
Abstract
High-mobility group box-1 (HMGB1) was originally identified as a ubiquitously expressed, abundant, nonhistone DNA-binding protein. It has well-established functions in the maintenance of nuclear homeostasis. The HMGB1 can either be passively released into the extracellular milieu in response to necrotic signals or actively secreted in response to inflammatory signals. Extracellular HMGB1 interacts with receptors, including those for advanced glycation endproducts (RAGEs) as well as Toll-like receptor 2 (TLR2) and TLR4. The HMGB1 functions in a synergistic manner with other proinflammatory mediators and acts as a potent proinflammatory cytokine-like factor that contributes to the pathogenesis of diverse inflammatory and infectious disorders. Numerous reports point to HMGB1 as a novel player in the ischemic brain. This review provides an appraisal of the emerging roles of HMGB1 in cerebral ischemia injury, highlighting the relevance of HMGB1-blocking agents as potent therapeutic tools for neuroprotection.
Introduction
High-mobility group box-1 (HMGB1) protein was originally described 30 years ago as a nonhistone DNA-binding protein with high-electrophoretic mobility (Goodwin et al, 1973). The HMGB1, also known as amphoterin, has been implicated in the stabilization of nucleosomal structure and the facilitation of gene transcription. The HMGB1 binds without sequence specificity to the minor groove of linear DNA and thereby facilitates assembly of nucleoprotein complexes and recruitment of transcription factors (Goodwin et al, 1973; Bustin, 2001). Moreover, HMGB1 is a key regulator of enhanceosome organization and overall activation of the basal transcriptional machinery (Ellwood et al, 2000; Verrijdt et al, 2002).
In addition to its role in the maintenance of nuclear homeostasis, HMGB1 recently emerged as an extracellular signaling factor with important roles in cell proliferation, differentiation, and disease pathogenesis (Hock et al, 2007). The HMGB1 was originally found within the cells; however, several studies have reported that HMGB1 also functions extracellularly (Rauvala and Pihlaskari, 1987; Merenmies et al, 1991; Wang et al, 1999a; Rouhiainen et al, 2004; Ulloa and Messmer, 2006). When present in the extracellular milieu, it can serve as an ‘necrotic marker,’ activating the innate immune system and mediating a wide range of physiological and pathological responses (Yamada and Maruyama, 2007). Extracellular HMGB1 contributes to the pathogenesis of disorders of the liver, lung, gut, and joints as well as septic shock and cancer (Dumitriu et al, 2005; Lotze and Tracey, 2005; Ulloa and Messmer, 2006; Yamada and Maruyama, 2007).
Moreover, in the brain, HMGB1 is released after cytokine stimulation and appears to be involved in the inflammatory process (Wang et al, 1999b; Agnello et al, 2002; Kim et al, 2006, 2008; Muhammad et al, 2008; Qiu et al, 2008). Interestingly, HMGB1, in turn, increases brain levels of tumor necrosis factor α (TNF-α) and interleukin-1β (IL-1β) induces anorexia and the loss of body weight in the mouse, and causes fever and allodynia in the rat (Agnello et al, 2002; O'Connor et al, 2003). A large body of information indicates that HMGB1's involvement in inflammatory reactions are of significance to progression of ischemic brain injury (Kim et al, 2006, 2008; Muhammad et al, 2008; Qiu et al, 2008). Thus, HMGB1 is a key regulator of the neuroimmune system and is involved in progression of neurologic disorders. This review aims to provide an appraisal of current knowledge of the role of HMBG1 in the brain with a focus on the relevance of HMGB1 in the pathogenesis of the cerebral ischemia and its potential as a novel therapeutic target.
Structure and functional domains of high-mobility group box-1
Structurally, HMGB1 has 215 residues organized into two basic DNA-binding domains, named A box and B box, and a negatively charged C-terminal tail (Landsman and Bustin, 1993) (Figure 1). Two nuclear localization signal sequences (NLS) are present in the protein at amino acids 28 to 44 and amino 180 to 185 (Bonaldi et al, 2003). The basic region contains two homologous HMG boxes, composed of α-helical structures implicated in DNA binding. In the nucleus, HMGB1 binds to the minor groove of linear DNA and bends it into a helical structure (Hock et al, 2007). After binding to DNA, HMGB1 interacts with and recruits various transcription factors, including p53, nuclear factor-κB (NF-κB), homeobox-containing proteins, recombination activating gene 1/2 proteins, and steroid hormone receptors (Hock et al, 2007), thereby altering chromatin architecture.
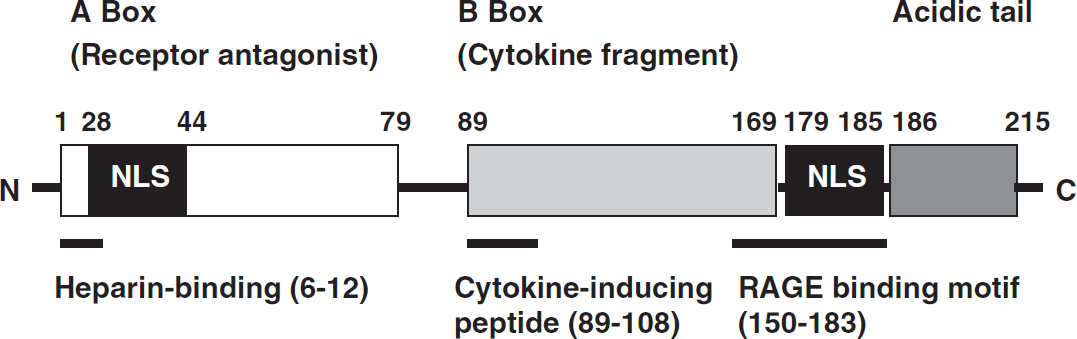
Schematic structure of HMGB1. The basic fragment comprises Box A and B, which can function as a RAGE receptor antagonist and cytokine, respectively. The two nuclear localization signals (NLS) are also depicted. The NLS contains the hyperacetylation sites targeted by acetyltransferases that is responsible for cytoplasmic accumulation of HMGB1.
The N-terminal region of HMGB1 contains a consensus sequence at amino acids 6 to 12 found in various heparin-binding proteins (Cardin and Weintraub, 1989) that likely contributes to the heparin/heparin sulfate binding capacity of HMGB1 (Huttunen and Rauvala, 2004). Other functional domains include the N-terminal amino acids 12 to 27 in the A box that are implicated in amyloidogeneis (Kallijarvi et al, 2001), a region in the A box relevant to antiinflammatory action (Yang et al, 2004), and proinflammatory cytokine (Lotze and Tracey, 2005), and amino acids in the B box important for cell differentiation (Sparatore et al, 2001).
A C-terminal region of HMGB1 at amino acids 150 to 183 mediates the receptor for advanced glycation endproducts (RAGEs) (Hori et al, 1995; Huttunen et al, 2002) and Toll-like receptors (TLRs) binding (Park et al, 2004). The RAGE is a cell-bound receptor of the immunoglobulin superfamily that is activated by a variety of proinflammatory ligands, including HMGB1, advanced glycation endproducts (AGE), members of the S100 family of proteins, and amyloid β-peptide (Bierhaus et al, 2005). Signal transduction through HMGB1-RAGE leads to activation of kinases such as ERK1/2 and p38, which in turn lead to activation of transcription factors, including NF-κB (Rauvala and Rouhiainen, 2007). The HMGB1 binding to TLR2 or TLR4 promotes transcriptional activity of NF-κB by activating myeloid differentiation primary response protein 88 (MyD88) (Park et al, 2004).
Release and activity of high-mobility group box-1
The HMGB1 has been highly conserved during evolution and is present in most eukaryotic cells. The HMGB1 also functions extracellularly (Wang et al, 1999a; Rouhiainen et al, 2004), though it lacks a classical secretion signal. The subcellular localization of HMGB1 depends on the type and activation state of the cell. The HMGB1 is usually localized in the cell nucleus and diffusely distributed in the cell cytoplasm (Bustin and Neihart, 1979). The HMGB1 can be released from the nucleus to the extracellular milieu in response to different stimuli (Figure 2).
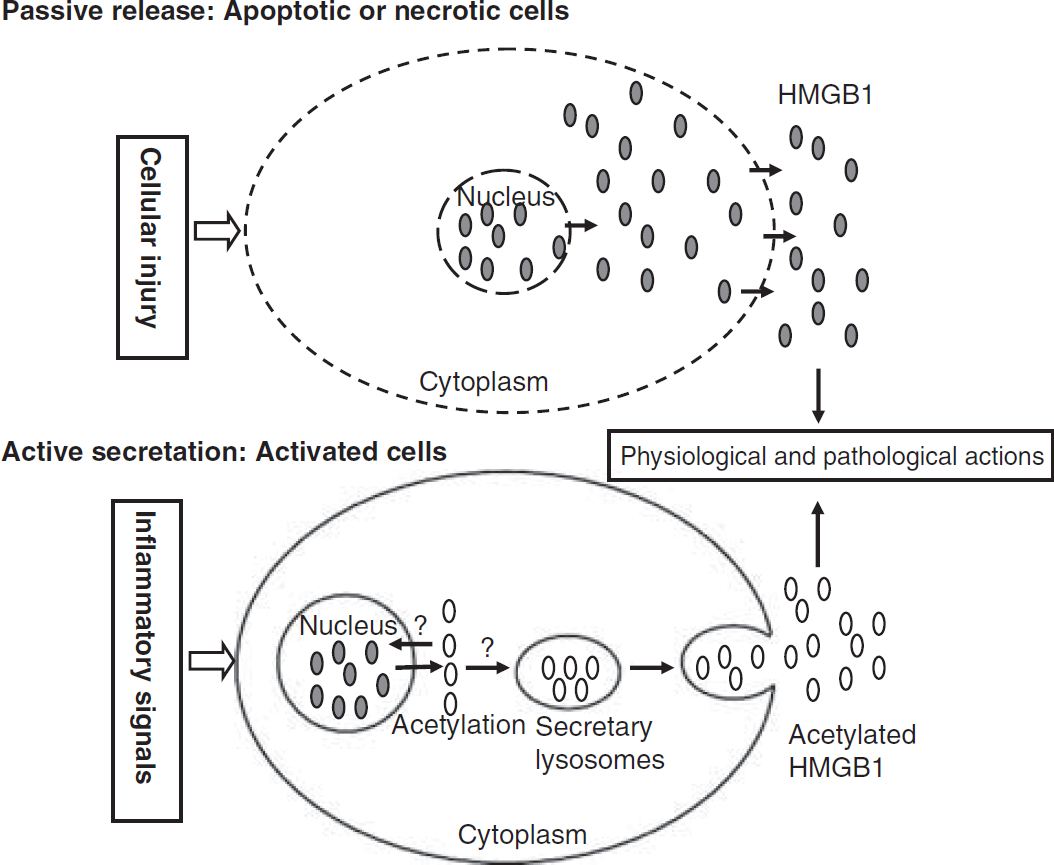
Pathways of HMGB1 secretion. There are two mechanisms used by cells to liberate HMGB1 into the extracellular milieu. Somatic cells contain large amounts of HMGB1 that is ‘passively released’ into the extracellular milieu during cellular apoptosis or necrosis. A second mechanism is the ‘active secretion’ of HMGB1 from activated immune cells or neuronal cells. The HMGB1 becomes acetylated on lysine residues within nucleus, which is thought to inhibit nuclear HMGB1 translocation, and thus, hyperacetylated HMGB1 isoforms accumulate in the cytosol where they are packaged into secretory lysosomes. The fusion of secretory lysosomes with the plasma membrane liberates HMGB1 into the extracellular environment. The molecular mechanisms underlying nuclear retention of HMGB1 and the trafficking of acetylated-HMGB1 into secretory lysosomes are still unclear. It is noteworthy that actively secreted hyperacetylated HMGB1 is molecularly different from passively released HMGB1.
At 25,000 daltons, HMGB1 is small enough to diffuse through nuclear pores. It contains nuclear localization signals and binds to chromatin in a more or less stable way, depending on the acetylation state of the chromatin (Scaffidi et al, 2002). The HMGB1 secretion is not fully understood. Gardella et al (2002) previously reported that the secretion of HMGB1 requires at least three steps: (1) exit from the nucleus into the cytoplasm, (2) translocation from the cytosol into cytoplasmic organelles, and (3) exocytosis. In normal conditions, HMGB1 protein is translocated from the cytosol into the nucleus where it binds to DNA and regulates transcription (Bianchi, 2004). Nuclear translocation of HMGB1 is controlled by least NLS1 and NLS2 and perhaps other signals (Bonaldi et al, 2003). Examination of the acetylated cluster suggests that the lysines between residues 27 and 43 might represent a classical bipartite NLS (Cokol et al, 2000). The HMGB1 becomes extensively acetylated on lysine residues on activation in monocytes and macrophages (Bonaldi et al, 2003). Once hyperacetylated, the NLS is masked and HMGB1 nuclear import is prevented, thereby leading to the cytoplasmic accumulation of the protein. The ultrastructural features of HMGB1-containing organelles suggest that HMGB1 is loaded into secretory lysosomes (Gardella et al, 2002; Bonaldi et al, 2003). The fusion of these secretory lysosomes with the plasma membrane liberates HMGB1 into the extracellular environment (Parrish and Ulloa, 2007) (Figure 2). The molecular mechanisms underlying nuclear retention of HMGB1 and the trafficking of acetylated-HMGB1 into secretory lysosomes are still unclear and deserve further investigation. In addition to acetylation, HMGB1 also undergoes different posttranslational modifications such as methylation, glycosylation, poly (ADP)-ribosylation, phosphorylation, and oxidation, which likely regulate its extracellular release and biological functions (Hoppe et al, 2006; Ulloa and Messmer, 2006).
As a nuclear protein, HMGB1 participated in chromatin architecture and transcriptional regulation as well as modification chromatin. In recent years, there is accumulating evidence that HMGB1 displays a wide range of activity of lipopolysaccharide (LPS) itself and other cytokines such as TNF-α. In addition to its effects, HMGB1 may have an important reparative as well as pathogenetic role in angiogenesis, myogenesis, and skeletal muscle function. These activities have been demonstrated in vitro and in vivo researches. In a word, to our knowledge, the intracellular and extracellular sources and targets of HMGB1 are summarized in Table 1.
Intracellular and extracellular sources, targets, and effects of HMGB1
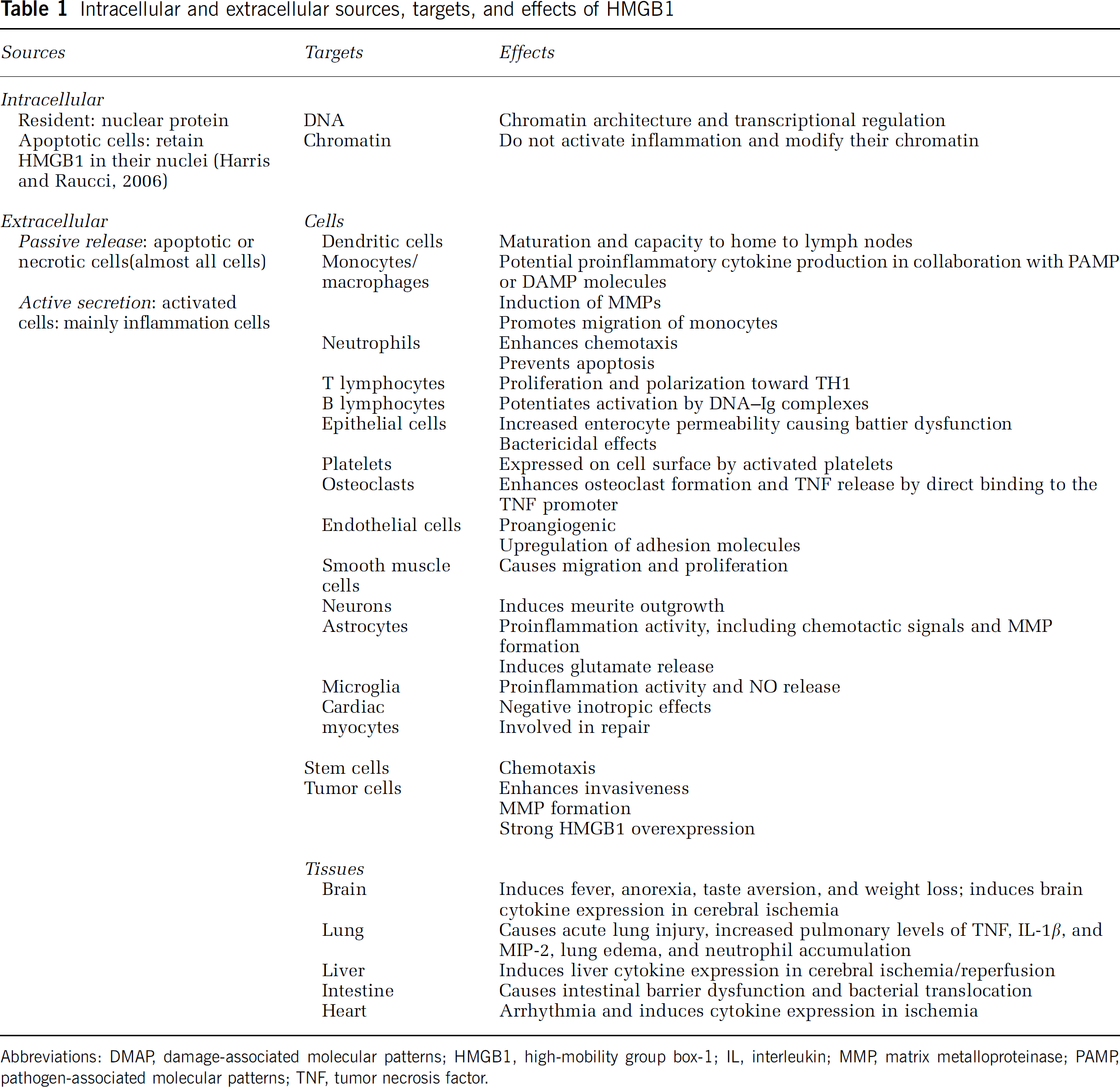
Abbreviations: DMAP, damage-associated molecular patterns; HMGB1, high-mobility group box-1; IL, interleukin; MMP, matrix metalloproteinase; PAMP, pathogen-associated molecular patterns; TNF, tumor necrosis factor.
High-mobility group box-1 receptors
The HMGB1 itself may signal through the receptors such as RAGE, TLR2, and TLR4, as well as other as yet unidentified receptors (Figure 3). Activation of these receptors results in the activation of NF-κB, which induces the upregulation of proinflammatory cytokines, thereby promoting inflammation. Furthermore, HMGB1-induced activation of NF-κB can induce the expression of HMGB1 receptors. Thus, HMGB1 can mediate amplification of inflammation through increased secretion of HMGB1 and increased expression of its receptors (Figure 3).
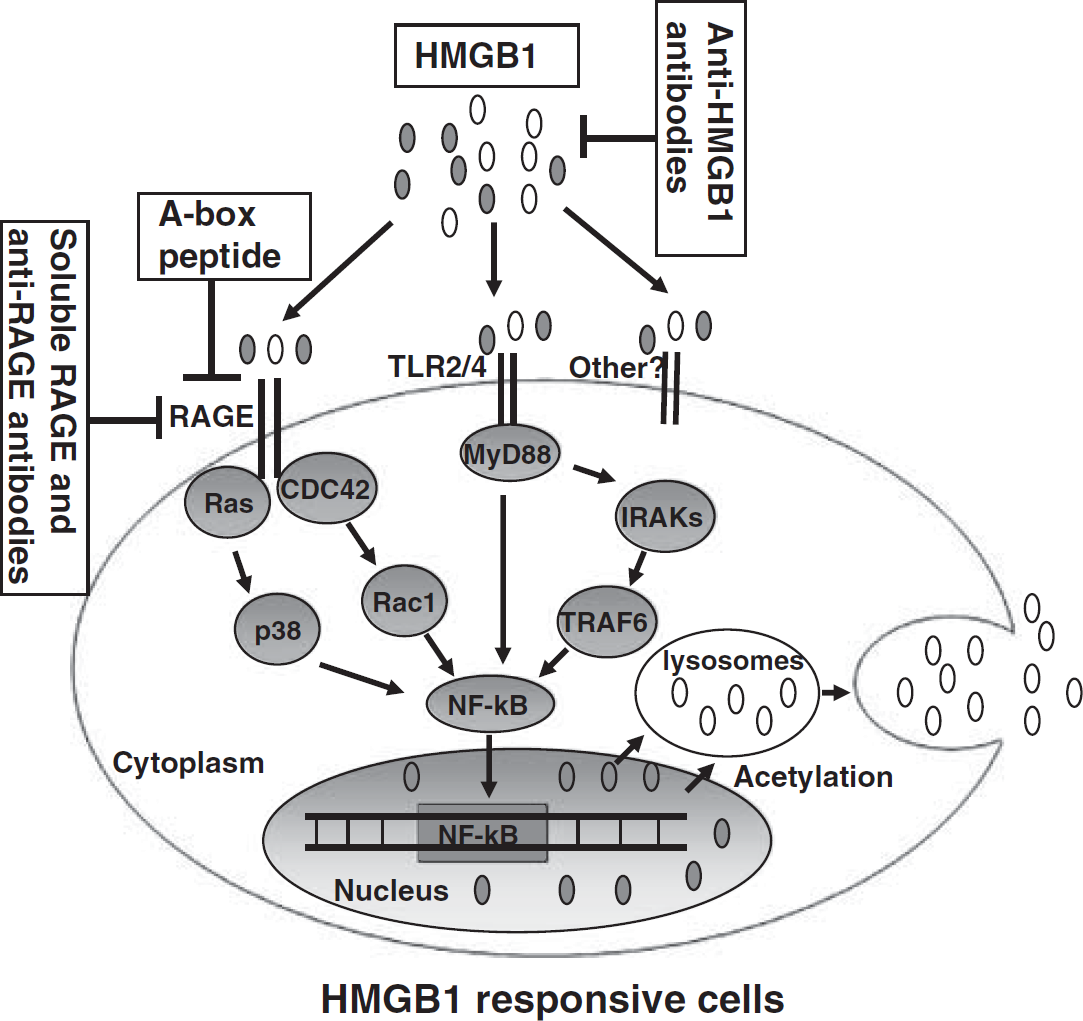
Schematic representation of HMGB1 signaling events mediated by RAGE and TLR2/4 and therapeutic strategies for cerebral ischemia. The HMGB1 is passively released into the extracellular space, thereby allowing its interaction with RAGE and TLR2/4, as well as other unknown receptors. The TLR2/4 can then signal via MyD88, IL-1R-associated kinase 1 (IRAK), and TNF-receptor-associated factor 6 (TRAF6) to NF-κB. The RAGE can activate the Rac1 and CDC42, as well as Ras and p38. The common signaling pathway involves activation of NF-κB to induce gene expression, including that of HMGB1. The HMGB1 may be a therapeutic target for the treatment of cerebral ischemia. Therapeutic strategies include anti-HMGB1 antibodies, HMGB1 A box protein, and soluble RAGE and anti-RAGE antibodies. Open ovals indicate ‘actively secreted’ acetylated HMGB1 and filled ovals indicate ‘passively released’ HMGB1.
Receptor for Advanced Glycation Endproduct
The RAGE is a multiligand signal transduction receptor of the immunoglobulin superfamily of cell surface molecules. The RAGE has been implicated in the pathogenesis of diabetic complications, neurodegenerative diseases, inflammatory disorders, and cancer (Rauvala and Rouhiainen, 2007). The RAGE is also one of the primary receptors for HMGB1 (Hori et al, 1995). The RAGE expression is detected on monocytes, macrophages, neurons, and endothelial cells, as well as on a variety of tumor cells (Yang et al, 2005).
The first evidence connecting HMGB1 to RAGE came from studies searching for RAGE ligands. Sequencing of proteins isolated with a RAGE affinity column identified the binding protein as HMGB1 (Hori et al, 1995). The HMGB1 binds to RAGE with a Kd of 5 to 10 nmol/L. Moreover, anti-RAGE antibodies inhibit neurite extension in cortical neurons induced by HMGB1 and RAGE and HMGB1 colocalize in the brain (Hori et al, 1995). Recently, Muhammad et al (2008) published evidence linking the functional role of RAGE to HMGB1 in ischemic mice model.
Activation of RAGE by HMGB1 can activate two major pathways, one encompassing CDC42/Rac and the other diverse mitogen-activated protein kinase (MAPKs) that finally lead to cytoskeletal changes and NF-κB activation, respectively. Expression of dominant-negative (dn) RAGE eliminates both NF-κB activation and neurite outgrowth. Moreover, dnCDC42 and dnRac1 but not dnRas inhibit neurite outgrowth induced by HMGB1. However, dnRas, but not dnCDC42 or dnRac1, inhibited NF-κB activation (Huttunen et al, 1999). Therefore, HMGB1-induced activation of RAGE is responsible for both NF-κB activation and neurite outgrowth via separate signaling pathways. Interestingly, the presence of NF-κB-binding sites in the RAGE promoter creates a positive feedback loop (Yan et al, 1994). Thus, HMGB1-mediated stimulation of RAGE may amplify this response by up-regulating RAGE expression. Typical NF-κB responsive genes are involved in inflammation (Fiuza et al, 2003). Thus, HMGB1-induced activation of RAGE can both initiate and sustain a proinflammatory phenotype.
The relationship between the biology of RAGE and HMGB1 is not straightforward. The RAGE knockout mice appear healthy without any overt phenotype (Liliensiek et al, 2004), whereas HMGB1 knockout mice live only until the early postnatal stage and suffer from problems of glucose homeostasis and multiorgan failure (Calogero et al, 1999). It is therefore expected that HMGB1 has other binding receptors in addition to RAGE.
Toll-like Receptor 2
The TLR2 mediates cell responses to lipoproteins and lipoteichoic acid from Gram-positive bacteria and mycobacteria (Means et al, 2001; Knapp et al, 2004) and to some rare LPS species (Martin et al, 2001). A direct interaction of HMGB1 with TLR2 has recently been demonstrated by fluorescence resonance energy transfer and immunoprecipitation in RAW264.7 macrophages (Park et al, 2006). The HMGB1 stimulation increases TLR2-mediated NF-κB activation in human embryonic kidney 293 (HEK293) cells (Park et al, 2006). Expression of dnTLR2 attenuates HMGB1-induced and NF-κB-driven luciferase expression in RAW264.7 cells (Park et al, 2004). In addition, expression of dn forms of the downstream TLR signaling regulators such as MyD88, TAK1 (TGF-β activated kinase 1), TAB2 (TAK1-binding protein 2), and p38 also inhibit HMGB1-induced NF-κB activation (Park et al, 2004). The HMGB1 induces significantly less TNF-α release in primary macrophages obtained from MyD88 and TLR4 knockout mice than from TLR2 knockout or wild-type controls (Yu et al, 2006). However, in HEK293 cells transfected with TLR2, HMGB1 effectively induces IL-8 release only from TLR2 over-expressing cells (Yu et al, 2006). Consistent with this, anti-TLR2 antibodies dose-dependently attenuate HMGB1-induced IL-8 release in TLR2-expressing HEK cells and markedly reduce HMGB1 cell surface binding activity on RAW 264.7 cells (Yu et al, 2006). Moreover, anti-TLR2 antibodies inhibit HMGB1-induced IL-8 and TNF-α secretion in several cell lines (Yu et al, 2006). Taken together, these data indicate that HMGB-1 induces the release of cytokines via a mechanism that at least in part depends on TLR2.
Toll-like Receptor 4
The TLR4 mediates many of the extracellular functions of HMGB1 (Park et al, 2004; Yu et al, 2006; Kruger et al, 2009; van Zoelen et al, 2009). Recently, a novel study was performed to investigate the role of TLR2, TLR4, and RAGE in in vivo responses to HMGB-1 (van Zoelen et al, 2009). The HMGB1-induced time-dependent elevations of TNF-α and IL-6 levels in peritoneal lavage fluid and plasma (van Zoelen et al, 2009). Compared with wild-type mice, both TLR4 and RAGE knockout mice display lower plasma levels of TNF-α and IL-6 at 2 h in their peritoneal lavage fluid after intraperitoneal injection of HMGB-1. In contrast, TLR2 knockout mice showed increased levels of TNF-α and IL-6 in their peritoneal cavity relative to wild-type mice (van Zoelen et al, 2009). These data suggest that HMGB1 induces the release of cytokines via a mechanism that depends on TLR4 and RAGE and that there is a differential effect of TLR2 and TLR4 on HMGB1 signaling.
How activation of TLR4 mediates the pleiotypic effects of HMGB1 is still unclear. The HMGB1 may directly interact with TLR4, causing cell activation and the NF-κB-dependent transcription of proinflammatory cytokines (Yu et al, 2006). Moreover, HMGB1 binds to cytokines, in particular IL-1β (Sha et al, 2008). The HMGB1 also catalytically disaggregates LPS and transfers it to soluble CD14 and to leukocytes. This enhanced access sustains a positive feedback amplificatory loop, facilitating TLR4-mediated inflammatory responses (Youn et al, 2008).
Toll-like Receptor 9
Notably, HMGB1 is reported to be an essential component of DNA-containing immune complexes that stimulate cytokine production through a TLR9-MyD88 pathway involving the multivalent receptor RAGE (Tian et al, 2007). Binding of HMGB1 to class A CpG oligodeoxynucleotides (ODN) considerably augments cytokine production by means of TLR9 and RAGE (Tian et al, 2007). The TLR9 uses HMGB1 as a cofactor and binds to both microbial DNA and RAGEs. The CpG-DNA-TLR9 complex gains access to the endoplasmic reticulum-Golgi intermediate compartment, thus accelerating the delivery of microbial DNA to TLR9 (Ivanov et al, 2007). CpG-ODN stimulates macrophages and dendritic cells to secrete HMGB1; in turn, extracellular HMGB1 accelerates the delivery of CpG-ODNs to its receptor, leading to a TLR9-dependent augmentation of IL-6, IL-12, and TNF-α secretion (Ivanov et al, 2007). Loss of HMGB1 leads to a defect in the IL-6, IL-12, and TNF-α response to CpG-ODN. However, lack of intracellular TLR9-associated HMGB1 can be compensated by extracellular HMGB1. Thus, the DNA-binding protein HMGB1 shuttles in and out of immune cells and regulates inflammatory responses to CpG-DNA (Ivanov et al, 2007) and is considered as an accelerator of TLR9 response with CpG-DNA (Tian et al, 2007).
Role of high-mobility group box-1 in the brain
The role of HGMB1 in the diseases of central nervous system (CNS) is under appreciated. It is notable that the extracellular presence of HMGB1 was first discovered in the brain (Goodwin et al, 1973). The HMGB1 was originally isolated from the brain and named p30; it was shown to have potent neurite outgrowth promoting activity (Rauvala and Pihlaskari, 1987). Since its discovery, a great deal of interest focused on the role of HMGB1 roles in CNS functioning. Several studies highlight a role of HMGB1 in CNS function. The HMGB1 is expressed in the nucleus of neurons, astrocytes, and pituicytes of the mouse brain (Passalacqua et al, 1998; Faraco et al, 2007; Qiu et al, 2008).
High-Mobility Group Box-1 Mediates Neuritic Outgrowth
Studies on rodent brain development suggest that HMGB1 has a role in the regulation of proper neuronal migration and sprouting (Rauvala and Pihlaskari, 1987; Merenmies et al, 1991; Guazzi et al, 2003). In N18 neuroblastoma cells that are at an active stage of flattening and initiating neurite, HMGB1 localizes to the advancing plasma membrane of filopodia at the leading edges in motile cells, which in turn promotes the outgrowth of neuritis (Rauvala and Pihlaskari, 1987; Merenmies et al, 1991). The HMGB1 is differently expressed in the mouse brain at various steps of development and tends to be downregulated in the adult animal (Guazzi et al, 2003). Moreover, the expression of and HMGB1 and RAGE colocalize at precise regions of the developing mouse cortex and cerebellum in a timely restricted manner (Chou et al, 2004). The RAGE expression levels are not affected in the adult mouse brain but anti-RAGE antibodies inhibit neurite outgrowth and neuronal migration (Chou et al, 2004). These findings strengthen the relevance of HMGB1 and its receptor RAGE to brain development.
High-Mobility Group Box-1 Regulates Neuroendocrine Response
Agnello et al (2002) reported that intracerebroventricular (ICV) injection of HMGB1 increased brain levels of TNF-α and IL-6 and induced anorexia and loss of body weight, as well as taste aversion with potencies equivalent to LPS, indicating that HMGB1 regulates the neuroendocrine response to immune stimuli. Rats develop fever and show increased TNF-α and IL-1β levels in various brain regions after ICV injection of HMGB1 (O'Connor et al, 2003). Moreover, allodynia occurs in rats treated with HMGB1 in the subarachnoid space at the lumbosacral level (O'Connor et al, 2003). Overall, data point to a key role of HMGB1 in triggering production of proinflammatory mediators typically associated with activation of the neuroimmune responses.
High-Mobility Group Box-1 Regulates Glutamate Release
Recently, two studies indicated that HMGB1 could act as a modulator of glutamate homeostasis in the brain (Pedrazzi et al, 2006; Bonanno et al, 2007). Pedrazzi et al (2006) reported that HMGB1 induced the release of a stable glutamate analogue, [3H]-D-aspartate and endogenous glutamate, from gliosomes, whereas nerve terminals were insensitive to the protein. Similarly, Bonanno et al (2007) demonstrated that HMGB1 induced glutamatergic release from glial (gliosomes) but not neuronal (synaptosomes) resealed subcellular particles isolated from mouse cerebellum and hippocampus. In a search for the mechanisms underlying the effect of HMGB1 on gliosomes, Pedrazzi et al (2006) found evidence that HMGB1-induced glutamate release is due to an interaction between HMGB1, RAGE, and the glial glutamate transporter. In addition, Ca2+ regulates HMGB1-dependent stimulation of glutamate release by facilitating HMGB1 binding to RAGE (Pedrazzi et al, 2006). Thus, HMGB1 has a role in the modulation of glutamate release and may be involved in the deleterious effects of glutamate-mediated excitotoxicity after the initial insult.
High-Mobility Group Box-1 Is Associated with Amyloidgenesis
Beyond the involvement of HMGB1 in the maintenance of brain homeostasis, HMGB1 participates to neuropathology. Takata et al (2003) found that HMGB1 was associated with senile plaques and was increased in brains affected by Alzheimer's disease. The HMGB1 immunoreactivity is increased in the hippocampi of kainic acid- or β-amyloid-microinjected mice (Takata et al, 2004). More interestingly, injection of β-amyloid together with HMGB1 delayed fibril clearance and increased their neurotoxic potential (Takata et al, 2004). Together, these findings suggest that HMGB1 promote amyloidgenesis.
Role of high-mobility group box-1 in cerebral ischemia
The HMGB1 has also been implicated in the mechanism of ischemic brain damage (Goldstein et al, 2006; Kim et al, 2006, 2008; Faraco et al, 2007; Liu et al, 2007; Muhammad et al, 2008; Qiu et al, 2008). In a stroke model, short hairpin (sh)RNA-mediated HMGB1 down-regulation in the brain reduces the severity of lesions (Kim et al, 2006). Notably, intravenous injection of anti-HMGB1 monoclonal antibody (mAb) causes a dramatic reduction in infarct size in stroke model (Liu et al, 2007; Mori et al, 2009). These indicate that HMGB1 has a critical role in cerebral ischemia.
Release of High-Mobility Group Box-1 in Cerebral Ischemia
Numerous studies have reported that HMGB1 is released early after ischemic injury from neurons. In patients with ischemic stroke, the serum or plasma levels of HMGB1 are dramatically higher than those in age- and sex-matched controls (Goldstein et al, 2006; Muhammad et al, 2008). In an ischemic stroke animal model, the serum level of HMGB1 is increased 4 h after ischemia (Kim et al, 2006, 2008). The HMGB1 is massively released into the extracellular space immediately after ischemic insult and it subsequently induces the release of inflammatory mediators in the post-ischemic brain (Kim et al, 2006). Furthermore, oxygen glucose deprivation, which mimics ischemia in vitro, induces the release of HMGB1 in the culture media of neurons (Faraco et al, 2007; Kim et al, 2008). Recent studies have revealed the relocation dynamics of HMGB1 in the neuronal cells. The HMGB1 is initially expressed in the nucleus of neurons and astrocytes of the mouse brain, predominantly translocates into the cytoplasm of neurons within the ischemic brain (Faraco et al, 2007; Qiu et al, 2008), then rapidly disappears from neurons (Kim et al, 2008; Qiu et al, 2008). HMGB-1 translocated from the neuron nuclei to the cytoplasm is subsequently depleted from neurons after 1 h of middle cerebral artery occlusion (Kim et al, 2008; Qiu et al, 2008).
High-Mobility Group Box-1 Contributes to Cerebral Ischemia
Three novel studies have indicated that HMGB1 has a pivotal role in ischemic brain injury. Firstly, Kim et al (2006) knocked down HMGB1 mRNA using a plasmid expressing an shRNA targeting the HMGB1 gene. The shRNA-mediated HMGB1 down-regulation in the post-ischemic brain suppressed infarct size. Importantly, reducing HMGB1 expression by shRNA reduced ischemia-dependent microglia activation and induction of inflammatory cytokines/enzymes (TNF-α, IL-1β, and inducible nitric oxide synthase (iNOS)) in the ischemic brain, indicating that HMGB1 has a pivotal role in the inflammatory process in ischemic brain (Kim et al, 2006). Secondly, treatment with neutralizing anti-HMGB1 mAb remarkably ameliorated brain infarction induced by 2 h occlusion of the middle cerebral artery in rats, even when the mAb was administered after the start of reperfusion (Liu et al, 2007). Furthermore, anti-HMGB1 antibody inhibited the activation of microglia, the expression of TNF-α and iNOS. In contrast, ICV injection of HMGB1 increased the severity of infarction and neuroinflammation (Liu et al, 2007). Finally, additional evidence indicating that HMGB1 is associated with ischemic brain injury comes from experiments showing that down-regulation of HMGB1 brain levels with rabbit polyclonal anti-HMGB1 antibody correlates with diminished infarct volumes (Muhammad et al, 2008).
Receptor for Advanced Glycation Endproduct Mediates High-Mobility Group Box-1-Induced Ischemic Brain Injury
A recent study assessing the contribution of RAGE activity to neural injury following cerebral ischemia in RAGE-targeted transgenic mouse indicates that RAGE directly contributes to pathology in cerebral ischemia (Hassid et al, 2009). Furthermore, the effect of HMGB1 box A, a functional antagonist of extracellular HMGB1 and HMGB1 interaction with RAGE, provides evidence that the effect of HMGB1 is mediated by RAGE (Bianchi and Manfredi, 2007). Muhammad et al (2008) reported that HMGB1 box A ameliorated ischemic brain damage. Interestingly, genetic RAGE deficiency and the decoy receptor soluble RAGE reduced the infarct size significantly. Moreover, in vitro data showed that the effect of HMGB1 depends on the expression of RAGE (Muhammad et al, 2008). However, this study did not exclude HMGB1 signaling through membrane receptors other than RAGE in cerebral ischemia. Our previous work has indicated that TLR4 may be involved in the pathophysiology of cerebral ischemia (Cao et al, 2007; Yang et al, 2008). However, it is not currently clear to what extent the TLR2 and TLR4 contribute to the HMGB1-initiated cerebral ischemic injury. Intriguingly, a recent study has questioned the above hypothesis, and showed that only TLR9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE (Tian et al, 2007).
Therapeutic interest of high-mobility group box-1 in cerebral ischemia
The HMGB1 has been proven to be an excellent therapeutic target in experimental models of infectious and inflammatory disorders, including sepsis, trauma, cancer, and rheumatoid arthritis (Ulloa, 2005; Ulloa and Tracey, 2005; Ulloa and Messmer, 2006). Importantly, HMGB1 is a ‘late’ appearing inflammatory cytokine that provides a wider time frame for clinical intervention against progressive inflammatory disorders (Ulloa and Messmer, 2006). The HMGB1 is a potential target for the treatment of cerebral ischemic injury. Therapies related to this disease include anti-HMGB1 antibodies, HMGB1 A box protein, and soluble RAGE and anti-RAGE antibodies (Figure 3).
Anti-High-Mobility Group Box-1 Antibodies
Although HMGB1 antibodies have not worked in clinical stroke patients so far, there is evidence that these antibodies improve the outcome in animal model of stroke (Liu et al, 2007; Muhammad et al, 2008; Mori et al, 2009). The HMGB1 mAb evaluated in a rat ischemic model is directed to the repetitive C-terminal tail of HMGB1 (Liu et al, 2007). Most recently, Mori et al (2009) reported that treatment with HMGB1 mAb remarkably ameliorated brain infarction in rats, even when the mAb was administered after the start of reperfusion. Furthermore, the accompanying neurologic deficits in locomotor function were significantly improved. In addition, some biochemical markers such as permeability of the blood–brain barrier, the expression of TNF-α, iNOS, and matrix metalloproteinase-9 were altered by mAb injection (Mori et al, 2009). Interestingly, anti-HMGB1 antibodies inhibit HMGB1-induced TNF-α and IL-6 production, but do not affect the ability of TNF-α to induce IL-6 release in macrophages (Wang et al, 1999a). These results indicate that anti-HMGB1 antibodies specifically inhibit extracellular HMGB1 activity through its membrane receptors rather than block the ability of cells to respond to immune stimuli (Wang et al, 1999a).
Similarly, intervention with neutralizing polyclonal anti-HMGB1 antibodies can prevent the progression of established stoke in rodents (Muhammad et al, 2008). Indeed, HMGB1 in vitro avidly binds different molecules, thereby preventing HMGB1-specific mAbs from recognizing any given epitope of the HMGB1 molecule. Moreover, polyclonal antibodies reacting with multiple epitopes may circumvent the problem resulting from occupancy of binding sites by ligated determinants. Interestingly, neutralizing strategies specifically inhibit extracellular HMGB1 activity rather than prevent its secretion (Wang et al, 1999a); therefore, current efforts are focused on developing anti-inflammatory strategies that inhibit HMGB1 secretion.
High-Mobility Group Box-1 A Box Protein
The HMGB1 contains two DNA-binding domains (box A and box B) and an acidic C-terminal tail (Bianchi and Manfredi, 2007). The proinflammatory property of HMGB1 may be reproduced by the B domain. The HMGB1 box A is a functional antagonist of the proinflammatory effects mediated by full-length HMGB1 protein through competitive binding to RAGE (Yang et al, 2004). The HMGB1 box A has been used as a pharmacological tool to inhibit the effects of HMGB1 that are mediated through RAGE (Yang et al, 2004; Sitia et al, 2007). A box protein therapy confers significant clinical protection in experimental inflammatory conditions, including lethal endotoxemia and arthritis (Kokkola et al, 2003; Yang et al, 2004). Thus, there may be therapeutic potential in using genetically engineered peptides to decrease inflammation. In a study of stroke, systemic administration of HMGB1 box A protein significantly ameliorated ischemic brain injury (Muhammad et al, 2008), suggesting HMGB1 box A may provide a tool for therapy in stroke.
Soluble Receptor for Advanced Glycation Endproduct and Anti-Receptor for Advanced Glycation Endproduct Antibodies
The HMGB1 can signal through RAGE and this interaction has an important role in brain ischemia (Muhammad et al, 2008). Inhibition of the HMGB1–RAGE interaction suppresses activation of NF-κB, which induces the down-regulation of proinflammatory cytokines (Rauvala and Rouhiainen, 2007). The RAGE has a secretory, alternatively spliced isoform, sRAGE, which lacks the transmembrane domain and circulates in plasma. By competing with cell surface RAGE for ligand binding, sRAGE may act as a decoy receptor and remove circulating RAGE ligands (Schmidt et al, 1994). The sRAGE provides a pharmacological tool to translate results obtained in RAGE knockout mice into a more clinical setting (Schmidt et al, 1994). Treatment with sRAGE protein ameliorates both inflammation and infarct size in the brain of rat (Muhammad et al, 2008). Furthermore, anti-RAGE-specific mAb is effective in an experimental model of sepsis (Lutterloh et al, 2007), supporting this pathway as a target for therapies.
Summary of high-mobility group box-1 work in central nervous system
To date, the majority of information on HMGB1's function has been gathered from studies focused on peripheral organs. A few results indicate HMGB1 may have important role in disease's of CNS, especially in cerebral ischemia. But, the protein's role in diseases of the CNS is less appreciated. In fact, in the CNS, the biological property of HMGB1 remains unclear and the mechanism by which HMGB1 acts in the brain has been difficult to define. Since its discovery, a great deal of interest focused on the role of HMGB1 roles in CNS functioning, therefore, increasing efforts will help elucidating the relevance of HMGB1 to the pathogenesis of CNS diseases, as well as providing new tolls to therapeutic interventions. In a word, to our knowledge, the targets and effects of approaches that modulate HMGB1 in CNS, including ischemia stroke, are summarized in Table 2.
The targets and effects of approaches that modulate HMGB1 in CNS
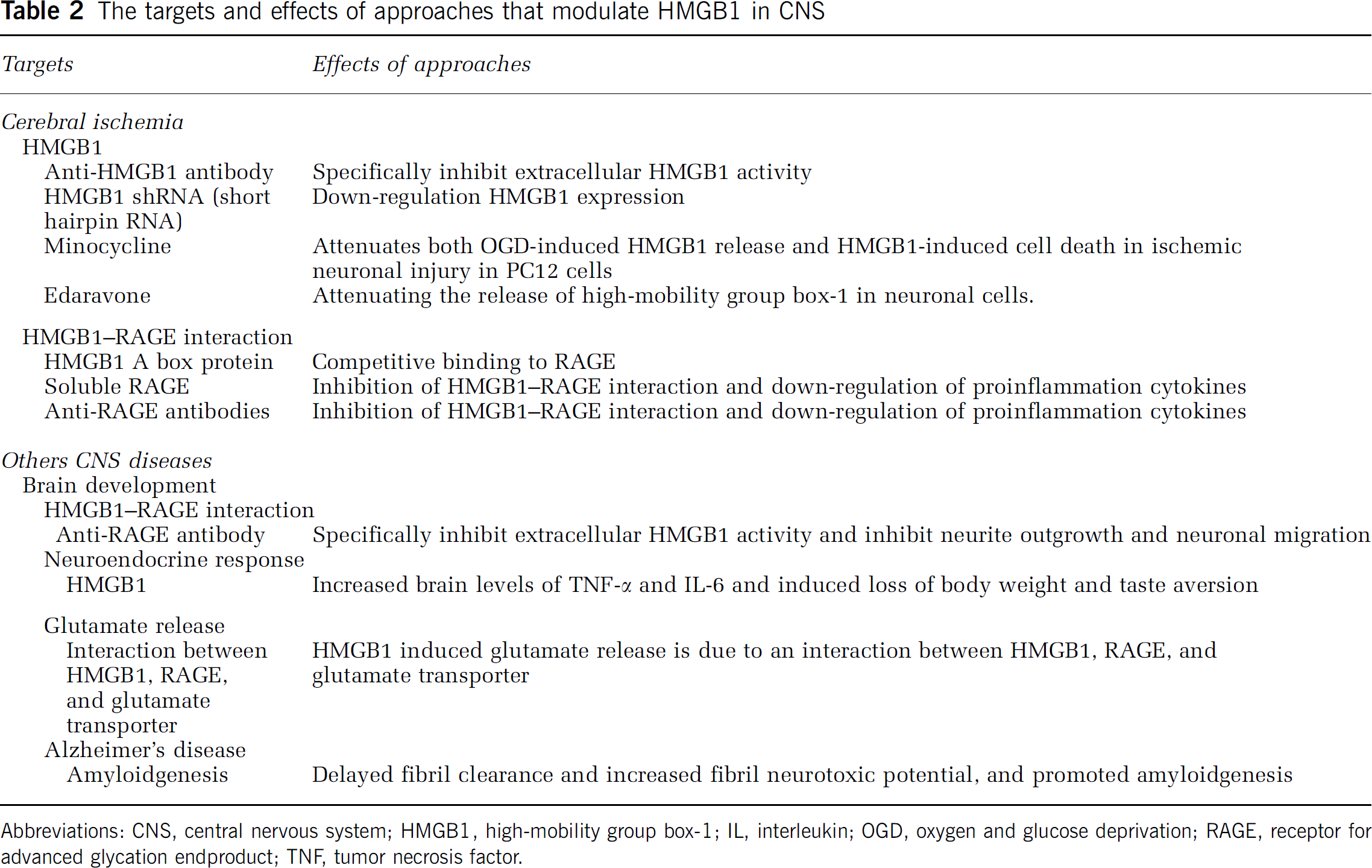
Abbreviations: CNS, central nervous system; HMGB1, high-mobility group box-1; IL, interleukin; OGD, oxygen and glucose deprivation; RAGE, receptor for advanced glycation endproduct; TNF, tumor necrosis factor.
Unanswered questions
A growing body of evidence indicates that HMGB1 represents a comprehensive cytokine able to orchestrate the regulation of inflammation (Yang et al, 2005; Ulloa and Messmer, 2006). Accordingly, HMGB1 can represent a potential target for inflammatory conditions. However, a major limitation in designing pharmacological strategies against HMGB1 is that its mechanism of secretion from cells remains unknown. There are a number of ‘holes’ in our knowledge of HMGB1. For instance, where is HMGB1 synthesized? Why is HMGB1 consolidated in lysosomes? Is there more than one pool of HMGB1 in cytoplasm? By what mechanism is HMGB1 transported into the nucleus? How is HMGB1 metabolized after release from cells? How does HMGB1 translocate into the brain? The nature of HMGB1 release, whether lysosome release, secretion, or release on cell demise, needs to be well determined before HMGB1 can be used as target for therapies.
Conclusions
The HMGB1 has been mainly studied in the context of a wide variety of inflammatory conditions in peripheral organs. Emerging evidence indicates that HMGB1 has a key role in homeostatic regulation of brain functions and actively participates in neuropathology and neuroinflammatory responses. Several studies highlight HMGB1's role as a novel mediator of cerebral ischemia injury. Targeting HMGB-1 signaling may be an effective therapeutic strategy for ischemic stroke. Efficient inhibition of HMGB1 signaling has been achieved by using HMGB1 box A as a decoy agent or anti-HMGB1 antibodies in the brain ischemia models.
Footnotes
Acknowledgements
This work was supported in part by a grant from the National Natural Science Foundation of China (No. C30500640, C30870859), the Chongqing Natural Science Foundation (CSTC, 2008BB5279), and a grant from the Science Funds of the Third Military Medical University (No. 06105).
The authors declare no conflict of interest.