doses) (Figures 7A and 7B). COX-2 was mainly detected in neurons (Figure 7C) suggesting that oxidative/nitrosative stress was involved in triggering neuronal COX-2 induction and that the effect of CR-6 reached the neurons either directly or indirectly.
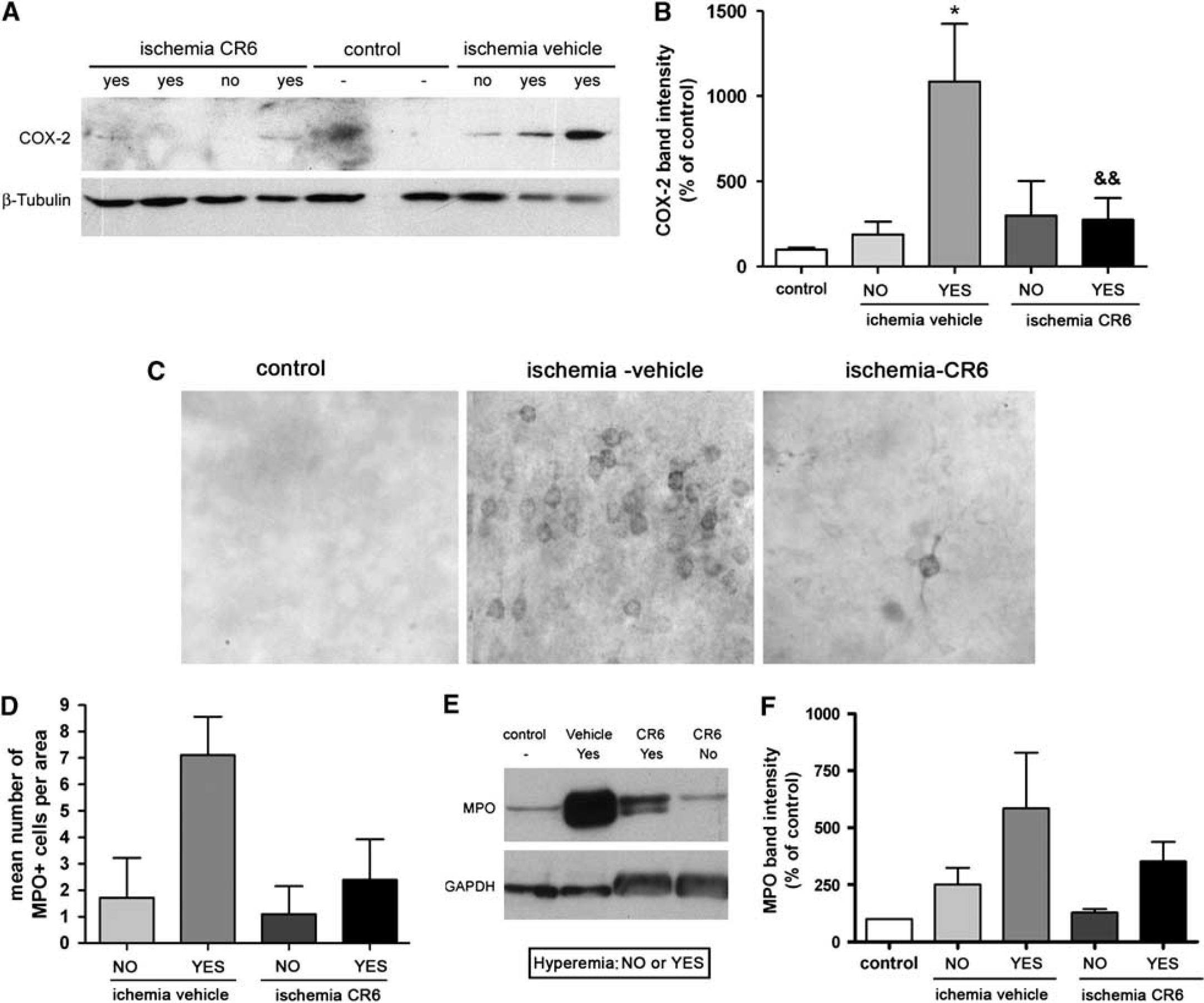
Expression of COX-2 and MPO. (
CR-6 Attenuates Neutrophil Accumulation after Ischemia
The expression of MPO in brain tissue was examined to assess the presence of neutrophils. After immunohistochemistry, the number of MPO + cells was counted in different areas and the mean number of cells per area was calculated. Hyperemic rats tended to show higher numbers of neutrophils than non-hyperemic rats (Figure 7D). CR-6 (two doses) showed a tendency to attenuate the numbers of neutrophils in hyperemic rats (Figure 7D). By western blotting, lower MPO expression was observed in hyperemic rats treated with CR-6 than in rats receiving the vehicle (Figure 7E). Semi-quantification of western blotting band intensity confirmed the tendency of CR-6 to attenuate MPO expression (Figure 7F).
CR-6 in Plasma and Brain Tissue
In view of the above findings, we set up a technique for detection of CR-6 in biological samples to find out whether CR-6 enters the brain after oral administration. Blood samples were withdrawn at several time points ranging from 15 mins to 4 h after CR-6 administration. A group of animals was killed at 15 mins (n = 4) and another group was killed at 4 h (n = 3). Rats were anesthetized and perfused through the heart with saline to remove blood from brain tissue. CR-6 was detected in plasma and brain samples by HPLC with fluorimetric detection (Figure 8A). CR-6 showed a peak in plasma between 15 and 30 mins (Figure 8B), and levels were highly reduced from 1 h onwards. The mean ± s.e.m. CR-6 plasma concentration at 15 mins was 359 ± 162 ng/mL. CR-6 was also detected in brain tissue at 15 mins but not at 4 h (Figure 8B). At 15 mins, brain CR-6 concentration (ng/g of tissue) was four-fold higher (Figure 8B) than that in plasma (ng/mL) in all the samples, and no differences in CR-6 concentration were observed between the contralateral (1,384 ± 618 ng/g) and ipsilateral (1,259 ± 574 ng/g) hemispheres in ischemic rats (n = 3), or between ischemic and non-ischemic animals. The brain/plasma ratio (mean ± s.e.m.) at 15 mins was 4.2 ± 0.6, indicating good delivery of CR-6 from blood to brain.
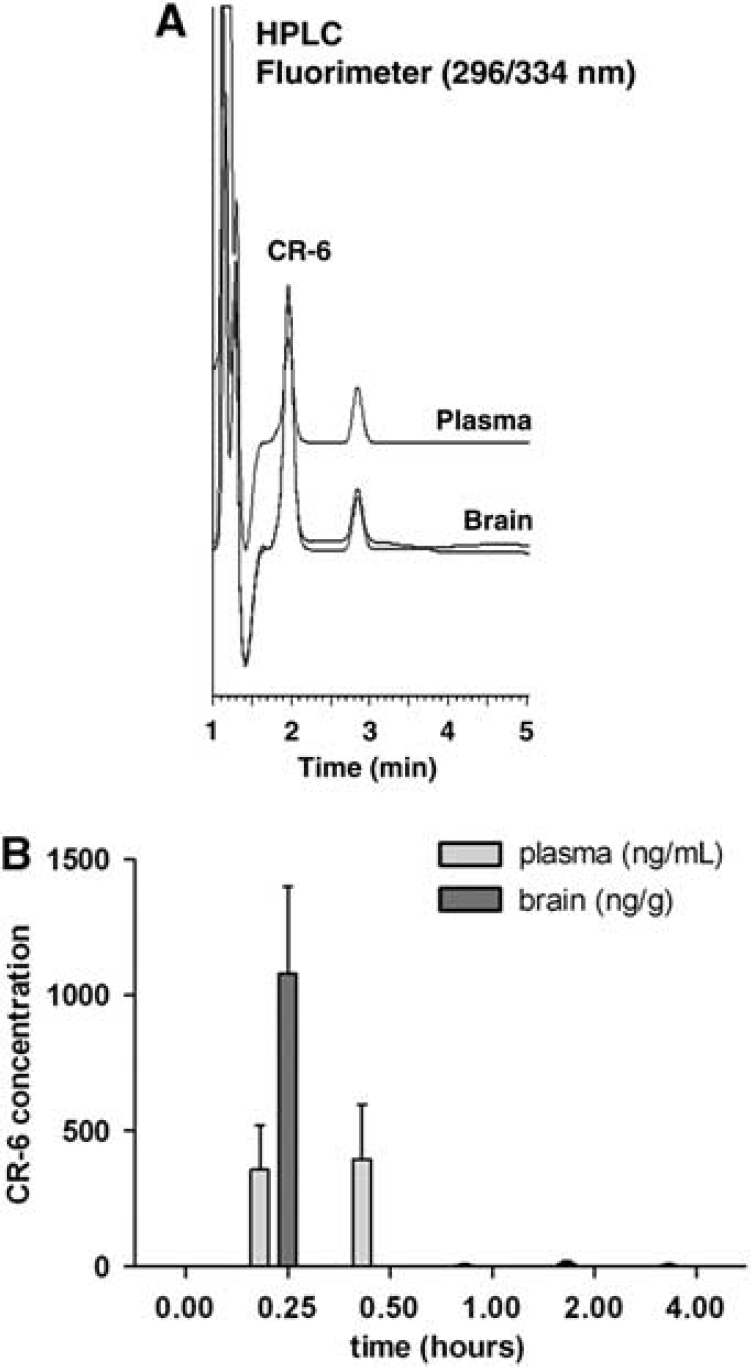
Detection of CR-6 in plasma and brain. CR-6 was extracted from plasma and brain, and the concentration was quantified by an HPLC method using fluorimetric detection. (
Discussion
CR-6 is a synthetic, structurally simpler, vitamin-E analogue that was previously used in a few studies in cells and animals subjected to conditions involving oxidative stress (Montoliu et al, 1999; Sanvicens et al, 2006; Miranda et al, 2007). This work is the first to investigate the action of CR-6 in animal models of stroke. The results show that CR-6 enters the brain after oral administration and exerts a protective action against cerebral ischemia/reperfusion, but not against permanent ischemia. We noticed that signs of cortical reperfusion injury became apparent in certain, but not all, rats subjected to ischemia/reperfusion. Rats with signs of reperfusion injury had a poorer outcome, and the beneficial effect of CR-6 was more powerful in this subpopulation of individuals. Indeed, after continuous recording of cortical CBF, we evidenced that some of the rats had reactive hyperemia immediately after reperfusion. These animals developed larger cortical infarcts and had worse neurological deficits than rats not showing hyperemia. On the basis of these findings, we can disclose that reactive hyperemia at early reperfusion is a sign of reperfusion injury. Nonetheless, we cannot exclude that other events associated to reactive hyperemia, such as the reported subsequent hypoperfusion (Traupe et al, 1982), were responsible for exacerbating ischemic damage. Solid lines of evidence support that hyperemia at the time of reperfusion is associated with worst brain injury (Tamura et al, 1980; Moskowitz et al, 1990; Macfarlane et al, 1991), and several studies showed that suppression of this effect was protective (Yamagami et al, 1998; Kim et al, 2006). Reactive hyperemia is induced by dilation of brain blood vessels due to decreased vascular resistance during the period of ischemia (Gourley and Heistad, 1984). Neurogenic mechanisms seem to be involved in the development of post-ischemic hyperemia (Macfarlane et al, 1991). Also, a previous study showed that administration of the NOS inhibitor L-NAME prior to ischemia prevented reactive hyperemia (Humphreys and Koss, 1998), suggesting that NO was involved in this effect. NO can react with superoxide generated at reperfusion to form peroxynitrite (Beckman et al, 1990), a very reactive species that induces irreversible protein nitrotyrosination and can cause tissue damage. Here we found that protein nitrotyrosination after ischemia was exacerbated in hyperemic rats and that this effect was attenuated by CR-6, likely through scavenging NO and peroxynitrite (Montoliu et al, 1999). The reaction of CR-6 with active nitrogen species renders the 5-nitroderivative of CR-6 as the main by-product. CR-6 also inhibits lipid peroxidation (Casas et al, 1992; Yenes et al, 2004) and previous studies indicate that CR-6 acts on radical oxygen species by a monoelectron transfer through the homolytic break of the phenolic hydroxyl group (Yenes et al, 2004). All these properties of CR-6 might contribute to the observed benefit.
Hyper-perfusion at reperfusion is not a phenomenon occurring in rats only, as there is evidence that some patients can develop hyper-perfusion shortly after thrombolysis with rtPA (Kidwell et al, 2001). Further investigation of the signs of reperfusion injury in reperfused patients would be necessary to find out whether this effect might be associated with poorer clinical outcome. Furthermore, our experimental study supports that, those individuals developing hyperemia at reperfusion are susceptible to respond better to antioxidant treatment, as a significant beneficial effect of CR-6 on the neurological score and infarct volume was precisely found in those animals, but not in animals not developing hyper-perfusion or in non-reperfused animals.
CR-6 attenuated the expression of COX-2 induced in neurons by ischemia/reperfusion, thus suggesting that oxidative/nitrosative stress underlies the induction of COX-2 under this condition and that CR-6 treatment directly or indirectly affected the neurons. Several lines of evidence support that COX-2 exerts detrimental effects in transient brain ischemia, as deficient mice show less damage than wild-type animals (Iadecola et al, 2001). Also, COX-2 inhibition is protective (Doré et al, 2003; Candelario-Jalil et al, 2007) and it reduces BBB breakdown and leukocyte infiltration (Candelario-Jalil et al, 2007), whereas overexpression of COX-2 worsens the outcome of ischemia (Doré et al, 2003). The harmful effect of COX-2 after ischemia is attributed to the production of prostaglandin-E2 rather than to generation of oxidative stress (Kunz et al, 2007). As COX-2 is also induced after very short episodes of ischemia causing no major brain damage (Planas et al, 1999), it is possible that the biochemical cascade activated by COX-2 needs interaction with other signals generated after severe ischemia to exert detrimental effects.
We investigated whether there were signs of oxidative stress in the MCA isolated from our animals. Generation of superoxide radicals was detected in the MCA after ischemia/reperfusion in rats receiving the vehicle. This effect was prevented in animals treated with CR-6. As this compound also attenuated ischemia-induced BBB dysfunction, it is feasible that CR-6 had some advantageous effect on the vasculature. Thus, the results support a beneficial vascular effect of CR-6, which may contribute to the protection observed in the ischemic brain tissue.
Taken together, these findings show that the antioxidant CR-6 reaches the rat brain tissue after oral administration and is protective after ischemia/reperfusion; protection is better manifested in a subgroup of animals that develop hyperemia at the time of reperfusion; brain damage is exacerbated in these animals as compared with that in animals not showing hyperemia; and CR-6 confers benefits at the interface between blood and brain by preventing generation of oxidative stress in the vessels and by attenuating BBB breakdown. The results also evidence that reperfusion injury may selectively develop in certain individuals, which would be the target of specific antioxidant drugs. This finding suggests that signs of reperfusion injury should be better investigated in reperfused stroke patients. In conclusion, this experimental study supports further exploration of the use of CR-6 for the treatment of stroke to attenuate possible negative effects associated with reperfusion.
Footnotes
Acknowledgements
We thank Ms Noelia Montoya and Mr Luca Maggioni for excellent technical assistance.
FJPA is a postdoctoral fellow of the ‘Juan de la Cierva’ program. The remaining authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.