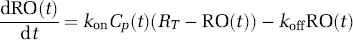542. Oral methylphenidate (Ritalin) displays high potency to block norepinephrine transporters: a PET study with (S,S)-[ 11C]MRB in healthy subjects
Y.-S. Ding1, J. Hannestad2, B. Planeta-Wilson1, J.-D. Gallezot1, S.-F. Lin1, J. Ropchan1, D. Labaree1, W. Williams2, R. Carson1 and C. Van Dyck2
1Diagnostic Radiology; 2Psychiatry, Yale University School of Medicine, New Haven, Connecticut, USA
Objectives: ADHD is a major psychiatric disorder with onset in childhood that continues into adulthood. The neurochemical mechanisms of ADHD are poorly understood. Methylphenidate (MP, Ritalin), the most commonly used drug for the treatment of ADHD, significantly blocks dopamine (DA) transporters at 60 mg oral dose;1 however, MP binds to the norepinephrine transporter (NET) with a higher inhibitory effect in vitro on NE uptake than on DA uptake (IC50 37.7 versus 193 nM). We report the first PET imaging study in healthy subjects using (S,S)-[11C]methylreboxetine (MRB), a promising NET ligand, to determine the duration and magnitude of NET occupancy by oral MP.
Methods: For the duration study, 4 scans were performed on 3 days, with subjects receiving placebo or oral MP (40 mg) 75, 150, and 225 mins before a scan. For the occupancy study, each subject had 4 PET scans after oral administration of single-blind placebo or MP 2.5 mg, 10 mg, and 40 mg. After injection of 11C-MRB (740 MBq), 2 h list mode data were acquired with the HRRT. Parametric non-displaceable binding potential (BPND) images were computed using the multilinear reference tissue model (MRTM2) with occipital cortex as the reference region. Regions of interest (ROIs) from the AAL template were analyzed; small regions, such as locus coeruleus (LC), brainstem, hypothalamus, and thalamic subnuclei, were also defined. BPND and IC50 values were estimated.
Results: From the duration study, there was no significant difference in the BPND values at 75 mins compared to 150 or 225 mins; thus, we chose 75 mins as the timing for the occupancy study. In the occupancy study, BPND was reduced by MP in a dose-dependent manner in all NET-rich regions. The avg. IC50 was 10 mg (range 6 to 15 mg, across all subjects [n = 6] and all ROIs9). At 40 mg MP, complete displacement was observed in LC, raphe and hypothalamus, whereas, 50% to 70% displacement was achieved in thalamus and its subnuclei.
Conclusions: Oral MP reached peak NET occupancy at approx. 75 mins (duration at least 3 h), occupying NET in a dose-dependent manner. Our data indicate that oral MP significantly blocks NET at clinically relevant doses; more importantly, these results demonstrate an IC50 for oral MP at NET (∼0.14 mg/kg) comparable to that previously reported at DAT (0.25 mg/kg), strongly suggesting a crucial role of NET in the treatment and pathophysiology of ADHD.
885. Investigation of atomoxetine occupancy of serotonin transporters
M. Naganawa1,2, D. Weinzimmer1, S.-F. Lin1, C. Sandiego1, J.-D. Gallezot1, Y. Huang1, R.E. Carson1, E.A. Rabiner3, M. Laruelle3 and Y.-S. Ding1
1Yale PET Center, Yale University School of Medicine, New Haven, Connecticut, USA; 2Molecular Imaging Center, National Institute of Radiological Sciences, Chiba, Japan; 3GlaxoSmithKline, London, UK
Objectives: Atomoxetine (ATX) is a putative selective norepinephrine transporter (NET) reuptake inhibitor and is used for treatment of depression and attention-deficit/hyperactivity disorder. We have shown that ATX displayed a dose-dependent occupancy on NET using PET and [11C]MRB, a selective NET radioligand.1 Our previous study also indicated that ATX (1.5 mg/kg) blocked the binding of the SERT ligand [11C]DASB in the baboon brain, an effect similar to that of fluoxetine (an SSRI).2 The purpose of this study was to determine if ATX, at clinically relevant doses, occupied SERT in a dose-dependent fashion in rhesus monkeys, using PET and [11C]AFM, a highly selective radioligand for SERT.3
Methods: Following a similar scanning paradigm as our previous study using [11C]MRB,1 rhesus monkeys were scanned four times with [11C]AFM (baseline & medium dose of ATX on day 1; low and high doses of ATX on day 2). ATX or saline infusion began 2 h before each scan, lasting until the end of the 2 h scan, to mimic the human oral dose PK profile. Infusion rates ranged 0.045 to 1.054 mg/kg/h. ATX plasma levels and arterial input functions were measured. Distribution volumes (VT) were estimated by one-tissue compartment model and ATX IC50 values were calculated.
Results: In baseline scans, regional brain [11C]AFM VT [mL/cm3] reflected the known distribution of SERT, with high binding in the brainstem (VT = 127±52) and thalamus (VT = 114±23), intermediate binding in temporal cortex (VT = 73±13), and lowest binding in the cerebellum (VT = 36±0.6). VT in the cerebellum was reduced by up to 32% with increasing ATX dose, suggesting that there was some contribution of specific binding to the cerebellum signal. Receptor occupancy (r) and non-displaceable volume of distribution (VND) were calculated from the occupancy model in the absence of an ideal reference region:4 VT (baseline)−VT (post-drug) = r × (VT (baseline)–VND). After administration of ATX, a dose-dependent occupancy from 49 to 90% was observed. The IC50 was estimated to be 167±16 ng/mL of plasma ATX concentration (corresponding to an infusion rate of 0.126±0.013 mg/kg/h). At a therapeutic ATX dose (1.8 mg/kg, ∼600 ng/mL plasma) ATX would have occupied ∼80% of SERT.
Conclusions: This study demonstrated that ATX inhibited [11C]AFM binding in rhesus monkey brain in a dose-dependent fashion. Compared with our reported ATX IC50 value for NET with [11C]MRB,3 the ATX in vivo IC50 ratio of SERT to NET was ∼6, consistent with the reported in vitro affinity (Kd) ratio of ∼4.5 (8.9 and 2 nmol/L for SERT and NET, respectively).5 Our comparative studies of ATX effects on NET and SERT suggest that ATX at clinical doses occupies both transporters. Thus, PET occupancy studies are important in clarifying the mechanism of ATX therapeutic action.
164. Characterising the relationship between plasma pharmacokinetics and occupancy following single dose allows prediction of repeat dose occupancy
S. Abanades1,2, J. Van Der Aart1, J. Barletta1, C. Marzano1, G. Searle1, J. Ahmad1, S. Zamuner3, V. Cunningham1, E. Rabiner1, M. Laruelle4 and R. Gunn1,5
1GSK, Clinical Imaging Center; 2Department of Experimental Toxicology, Imperial College, London, UK; 3GSK, CPMS, Verona, Italy; 4GSK, Schizophrenia & Cognition DPU, Harlow; 5Department of Engineering Science, University of Oxford, Oxford, UK
Objectives: PET receptor occupancy (RO) studies provide valuable information for dose selection and optimisation in clinical drug development. To date, occupancy data have mainly been analysed assuming a ‘direct’ relationship between plasma drug concentration and RO, however, these models may fail to characterise a system which is not in equilibrium. A more comprehensive modelling approach to link the pharmacokinetics of a drug in plasma (PK) and RO using an ‘indirect’ model is presented. The method is applied to predicting serotonin transporter (SERT) occupancy after repeat dosing (RD) of the SSRI duloxetine, from PET data acquired after a single dose (SD) of duloxetine.
Methods: Four healthy male volunteers were scanned 4 times with [11C]DASB on a Siemens HiRez scanner: baseline, two time points following SD of 20 mg duloxetine (times selected using an adaptive design in the interval 0 to 72 h), and once after RD (20 mg dose given daily for 4 days). Duloxetine plasma concentration was measured throughout the study. Regions of interest (ROI) were delineated on a coregistered MR-T1 image for the striatum, thalamus and midbrain, with the cerebellum as the reference region. ROI were applied to the PET data to generate time activity curves and the simplified reference tissue model was used to derive regional estimates of binding potential (BPND) allowing for the calculation of RO in scans 2, 3 and 4. Plasma duloxetine data following SD was fitted to a one compartment model with a first order absorption rate constant to derive continuous duloxetine SD plasma data and predicted RD plasma data. An indirect PK/RO model was fitted to the measured SD PET occupancy data to characterize the relationship between PK and RO.
Indirect PK/RO Model.
CP(t) is the duloxetine plasma concentration, kon and koff are the receptor association and dissociation rate constants, RO(t) is the RO time-course and RT is the maximum RO. This model was used to predict the duloxetine RD occupancy by applying it to the predicted RD PK time-course.
ResultsConclusions: Characterizing the relationship between plasma PK and RO of a drug following SD allows for prediction of RD occupancy.
Measured and Predicted RO for duloxetine at RD
Subject
Single dose
Repeat dose
Time (h)
Measured RO
Time (h)
Measured RO
Predicted RO
1
6.3
0.70
7.0
0.71
0.77
26.1
0.49
2
6.0
0.74
6.0
0.78
0.79
26.3
0.49
3
8.0
0.79
6.1
0.72
0.91
32.0
0.72
4
18.9
0.65
7.1
0.78
0.80
44.0
0.43
Mean injected activity = 235.5+−49.9MBq, Duloxetine plasma t1/2 = 11.1+−3.6 h.
232. Brain serotonin transporter occupancy by oral sibutramine dosed to steady state: a PET study using 11C-DASB in healthy humans
P.S. Talbot1, S. Bradley2, C.P. Clarke2, K.O. Babalola3, A.W. Philipp2, G. Brown1, A.W. Mcmahon1 and J.C. Matthews1
1Wolfson Molecular Imaging Centre, University of Manchester; 2Icon Development Solutions; 3Division of Imaging Science & Biomedical Engineering, University of Manchester, Manchester, UK
Background and aims: Sibutramine HCl is a monoamine reuptake inhibitor prescribed in a dose of 10 to 15 mg/day as an appetite suppressant in the clinical management of obesity. In vitro and in vivo animal data suggest that efficacy is mediated by its metabolites desmethylsibutramine (M1) and didesmethylsibutramine (M2) which have approximately 100-fold higher affinity for the 5-HT and norepinephrine reuptake transporters (SERT and NET, respectively) than the parent compound. However, there is a paucity of in vivo data in humans about mechanisms underlying:
clinical efficacy; and
the dose-independent non-response observed in a significant minority of patients. We present the first PET study investigating central actions of sibutramine at the SERT in humans.
Methods and purpose: 12 consenting, normal-weight, healthy males (mean age 40.7 yr; range 30 to 50 yr) completed a double-blind, placebo-controlled, balanced-order, within-subject crossover investigation of the central SERT occupancy (measured with 11C-DASB) associated with sibutramine 15 mg/day dosed to steady state over 5 days. Exploratory secondary analyses investigated correlations between SERT occupancy and
plasma concentrations of sibutramine, M1 and M2;
food intake as measured by a standardised test meal.
For the PET scans (2 per subject, on placebo and sibutramine), emission data were acquired for 100 mins on the High Resolution Research Tomograph (HRRT) following i.v. injection of 11C-DASB (498.5±105.8 MBq [13.5±2.9 mCi]); injected mass 4.5±2.6 μg; radiochemical purity 96.7%±1.1%. For each subject, ROIs (putamen, caudate, thalamus and brainstem [BS]) were extracted from a T1-weighted MRI using an automated method. Binding potentials (BPND) were calculated by Logan reference tissue method, using cerebellum as reference region. SERT occupancy was defined as % change in BPND between placebo and sibutramine conditions.
Results: Mean±s.d. baseline (placebo) BPND values across regions were: BS 0.60±0.09; caudate 0.82±0.24; putamen 1.22±0.32; thalamus 1.25±0.31. Mean occupancy across all regions and subjects was 30.0% ± 10.2% (range ∼10% to 50%), with a significantly greater proportion of the variability across subjects (ANOVA 63%) than regions (8%). There was no significant relationship between SERT occupancy and plasma concentration of either parent sibutramine or M1, and a positive correlation (trend; P = 0.09) between occupancy and M2. All subjects with significant appetite suppression (n = 5) had SERT occupancy in the upper range (∼25% to 50%). Non-responders (n = 7) had occupancy across the whole range. Very weak correlations were found between plasma concentrations and effect on appetite, mainly due to the high levels of variability in eating response across the group.
Conclusions: Our data would support the following:
SERT occupancy by clinical doses of sibutramine can be measured accurately by 11C-DASB PET and is of modest magnitude compared to clinically effective doses of SSRI antidepressants (∼80%);
SERT occupancy is predominantly mediated by M2 in humans;
SERT occupancy may be necessary but not sufficient for efficacy in humans, supporting preclinical data suggesting that the hypophagic effect of sibutramine requires the co-inhibition of both SERT and NET.
272. Fenfluramine decreases 5-HT1B binding OF [11C]AZ10419369 in the primate brain
S.J. Finnema1, A. Varrone1, T.J. Hwang1, B. Gulyás1, E. Pierson2, L. Farde1 and C. Halldin1
1Clinical Neuroscience, Karolinska Institutet, Stockholm, Sweden; 2CNS Discovery, AstraZeneca, Wilmington, Delaware, USA
Objectives: We recently reported an initial PET-study using the selective serotonin 5-HT1B receptor radioligand [11C]AZ10419369.1 The 5-HT1B receptor has a role in the modulation of synaptic serotonin release. The aim of the present study was to assess the sensitivity of [11C]AZ10419369 binding to pharmacological manipulation of endogenous serotonin levels in cynomolgus monkeys.
Methods: A total of 12 PET measurements were conducted on six experimental days in four cynomolgus monkeys. On each day two measurements were performed using i.v. bolus administration of [11C]AZ10419369. A baseline measurement was followed by a displacement measurement in which fenfluramine (1.0 or 5.0 mg/kg) was infused i.v. between 15 and 20 mins after radioligand injection. The monkeys were anaesthetized with sevofluran (2% to 5%), except in two measurements where a mixture of ketamine and xylazine was used. Emission data were acquired for 123 mins using the HRRT PET-system. The specific binding ratio was calculated as the ratio of the area under the curve (45 to 123 mins) of the target region to the reference regions (cerebellum). The displacement effect was estimated as relative change (%) in specific binding ratio.
Results: Administration of fenfluramine had no evident effect on radioactivity in the reference region (cerebellum). After administration of fenfluramine (1.0 and 5.0 mg/kg), the respective binding ratios decreased in the occipital cortex by 33%±11% and 49%±19%, in striatopallidal complex by 18%±23% and 38%±9%, and in midbrain by 28%±18% and 49%±16%, respectively.
Conclusions: This preliminary study supports that the new 5-HT1B-ligand [11C]AZ10419369 is sensitive to endogenous serotonin levels in vivo. The radioligand may accordingly serve as a tool to further examine serotonin-related brain functions and psychiatric disorders, as well as effects of drugs on endogenous levels of serotonin in brain.
531. The serotonin 4 receptor PET-ligand [11C]SB207145: sensitivity to occupancy by unlabeled ligand and to endogenous serotonin
L. Marner1,2, N. Gillings2,3, K. Madsen1,2, D. Erritzoe1,2, W. Baaré2,4, C. Svarer1,2, S.G. Hasselbalch1,2 and G.M. Knudsen1,2
1Neurobiology Research Unit, University Hospital Rigshospitalet; 2Center for Integrated Molecular Brain Imaging; 3PET and Cyclotron Unit, Copenhagen University Hospital Rigshospitalet; 4Danish Research Center for Magnetic Resonance, University Hospital Hvidovre, Copenhagen, Denmark
Background and aims: The serotonin 4 (5-HT4) receptor is involved in learning and memory and is a potential target for treatment of Alzheimer′s disease and depression. [11C]SB207145 has emerged as a useful radiotracer for quantitative PET-imaging of the cerebral 5-HT4 receptors in humans.1 In this study we investigate the in vivo affinity, KDapp, and the radiotracer‘s susceptibility to changes in endogenous serotonin.
Methods: Sixteen healthy subjects (age—range 20 to 45 years, 8 males) underwent a 2-h dynamic [11C]SB207145 PET examination. Two scans were performed on the same day in 13 subjects, of which seven received pharmacological challenges consisting of a 3-day blockage of the 5-HT1A autoreceptors by a partial agonist/beta-adrenoceptor antagonist, pindolol, and a 60-mins infusion of the selective serotonin reuptake inhibitor, citalopram, initiated about 30 mins prior to the second injection of [11C]SB207145. Volumes of interest were delineated automatically on coregistered 3T magnetic resonance images and time-activity curves were extracted. Modeling of BPND was performed using simplified reference tissue model with cerebellum as reference region.
The concentration of free unlabelled ligand (F) was estimated from the cerebellar time activity curve (40 to 110 mins) as the mean radioactive concentration in cerebellum divided by the specific radioactivity. The bound ligand (B) was estimated as the difference between radioactive concentration in striatum and cerebellum divided by the specific radioactivity.
Results: The range in amount of injected unlabelled ligand enabled the estimation of a population-based KDapp of 1.2±0.53 nmol/L (±s.e.) as the negative inverse slope of a scatchard plot (Figure). Subsequently, the receptor occupancy (O) was estimated for each individual as: O = F/(F+KD) and the BPND was subsequently individually corrected by dividing by 1–O. An upper limit of 0.028 mg/kg (70 kg subject: 2.0 mg) of [11C]SB207145 per PET examination should ensure a receptor occupancy below 5%.
The occupancy was higher in females (11.9%) compared to the males (6.6%) (P = 0.046) that had a larger body distribution volume. Thus, we found a sex difference in non-corrected BPND (P = 0.02), which disappeared when correcting for occupancy. In spite of a significant increase in plasma prolactin level throughout the pharmacologically challenged scans as proxy for increased cerebral serotonin levels, BPND was unaltered in all tested regions (caudate nucleus, lentiform nucleus, insula, and hippocampus).
Conclusions: [11C]SB207145 is a valuable tool for non-invasive quantification of 5-HT4 receptors in the human brain. Due to its relatively high receptor affinity, KDapp = 1.2, and to a relatively low protein binding (fP = 0.25), a production with a relatively high specific radioactivity is required. The ligand is insensitive to acute changes in cerebral serotonin levels, which is an advantage when subjects are investigated under circumstances with possible fluctuations in the serotonin levels.