15. Hippocampal spreading depression is associated with a severe blood flow—glucose use mismatch but not with CA1 neuron injury
G. Mies
Max Planck Institute for Neurological Research, Cologne, Germany
Objectives: Prolonged cortical spreading depression (CSD) has not been associated with neuronal injury1 because of a close coupling between cerebral blood flow (CBF) and cerebral metabolic rate of glucose (CMRG). The hippocampal formation, however, is known for its selective vulnerability of CA1 neurons even after a brief episode of transient cerebral ischemia. It, therefore, was of interest to examine the hippocampal CBF/CMRG coupling and the histological outcome after repetitive hippocampal spreading depression (HSD).
Methods: Male BD IX rats (n = 21) were used for this investigation under pentobarbital anesthesia and mechanical ventilation. HSD was induced by micropipette injections of 4 Mol KCl into the CA1 layer of the hippocampus. Hippocampal EEG activity and DC waves were recorded from a neighbouring glass microelectrode attached to a calomel electrode. In 5 rats, double tracer autoradiography was applied during repetitive HSD using 14C-deoxyglucose (DG) for local CMRG starting at 15 mins after HSD onset and 131I-iodoantipyrine (IAP) at 44 mins for CBF. In other animals, CBF was examined at 15 mins and CMRG after 45 mins of continuous HSD. The histological outcome was examined in a separate group following 45 mins of several HSD waves after 7 days of survival.
Results: The most striking finding of this investigation was the absence of CA1 neuron loss at one week after 45 mins of repetitive HSD despite a pronounced mismatch of the early hippocampal CBF/CMRG coupling. After several evoked HSD-waves at 45 min, CMRG had increased by 66% (41±8 vs. 68±16 μmol/100g/mins; P<0.05) whereas CBF declined by 38% (78±14 vs. 56±8 ml/100g/min; P<0.05) (see Figure), accounting for a severe mismatch of the flow/metabolism relationship (0.9±0.3 vs. 1.9±0.3 ml/μmol; P<0.05). CMRG and CBF measured at 15 mins or 45 mins following HSDs were comparable. At day 7 after 60 mins of repeated HSDs, however, no significant difference in the number of hippocampal CA1 neurons was observed.
Conclusions: In this study, it has been shown for the first time that a severe mismatch in CBF/CMRG alone does not cause neuronal injury, even in most selectively ischemic vulnerable CA1 neurons. These findings suggests that not the transient decrease in CBF and/or the rise in CMRG trigger neuronal injury per se but a critical level of hypoxia and its duration seems to determine neuronal survival. Further studies are in progress to elucidate the observed pathogenetic differences of hippocampal CA1 injury in transient ischemia and HSD.
82. Transient hemiparesis after topical application of 0.3 M KCl to the sensorimotor cortex in unrestricted awake mice
H. Hattori1, M. Tomita1, H. Toriumi1, Y. Tomita2, M. Unekawa1 and N. Suzuki1
1Department of Neurology; 2Department of Preventive Medicine for Cerebrovascular Disease, Keio University School of Medicine, Tokyo, Japan
Purpose: Cortical spreading depression (CSD) is a fundamental process in brain pathology, but visualization of its occurrence is difficult without employing invasive techniques. Here we report a novel model in which CSD occurrence is noninvasively evaluated from objective clinical symptoms.
Method: We used sixteen male C57BL/6J mice, anesthetized with isoflurane. A pinhole was drilled above the left parieto-temporal cortex, 1 mm lateral from the sagittal suture and 1 mm posterior from the bregma and 10 ml of 0.3 M KCl solution was injected through the hole onto the dura of the left cerebral cortex (n = 8). The same amount of saline was injected in control mice (n = 6). In 2 additional mice, DC potential was continuously recorded to confirm CSD occurrence with an enamel-covered 150 mm platinum electrode (implanted 300 mm deep into the cerebral cortex through another hole 3 mm anterior to the first hole), which was fixed tightly with dental cement to the skull bone. An Ag-AgCl electrode inserted into the space between the skull and scalp was used as the reference electrode. The wires were securely sutured to the back skin and were long enough to allow free movement. The mouse was released from the head holder and its behavior after recovery from anesthesia was videotaped and analyzed.
Results: Each mouse awoke within a few minutes, and attempted to stand up. In the CSD group the mouse showed a right-twisted posture with hemiparesis of the right extremities: the right fingers took a flexor position and the leg was loosely extended. Most mice struggled to lift their body, but were not able to do so because of the weakness of the right extremities. The mice could use only the left extremities, and so could only move in circles. Hemiparesis was observed in all KCl-treated mice. It was transient, lasting approximately 5 mins, and was followed by recovery. In the DC potential recording group, we detected negative repetitive deflections of ca. 20 mV in DC potential. When the mouse awoke, the base-line level of the DC potential slightly shifted up, but negative deflection was seen occasionally, overlapping on the baseline record. The alert mouse became quiet with eyes closed concomitantly with the negative deflections. In one mouse the DC potential was recorded for more than one hour, but the recording was terminated when the mouse pulled out the electrode from its brain with its hands.
Discussion: Such transient symptoms as abnormal posture and hemiparesis, comparable to those in cases of MCA occlusion, have not been reported previously, since the animals in previous CSD studies were anesthetized and were immobilized with the head fixed to a head holder. We are not certain yet that the hemiparesis observed here reflects the DC potential deflection alone. However, since the same amount of K was administered in exactly the same manner as in the previous reports, we consider that the clinical symptoms elicited were attributable to the CSD.
Conclusion: Although the approach used here is quite simple, the model may be useful to investigate CSD.
227. Cardiovascular response to asphyxia does not critically depend on generation of anoxic depolarization
F. Richter1, A. Lehmenkühler2, R. Bauer3 and H.-G. Schaible1
1Insitute of Physiology I/Neurophysiology, Friedrich Schiller University Jena, Jena; 2Pain Inst. & Ctr. for Med. Education, Düsseldorf; 3Institute of Molecular Cell Biology, Friedrich Schiller University Jena, Jena, Germany
Recently, we were able to exclude that transient asphyxia (up to 1 mins) conditions the adult rat brainstem for spreading depression (SD). Rather superfusing the brainstem with artificial cerebrospinal fluid (ACSF) in which 75% of the chloride ions were replaced by acetate and to which 10 mmol/L KCl and 10 mmol/L tetraethylammonium (TEA) were added was suitable to condition the brainstem for SD. Brainstem SDs were accompanied by transient increases in regional blood flow in the brainstem, but not in cerebral cortex. Now we tested, whether a transient asphyxia:
can induce DC shifts resembling SD in the adult brainstem, and
whether such DC shifts were accompanied by changes in regional blood flow or vegetative parameters.
Experiments were performed in 15 sodium thiopentone-anesthetized rats (100 mg/kg, i.p.), paralyzed with pancuronium bromide (2 to 4 mg/kg/h i.v.) and artificially ventilated. We recorded DC deflections at two sites in the brainstem close to the caudal trigeminal nucleus (lateral spacing 1 mm), and in the cerebral cortex. Both at the cerebral cortex and at the brainstem local blood flow was continuously recorded by Laser Doppler probes together with arterial blood pressure and heart rate. Blood gases and acid-base balance values were checked prior, during and after each period of asphyxia. Asphyxia was induced by stopping the respiratory pump.
In the native brainstem an asphyxia lasting about 50 to 60s resulted in a negative DC deflection of 5.5±5.4 mV (n = 10) (mean value±s.d., respectively). Systemic arterial blood pressure (ABP) declined to 25.5%±4.7% and heart rate to 55.3%±21.3%. The regional blood flow in the brainstem decreased to 48.9%±28.1% and in the cortex to 66.1%±24.1% of baselines. During reoxygenation ABP and heart rate normalized, but blood flow increased transiently in the brainstem to 395.1%±100.7%, and in cerebral cortex to 210.9%±82.6%. Superfusion of the brainstem with acetate-TEA-KCl-ACSF did not alter blood gases and acid-base balance values. After this pretreatment, 1-min-asphyxia caused negative DC deflections in the brainstem reaching 25.4±6.5 mV (n = 30), accompanied by reduced heart rates (45.2%±21.5% of baseline) and ABP (43.8%±16.4% of baseline). Accordingly, regional blood flow in the brainstem decreased to 71.7%±33.1% and in cerebral cortex to 66.7%±37.2%. During reoxygenation both heart rate and ABP normalized quickly, but similar as in controls, regional blood flow in the brainstem exceeded the baseline to 343.2%±135.2% and in cerebral cortex to 225.5%±95.1%. Systemic administration of MK-801 (3 mg/kg, i.p.) did neither influence the asphyxia-related DC shifts nor the observed changes in ABP, heart rate or regional blood flow.
In adult rats the conditioning of the brainstem with acetate-TEA-KCl-ACSF was a prerequisite to observe full-blown negative DC shifts during asphyxia. However, during transient asphyxia heart rate and ABP declined quantitatively similar whether anoxic depolarization was elicited or not. This was in contrast to spreading depression in the brainstem where DC shifts caused a transient increase in regional blood flow in brainstem and in ABP.1 Thus, the cardiovascular response to asphyxia does not critically depend on generation of anoxic depolarization.
334. Gabapentin acutely suppresses cortical spreading depression in rats
U. Hoffmann1, C. Kudo1,2, E. Dilekoz1, M.A. Moskowitz1 and C. Ayata1,3
1Radiology, Stroke and Neurovascular Regulation Lab, Massachusetts General Hospital, Harvard Medical School, Charlestown, Massachusetts, USA; 2Department of Dental Anesthesiology, Osaka University Graduate School of Dentistry, Osaka, Japan; 3Neurology, Stroke Service and Neuroscience Intensive Care Unit, Massachusetts General Hospital, Harvard Medical School, Charlestown, Massachusetts, USA
Objectives: Cortical spreading depression (CSD), a slowly propagating wave of neuronal and glial depolarization, is the putative mechanism of migraine aura, and may trigger headache as well. It can be evoked in experimental animals when extracellular K+ concentration exceeds a critical threshold upon chemical or electrical stimulation. Spontaneously arising CSD-like depolarizations have recently been demonstrated in injured human brain as well. We have previously shown that migraine prophylactic agents (e.g., valproate) suppress CSD upon chronic treatment. It is not known whether this mechanism is shared by other migraine therapeutics as well. Gabapentin, an antiepileptic which also shows efficacy in migraine in preliminary studies, inhibits Cav2.1 channels (formerly the P/Q type) by binding the α2δ-1 subunit. Pharmacologic inhibition and gain-of function mutations in Cav2.1 channels, suppress and enhance CSD, respectively. Therefore, we tested the efficacy of gabapentin on CSD.
Methods: CSDs were recorded from parietal and frontal cortex using glass micropipettes in intubated and mechanically ventilated (70%N2O/30%O2) isoflurane-anesthetized rats. Gabapentin was administered as a single intravenous dose; CSD susceptibility was determined 1 h after injection. In addition, gabapentin was administered chronically for 4 weeks in two divided daily oral doses; CSD susceptibility was determined ∼2 h after the last dose on the day of testing. Chronic valproate treatment (single daily intraperitoneal injections for 4 weeks) was used as a positive control. CSD susceptibility was determined by:
electrical threshold for inducing CSD (single square pulses with increasing intensity on the occipital cortex), and
the frequency of repetitive CSDs induced by topical KCl (1 M for 1 h on the occipital cortex).
Results: A single administration of gabapentin dose-dependently suppressed KCl-induced CSD frequency, and elevated the electrical CSD threshold. Chronic oral treatment using a lower dose was ineffective. Chronic valproate administration significantly reduced KCl-induced CSD frequency and elevated the electrical CSD threshold, as reported previously. CSD speed, duration and amplitude did not differ among groups.
Conclusions: In summary, gabapentin dose-dependently suppressed CSD susceptibility as determined by two independent methods. Unlike other migraine prophylactic agents previously tested, chronic treatment was not required for efficacy, nor enhanced it. These data provide a potential mechanism for gabapentin action in migraine. Furthermore, because chronic treatment was not a prerequisite for efficacy, gabapentin, with its favorable pharmacokinetic and safety profile, may have a role in suppressing the CSD-like injury depolarizations shown to be detrimental in stroke, trauma and subarachnoid hemorrhage.
Data are mean±s.d. or median (interquartile range). bid, twice a day; qd once a day,
P<0.05.
341. Facilitated subcortical propagation of cortical spreading depression in familial hemiplegic migraine type 1 mutant mice
K. Eikermann-Haerter1, Y. Wang1, M.D. Ferrari2, A.M.J.M. Van Den Maagdenberg2,3, M.A. Moskowitz1 and C. Ayata1,4
1Radiology, Massachusetts General Hospital, Harvard Medical School, Charlestown, Massachusetts, USA; 2Neurology, 3Human Genetics, Leiden University Medical Center, Leiden, The Netherlands; 4Neurology, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts, USA
Background and aims: Familial hemiplegic migraine (FHM) is a severe migraine variant. Characteristic are headaches, sometimes accompanied by transient but often severe motor deficits. FHM type 1 has been linked to missense gain-of-function mutations (e.g., S218L and R192Q) in the CACNA1A gene encoding the alpha1 subunit of neuronal Cav2.1 channels. FHM patients with the S218L mutation have a stronger clinical phenotype compared to those with R192Q mutation, and may develop seizures and coma in response to mild head trauma. Cortical spreading depression (CSD), the putative electrophysiological event underlying migraine, is a transient disruption of membrane ionic gradients and depolarization that slowly propagates across the cerebral cortex. Under favorable conditions, CSD can propagate into subcortical structures. We previously showed that FHM mutant mice exhibit increased susceptibility to CSD and its propagation into the striatum. Here we studied in detail the subcortical propagation of CSD into striatum, hippocampus, thalamus, brain stem and cerebellum in FHM type 1 mutant mice.
Methods: CSDs were induced every 15 mins by brief topical KCl application on the occipital cortex in pentobarbital-anesthetized, mechanically ventilated (70%O2/30%N2), and physiologically monitored female R192Q or S218L knockin mice, and recorded simultaneously from subcortical structures and cortex using glass micropipettes. The spatial extent of subcortical SD propagation was studied by c-fos immunohistochemistry, 3 h after 3 CSDs induced over 45 mins.
Purpose: To study subcortical propagation pattern of spreading depression in FHM mutant mice.
Results: In S218L mice, CSD regularly propagated into striatum, hippocampus and thalamus, exhibiting an allele-dosage effect. When CSD propagated into thalamus, it did cross the midline and spread to the contralateral thalamus as well; spread into the contralateral cortex was not observed when tested in 3 mice. In R192Q mice, CSD often propagated into striatum (less frequently than S218L) but not into hippocampus or thalamus. None of the mutant strains showed brain stem or cerebellar SD. c-fos expression was upregulated in cortex, striatum, hippocampus (particularly dentate gyrus) and thalamus in S218L, and in cortex and striatum in R192Q mice.
Conclusions: In summary, FHM type 1 mutations facilitate subcortical propagation of CSD. Both S218L and R192Q strains show CSD propagation into striatum, the structure likely responsible for post-SD hemiparesis in mice. In addition, the S218L mutation facilitates CSD propagation into hippocampus as well as bilateral thalami, which are potential mechanisms for seizures and altered level of consciousness and coma in these patients.
Strain
Genotype
N
Striatum
Hippocampus
Thalamus
S218 L
WT
6
32%/75%
0%/0%
0%/0%
S218 L
HET
5
100%/100%
24%/100%
0%/0%
S218 L
HOM
10
100%/100%
88%/100%
58%/86%
R192Q
WT
4
32%/100%
0%/0%
0%/0%
R192Q
HOM
4
71%/100%
0%/0%
0%/0%
N: # of mice tested. Data are expressed as % of CSDs/% of mice that show propagation into indicated subcortical structures.
432. Infant brainstem is prone to the generation of spreading depression during severe hypoxia
M. Müller, M. Kron, M. Dutschmann and F. Funke
Zentrum Physiologie und Pathophysiologie, Universität Göttingen, Göttingen, Germany
Background and aims: Spreading depression (SD) resembles a concerted, massive neuronal/glial depolarization. Being associated with cerebropathology it is well studied in cortex and hippocampus. Brainstem tissue, however, is considered to be quite resistant to the generation of SD. Nevertheless, there have been recent in vivo studies showing that – upon proper preconditioning—SD can also be induced in the brainstem of rats.1,2 We have now analyzed under which conditions hypoxic SD (HSD) can be induced in rat brainstem slices.
Methods: Acute brainstem slices (400 μm) were prepared from infant and adult rats and placed in an interface recording chamber (35°C to 36°C). HSD was induced by severe hypoxia (95% N2, 5% CO2) and was followed by extracellular DC potential recordings and optical imaging of the intrinsic optical signal (IOS), complemented by monitoring extracellular K+ changes and sharp-electrode recordings from single neurons.
Purpose: Detailed analysis of the spatiotemporal profile of HSD in acute rat brainstem slices.
Results: In brainstem slices (preconditioned by increasing extracellular K+ to 8 mmol/L) severe hypoxia triggered SD within a few minutes. The sudden HSD-related extracellular DC potential shift of approximately −20 mV showed the typical profile known from other brain regions and was accompanied by an IOS, i.e. an increase in light scattering within the tissue.3 Spatiotemporal IOS analysis revealed that in infant brainstem, HSD was preferably ignited within the spinal trigeminal nucleus (Sp5) and spread out mostly medially invading the hypoglossal nucleus, the nucleus of the solitary tract (NTS), and occasionally the ventral respiratory group (VRG). The neuronal hypoxic depolarizations underlying the generation of HSD were massive, but incomplete. At the peak of HSD Sp5 neurons maintained a membrane potential of −35 mV. The propagation velocity of HSD (3.1±1.5 mm/mins, n = 17) and the extracellular K+ peak level (49.5±19.4 mmol/L, n = 8) were less marked than in other brain regions. In adult brainstem HSD was mostly confined to the NTS. Its occurrence was facilitated by hypotonic solutions (15 mmol/L NaCl omitted) but not by glial poisoning (5 mmol/L fluoroacetate, 3 to 5 h) or block of GABAergic and glycinergic synapses (20 μmol/L bicuculline plus 0.5 μmol/L strychnine, 20 mins).
Conclusions: Brainstem tissue reliably generates propagating HSD episodes, which may be of interest for basilar-type migraine and brainstem infarcts. The preferred occurrence of HSD in the infant brainstem and its propagation into the VRG may be of importance for neonatal brainstem pathology such as sudden infant death syndrome.
Supported by the DFG (CMPB) and the BMBF (BCCN-Göttingen).
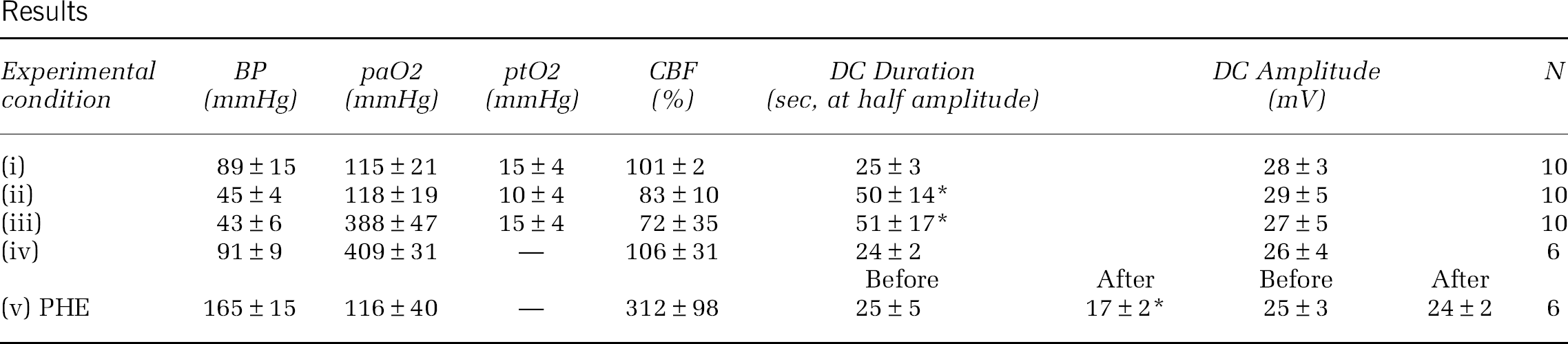
468. Cortical spreading depression recovery is modulated by tissue perfusion pressure rather than oxygenation
I. Sukhotinsky1, J. Xu1, J.R. Sims1 and C. Ayata1,2
1Stroke and Neurovascular Regulation Laboratory; 2Stroke Service and Neuroscience Intensive Care Unit, Massachusetss General Hospital/Harvard Medical School, Boston, Massachusetts, USA
Background and aims: We have recently shown that hypotension markedly prolongs the DC shift duration during CSD, while hypoxia under normotensive conditions had a much more modest effect, suggesting that perfusion pressure is a more important determinant of CSD recovery than tissue oxygenation. We hypothesized that the prolongation of DC shift during hypotension is due to tissue hypoxia, and tested whether normobaric hyperoxia restored the DC shift duration in hypotensive rats, and whether induced hypertension shortens the DC shift duration in normoxic rats.
Methods: Urethane-anesthetized rats were intubated and mechanically ventilated. Arterial blood pressure, gases and pH were monitored. CSD was induced by topical KCl (1 M) application, and recorded using intracortical glass microelectrodes. Associated CBF changes were recorded by laser Doppler flowmetry. Absolute tissue partial pressure of O2 (ptO2) was measured by phosphorescence lifetime using an intracortical electrode (OxyLite, Oxford Optronics). Experimental conditions were:
normoxic normotension,
normoxic hypotension,
hyperoxic hypotension,
hyperoxic normotension, and
normoxic hypertension.
Hypotension was achieved by controlled blood withdrawal, hypertension by intravenous phenylephrine infusion (PHE, 2 mg/ml, 3 ml/h), and normobaric hyperoxia by inhalation of 100% O2. Data are mean±SD. Conditions (i), (ii), (iii), and (iv) were studied within the same rat and compared among each other, while (v) was studied in a separate group of rats and data before and after PHE were compared. *P< 0.01 vs. control.
Results
Experimental condition
BP (mmHg)
paO2 (mmHg)
ptO2 (mmHg)
CBF (%)
DC Duration (sec, at half amplitude)
DC Amplitude (mV)
N
(i)
89±15
115±21
15±4
101±2
25±3
28±3
10
(ii)
45±4
118±19
10±4
83±10
50±14*
29±5
10
(iii)
43±6
388±47
15±4
72±35
51±17*
27±5
10
(iv)
91±9
409±31
—
106±31
24±2
26±4
6
Before
After
Before
After
(v) PHE
165±15
116±40
—
312±98
25±5
17±2*
25±3
24±2
6
Purpose: To test whether normobaric hyperoxia restores the DC shift prolongation during CSD under hypotensive conditions.
Results: Normoxic hypotension doubled the DC shift duration compared to normoxic normotensive rats, and transformed the CBF response by unmasking an initial hypoperfusion as reported previously. Normobaric hyperoxia normalized baseline ptO2 in hypotensive animals, but failed to restore the DC shift duration or the CBF response. Normobaric hyperoxia did not alter the DC shift duration or CBF response in normotensive animals either. Normoxic hypertension significantly increased baseline CBF (breakthrough hyperemia) and shortened the DC shift duration by ∼30% without changing the peak hyperemia during CSD.
Conclusion: Our data suggest that blood pressure is a critical determinant of CSD recovery and the CBF response to CSD. Hyperoxia did not reverse the hypotensive prolongation of CSD despite normalized ptO2, suggesting that the mechanism does not involve tissue hypoxia. Instead, the initial hypoperfusion and diminished hyperemia during CSD may play a role in delaying the DC shift recovery in hypotensive rats. The data also suggest that hypotension may be detrimental to tissue outcome in stroke, subarachnoid and intracerebral hemorrhage, and traumatic brain injury, where injury depolarizations have been observed.
596. ‘Normal’ and ‘inverse’ neurovascular coupling to cortical spreading depolarization (CSD) in the human brain
S. Major1,2,3, J. Woitzik4, C. Drenckhahn1,2,3, J.A. Hartings5, M. Fabricius6, A.J. Strong7 and J.P. Dreier1,2,3
1Neurology; 2Experimental Neurology; 3Center for Stroke Research (CSB) Berlin; 4Neurosurgery, Charité – University Medicine Berlin, Berlin, Germany; 5Neurosurgery, University of Cincinnati, Cincinnati, Ohio, USA; 6Clinical Neurophysiology, University of Copenhagen, Glostrup Hospital, Copenhagen, Denmark; 7Neurosurgery, King's College, London, UK
Background: CSD is a wave of neuronal depolarization which propagates in the cortex at a rate of ∼3 mm/mins. It induces vasodilation in healthy tissue. Hence, regional cerebral blood flow (rCBF) increases in response to CSD, a process termed ‘normal’ neurovascular coupling. Only after the neuronal repolarization, the vasodilation is followed by mild vasoconstriction resulting in spreading oligemia. The opposite of this ‘normal’ neurovascular response, termed ‘inverse’ neurovascular coupling, occurs when there is local dysfunction of the microvasculature. With ′inverse′ coupling, severe microvascular spasm instead of vasodilation is coupled to CSD (Dreier et al. (1998) J Cereb Blood Flow Metab, 18, 978–990). The resulting spreading perfusion deficit in turn prolongs the neuronal depolarization (as reflected by a prolonged negative shift of the direct current (DC) potential) since the oxygen-/glucose deprivation further reduces ATP availability. Thus, in animal experiments, a prolonged negative cortical DC shift, the defining electrophysiologic feature of ′inverse′ neurovascular coupling, indicates that the hypoperfusion is significant enough to produce a mismatch between neuronal energy demand and supply. Accordingly, the term ′cortical spreading ischemia′ describes the CSD-induced initial perfusion deficit when it leads to a prolonged negative cortical DC shift. Here we studied the occurrence of ‘normal’ and ‘inverse’ coupling in aneurysmal subarachnoid hemorrhage (aSAH) patients.
Methods: A prospective study was performed in thirteen patients with aSAH using subdural opto-/electrodes for simultaneous laser-Doppler flowmetry and DC-electrocorticography (DC-ECoG) combined with measurements of tissue partial pressure of oxygen (ptiO2). CSDs occurring in temporal clusters and in isolation were analyzed separately. Of each patient the isolated CSD with the longest ECoG depression period was analyzed here.
Results: Twelve patients showed isolated CSDs with a propagation velocity of 2.1 (1.8, 2.9) mm/min. The DC potential showed a negative shift of −10.8 (−9.7, −13.7) mV (duration: 153 [123, 283] s). Four of 12 CSDs analyzed displayed ‘normal’ neurovascular coupling since the predominant response was the initial hyperemia (257 (211, 311)% lasting for 689 (512, 812) s) followed by spreading oligemia to 92 (79, 102)%. Another three of the 12 CSDs did not show any significant change in level of rCBF. This behavior was rated as neurovascular ‘uncoupling’. ‘Inverse’ coupling was observed in the remaining 5 of 12 isolated CSDs and was characterized by the following:
the DC potential change preceded the initial hypoperfusion by 65 (16, 83) s;
rCBF fell to 59 (31, 62)% lasting for 118 (88, 265) s;
the local hypoperfusion propagated with the cortical DC potential negativity in the tissue at a rate of 3.5 (1.6, 3.5) mm/mins.
Linear regression found that the durations of the initial hypoperfusion and the cortical DC potential negativity were significantly correlated (R = 0.92, P<0.001, n = 8). Moreover, linear regression demonstrated a significant correlation between initial changes in level of rCBF and ptiO2 in response to CSD (R = 0.84, P = 0.019, n = 7).
Conclusion: Similar to animal experiments, ‘normal’ and ‘inverse’ coupling occur in the human brain. Thus, ‘inverse’ coupling is identified as a novel human disease mechanism and potential target for therapeutic intervention.
652. Evolution of cortical spreading depression is altered by aging and chronic cerebral hypoperfusion
E. Farkas1, L. Lenti1, L. Kemény1, T.P. Obrenovitch2 and F. Bari1
1Department of Physiology, Faculty of Medicine, University of Szeged, Szeged, Hungary; 2Division of Pharmacology, School of Life Sciences, University of Bradford, Bradford, UK
Objectives: Cortical spreading depression (CSD) are propagating waves of cellular depolarization that are associated with classical migraine and acute brain lesions. Aging and chronic cerebral hypoperfusion (CCH) compromise neural integrity and cerebrovascular regulation, yet their effect on CSD generation has not been established. Our objective was to determine the impact of age and CCH on the evolution of experimentally induced CSD and corresponding changes of cerebral blood flow (CBF).
Methods: Adult male Wistar rats were divided into 3 groups. Group 1 consisted of 2 month-old naïve animals. Groups 2 and 3 underwent either sham-operation (SHAM) or permanent, bilateral common carotid artery occlusion (2VO; a widely used model for CCH), under chloral-hydrate anesthesia (400 mg/kg, i.p.) at the age of 6 to 8 weeks, and survived for 6 months after 2VO onset. In all animals, two craniotomies were created on the right parietal bone (5 mm apart) under halothane anesthesia (in N2O:O2 = 2:1), at the age of 2 months (group 1, n = 6) or 8 months (group 2, n = 6; and group 3, n = 5). The rats were freely breathing during surgery and subsequent data acquisition. Mean arterial pressure was monitored continuously through a catheter inserted into the left femoral artery. The anterior open cranial window incorporated a glass capillary electrode for the recording of EEG and direct current (DC) potential, and a laser Doppler probe for the monitoring of cortical CBF. The posterior craniotomy was used to trigger CSD by the application of 1 M KCl, which was left in the window after the first application, and refreshed every 15 mins during recording. The variables were continuously acquired for 2 h. Each experiment was terminated by cardiac arrest, achieved by the injection of 1 ml air through a venous line in the left femoral vein.
Results: The frequency of CSD generation seen on the EEG recording as silencing of the signal was significantly lower due to aging (group 1: 9±6.0/2 h, group 2: 2±1.0/2 h, group 3: 3±2.0/2 h, median±s.d., Kruskall-Wallist test: P<0.033*). The occurrence of CSD was progressively more frequent toward the end of the 2-h recording period in all 3 groups. The degree of CSD-associated hyperemia showed an age-related tendency to decrease (group 1: 53%±7.4%, group 2: 47%±8.7%, group 3: 48%±10.4%, mean±s.e.m.). The sudden, transient drop of CBF typically preceding the CSD-associated hyperemia was less prominent in the older rats, especially under CCH (1st CSD: group 1: −34%±9.5%, group 2: −20%±6.1%, group 3: −13%±4.8%, mean±s.e.m.).
Conclusions: This study so far demonstrates that K+-induced CSD evolve less likely in older than in younger adult rats, implying a reduced sensitivity or higher threshold for CSD elicitation with aging. The impact of aging and CCH appears most obvious on the CSD-associated changes in CBF, which probably points to a less dynamic regulation of cerebrovascular reactivity. These findings prompt further analysis of CSD evolution in the aging brain, since CSD is known to occur during and after acute cerebrovascular events (e.g. subarachnoid hemorrhage, stroke), which are conditions more prevalent in later life.
Supported by the Hungarian Scientific Research Fund (OTKA; nr. K63401).
687. Does endothelin-1 induce cortical spreading depolarization (CSD) via a direct effect on the vasculature?
A.I. Oliveira-Ferreira, D. Milakara, D.V. Jorks, M. Alam, S. Major and J.P. Dreier
Center for Stroke Research Berlin, Charité – Universitätsmedizin Berlin, Berlin, Germany
Objectives: Endothelin-1 (ET-1) has attracted increasing interest since its discovery by Yanagisawa in 1988. Recognized as a neuropeptide with neurotransmitter/neuromodulator functions, ET-1 is also a potent vasoconstrictor. Moreover, it potently induces CSD in vivo. ET-1 has been related to several pathophysiogical states including subarachnoid hemorrhage, ischemic stroke and traumatic brain injury. The mechanism by which ET-1 induces CSD is not fully understood. Possibly ET-1-induced vasoconstriction leads to ischemia which causes CSD. Here we further analyzed whether ET-1-induced CSD is preceded by significant pial vasoconstriction and characterized pH changes and electrocorticogram (ECoG).
Methods: We used a two cranial window model in rats (n = 11). ET-1 was brain topically applied in one window at 100 nM and 1 μmol/L. A second window served as control. DC/AC-ECoG and laser-Doppler flowmetry were used to detect CSD. The pial arterioles were imaged onto a camera to assess whether significant vasoconstriction precedes ET-1-induced CSD. In the sequence of consecutive recorded images we marked our region of interest (ROI) at the right angle to the longitudinal axis of a pial arteriole. A single ROI is a line represented in MATLAB as a diagram with curves for the red, green and blue channels, where the x-axis represents length in pixel and the y-axis brightness. The blood vessel led to a clean valley with low noise in the green channel. The square root of absorbance between two inflection points was used to calculate the diameter value and diameter change over time. In another six experiments we recorded the pH changes associated with ET-1-induced CSD using pH-sensitive microelectrodes (comparison with CSDs in five control experiments).
Results: We observed a cluster of recurrent CSDs starting from the ET-1 superfused window and propagating to the control window. The cluster was associated with a negative DC shift of −2.6±2.2 mV on which transient negative DC shifts of CSDs were riding. This was accompanied by a positive DC shift of 0.9±0.9 mV in the control window superimposed with transient negative DC shifts. ET-1 induced significant vasoconstriction in large, medium and small pial arterioles before the first CSD (Wilcoxon Signed Rank Test). The magnitude of vasoconstriction was significantly more pronounced in medium and small compared to large arterioles (Repeated Measures ANOVA with Bonferroni Post-hoc test, Figure). Only in the presence of ET-1 a pH reduction to 7.20±0.07 preceded the sharp alkaline shift of the CSD in all experiments. The sharp alkaline shift coincided with the negative DC shift of CSD.
Conclusions: The pattern of the DC potential changes, the arteriolar constriction and pH reduction prior to the first cluster of CSDs support the notion that ET-1 induces CSDs due to its vasoconstrictive action. Our data may have implications for clinical conditions ranging from migraine to subarachnoid hemorrhage and ischemic stroke.
692. Propagation patterns of peri-infarct depolarization around ischemic foci in rat brain assessed by laser speckle flowmetry
T. Kumagai1, H. Nakamura1, M. Walberer2, S. Vollmar1, M. Sué1, H. Endepols1, G. Mies1, M. Schroeter2 and R. Graf1
1Max-Planck-Institute for Neurological Research; 2Department of Neurology, University of Cologne, Cologne, Germany
Objectives: Cortical spreading depression (CSD) and peri-infarct depolarization (PID) contribute to infarct expansion, however, the temporal and spatial aspects of this process are not fully elucidated. Alterations of cerebral blood flow (CBF) coupled to CSD/PID can be measured as a surrogate to assess CSD/PID propagation patterns in the surrounding of ischemic foci. We here studied CBF using Laser speckle flowmetry (LSF) in the cerebral cortex of rats in an embolic (macrosphere) stroke model.
Methods: In 6 male isoflurane anesthetized Wistar rats, a catheter was inserted into the left internal carotid artery. The left cranial bone was exposed (about 11 × 6 mm) and thinned to a thickness transparent enough to measure indicative CBF using LSF (CBFLSF). The LSF system (laser diode & detecting camera) was placed over the exposed area to acquire images at a rate of 40 frames/min. TiO2 macrospheres (diameter 0.335 mm) were injected through the catheter until a detectable CBFLSF change occurred. CBFLSF recording was continued for up to 5 h. Post-experimental evaluations of LSF images allowed three-dimensional spatial processing of CBFLSF changes over time to assess hemodynamic patterns around the ischemic core. Spatiotemporal patterns of CBFLSF changes were analyzed in 25 regions of interest (ROI: 1.4 × 1.4 mm) covering the whole field of view over frontal, parietal and temporal lobes.
Results: In 5 out of 6 rats, macrospheres caused occlusion of the middle cerebral artery (MCAo), and in 1 rat occlusion of the anterior cerebral artery (ACAo). In MCAo, the ischemic territory varied to some extent in size and location. In all rats, CSD/PID was observed after arterial occlusion. 1–3 mins after the initial CBFLSF decrease caused by the injection of macrospheres, the first one or two CSD/PID waves propagated radially or concentrically outwards starting from a primary ischemic center. These first waves seemed to act almost like a ‘switch’ that consecutively left a not well differentiated ischemic center behind and established an ischemic core with clearer borders than those seen in the primary center. Subsequent waves of CSD/PID showed variable patterns depending on the severity of the developing ischemia: In cases of severe ischemia, waves of CSD/PID emerged at the rim of the ischemic core and propagated circumferentially along the rim. In cases of moderate to mild ischemia, in contrast, waves of CSD/PID emerged at multiple foci within a wide zone surrounding the ischemic core, and appeared as a rather chaotic pattern of CSD/PID propagation. This pattern was particularly pronounced in the single case of ACAo.
Conclusions: The results indicate that depending on the severity of CBF reduction variable patterns of CSD/PID propagation develop in the surrounding of ischemic lesions including concentric, multifocal and circumferential appearance. It is conceivable that the various CSD/PID patterns serve as one mechanism of infarct maturation.
790. Evidence that cortical spreading depolarization (CSD) propagates preferentially along gyri in the human brain
D. Milakara1, G. Bohner1, S. Major2 and J. Dreier2
Objectives: Observations in gyrencephalic animals suggest that CSDs do not spread concentrically but propagate predominantly along gyri (Smith et al.J Anat 2001;537–554). The relatively thin layer of grey matter lining the sulci may limit the spread of CSD between neighbouring gyri, thus preventing concentrical spread. Here, we developed a method to study whether CSD propagation in the human brain is consistent with a spread along gyri rather than a concentrical spread.
Methods: Eight CSDs were recorded in a patient with aneurysmal subarachnoid hemorrhage (aSAH) using subdural electrocorticography (ECoG). ECoG recordings were acquired continuously in 5 active channels from the 6-electrode (linear array) subdural strips. Electrode 1 served as ground while electrodes 2 to 6 (interelectrode distance 10 mm) were connected in sequential unipolar fashion to an amplifier, each referenced to an ipsilateral subgaleal platinum electrode. CSD was defined by the sequential onset in adjacent channels of a propagating slow potential change (SPC). A CT scan was performed during ECoG monitoring to visualize the platinum electrodes which are invisible in MRI scans. The electrode strip was then superimposed from the CT scan onto the 3D rendered brain surface of the MRI. If two electrodes were positioned on one gyrus, the shortest pathlength between the two electrodes was determined following the ridge of that gyrus. Then the CSD velocity was calculated from the latencies of the SPC onsets between electrode pairs divided by the measured pathlength. This MRI-corrected velocity was compared with the uncorrected velocity assuming an ideal linear spread between electrodes.
Results: The boxplots demonstrate that large differences of the propagation velocities were observed between different electrode pairs assuming a linear spread along the recording strip. Between subsequent CSDs propagation velocity was remarkably stable. If the velocities were corrected according to the anatomical brain surface assuming a preferential spread of CSD along a gyrus, the differences of the propagation velocities between different electrode pairs decreased significantly (difference between highest and lowest recorded speed: 1.0 (0.8, 1.1) versus 3.2 (3.1, 3.3) mm/min, paired t-test, n = 8, P<0.0001).
Conclusions: The data suggest that CSD spread is hindered by sulci in the human brain in a fashion similar to that in other gyrencephalic species. This has to be confirmed in a larger study.
CSD Velocity.
807. Shift of frequency distribution function of RBC velocity in parenchymal capillaries during potassium-induced cortical spreading depression in anesthetized rats
M. Unekawa1, M. Tomita1, Y. Tomita1,2, H. Hattori1, H. Toriumi1 and N. Suzuki1
1Neurology; 2Preventive Medicine for Cerebrovascular Disease, School of Medicine, Keio University, Tokyo, Japan
Object: Cortical spreading depression (CSD) is a mass depolarization of neurons, and vascular changes occur concomitantly with CSD. We have already reported a repetitive wave-ring spread of decrease and increase of microvascular flow during potassium-induced CSD in rats (Tomita M et al.JCBFM 2005;25:742). In this experiment, we focused on the velocity distribution of red blood cells (RBCs) in capillaries after application of potassium in cerebral cortex of anesthetized rat by employing an automated analyzing system developed by us (Tomita M et al.Microcirculation 2008;15:163).
Methods: In urethane-anesthetized male Wistar rats (n = 12), a cranial window was made in the left temporo-parietal region. FITC-labeled RBCs were injected through the femoral vein, and the labeled RBCs appearing in the region of interest (ROI) at 50 μm depth below the brain surface were tracked by means of high-speed camera (500 fps) laser-scanning confocal microscopy. Their velocities were calculated with Matlab-domain software, KEIO-IS2. Capillaries were defined as microvessels having a diameter of less than 10 μm. The frequency distribution function of RBC velocity was obtained in the control state and at 3 mins after KCl application by counting RBCs with velocities in each successive 0.5 mm/s range (Unekawa M et al.Asian Biomed 2008;2:203). CSD was induced by application of approximately 10 μl of KCl solution of 0.3 M or more on the brain surface around the ROI.
Results: Nine pairs (control and 3 mins after application) of microscopic images (3 to 10 s, or 1500 to 5000 frames each) were successfully obtained. From the data, 97 and 174 RBCs were defined as flowing in single capillaries among 3611 and 3894 detected RBCs, respectively. The average velocity decreased from 2.05±1.31 mm/s to 1.97±1.27 mm/s, but this decrease was not statistically significant. During CSD, there was transient flow cessation. However, it was too short to affect the average value. Analysis every 0.1 or 0.2 s revealed transient suppression of RBC appearance lasting 0.3 to 0.6 s in 4 rats. During the suppression period, the frequency distribution of RBC velocity in all vessels, including capillaries, arterioles, venules and veins, showed a notable decrease of RBCs with velocity in the range of 1.0 to 2.5 mm/s, but fast-flowing RBCs (more than 5 mm/s) persisted (Figure). During the same period, capillary RBCs disappeared or their velocity fell below the calculable limit of ca. 0.2 mm/s. Exceptionally, RBCs flow remained consistently fast in one capillary.
Conclusion: Potassium-induced CSD decreased the number of RBCs flowing in capillaries in the velocity range that was most frequently seen in the control, and a similar tendency was seen in non-capillary vessels.
Frequency distribution function of RBC velocity.
983. Cyclosporin a ameliorates prolonged oligemia in the wake of cortical spreading depression
H. Piilgaard1, P. Rasmussen1, B. Witgen1 and M. Lauritzen1,2
1Department of Neuroscience and Pharmacology, University of Copenhagen, Copenhagen N; 2Department of Clinical Neurophysiology, Glostrup Hospital, University of Copenhagen, Glostrup, Denmark
Introduction: Cortical spreading depression (CSD) is a slowly propagating depolarization wave of cerebral gray matter, which occurs in the cerebral cortex during migraine aura or traumatic brain injury. Associated to CSD are changes in Cerebral blood Flow (CBF): Initial vasoconstriction (not always seen) followed by vasodilation (during DC shift) followed by prolonged oligemia for up to 2 h. During an episode of CSD, neurons accumulate Ca2+ while free fatty acids and NO are released. This combination of events favors opening of the mitochondrial permeability transition pore (mPTP). Cyclosporin A (CsA) is a potent blocker of the mPTP and was used here to test the hypothesis that the mPTP was activated during CSD.
Methods: CBF changes were monitored through an open cranial window over the right somatosensory cortex by laser-Doppler flowmetry. Also tissue oxygenation (tpO2), local field potentials (LFP) and electrocorticogram (ECoG) were measured in the same area. Bipolar stimulation of transcallosal fibers projecting was performed in the homologous left somatosensory cortex by inserting a stimulation electrode directly into the cortex. Stimulations were given in 1 s lasting trains at 1, 6 and 20 Hz (1.5 mA, 0.1 ms lasting square pulses). Cerebral metabolic rate of oxygen (CMRO2) was calculated from the simultaneously recorded values of tpO2 and CBF as described by Gjedde1. Episodes of CSD were elicited by a brief needle stab to the right frontal cortex. After initial recordings of evoked responses, 0.1 mmol/L CsA was topically applied to the somatosensory cortex of the right hemisphere as desribed by Sullivan2 for 40 mins before repeating evoked responses and eliciting CSD. The impact of CsA on baseline CBF and CMRO2, and evoked synaptic, vascular, and metabolic responses after CSD (n = 8) was compared to the effect of CSD in untreated animals (n = 7).
Results: CsA augmented the maximal CBF response during CSD by 43.9% for the first few minutes (P = 0.04). CsA also reduced the maximum undershoot in CBF after CSD from 25.3±4.1% to 11. 9±2.1% (P = 0.02), an improvement of 53.1% without changing the time taken for CBF to return to normal. In comparison, baseline tpO2 and CMRO2 remained unchanged following CsA treatment. CsA itself had no influence on the evoked LFP, CBF, or CMRO2 response amplitudes following TC stimulation.
Conclusion: We conclude that CsA augments the acute CBF response during CSD and ameliorates the prolonged oligemia after CSD.
Perspectives: CSD causes CBF reduction by direct interaction with the blood vessels. We here hypothesise that this change of function relates mechanistically to activation of mPTP in the vessel walls. If so, this opens the possibility that blockers of mPTP may be useful for treating vascular dysfunction in neurological disorders in which CSD or CSD-like phenomena are implicated.
Grant support: Lundbeck Foundation via the Lundbeck Foundation Centre for Neurovascular Signalling (LUCENS), the NOVO Nordisk Foundation, and the Danish Medical Research Council.
References
1.
NedergaardM. Acta Neuropathol1987;73(3):267–74.
2.
Richter. JCBFM2008;28:984–94.
3.
RichterFRuprechtSLehmenkühlerASchaibleHG. Spreading depression can be elicited in brain stem of immature but not adult rats. J Neurophysiol2003;90:2163–70.
4.
RichterFBauerRLehmenkühlerASchaibleHG. Spreading depression in the brainstem of adult rat: Electrophysiological parameters and influences on regional brainstem blood flow. J Cereb Blood Flow Metab2008;28:984–94.
5.
MüllerMSomjenGG. Intrinsic optical signals in rat hippocampal slices during hypoxia-induced spreading depression-like depolarization. J Neurophysiol1999;82:1818–31.