
Abstract
Reactive oxygen species, derived from hypoxia and reoxygenation during transient focal cerebral ischemia (tFCI), are associated with the signaling pathway that leads to neuronal survival or death, depending on the severity and duration of the ischemic insult. The Akt survival signaling pathway is regulated by oxidative stress and is implicated in activation of nuclear factor-κB(NF-κB). Mild cerebral ischemia in mice was used to induce increased levels of Akt phosphorylation in the cortex and striatum. To clarify the role of Akt activation by NF-κB after tFCI, we injected the specific Akt inhibitor IV that inhibits Akt phosphorylation/activation. Inhibition of Akt phosphorylation induced decreases in sequential NF-κB signaling after 30 mins of tFCI at 1 h. Furthermore, the downstream survival signals of the Akt pathway were also decreased. Akt inhibitor IV increased ischemic infarct volume and apoptotic-related DNA fragmentation. Superoxide production in the ischemic brains of mice pretreated with the Akt inhibitor was higher than in vehicle-treated mice. In addition, those pretreated mice showed a reduction of approximately 33% in copper/zinc-superoxide dismutase expression. We propose that Akt signaling exerts its neuroprotective role by NF-κB activation in oxidative cerebral ischemia in mice.
Introduction
Oxidative stress is generated during cerebral ischemia and reperfusion and is associated with the signaling pathways that lead to neuronal survival or death. Regulation of the oxidant state is important in its survival after cerebral ischemia through modulation of key signaling pathways (Saito et al, 2005; Zhang et al, 2007). Copper/zinc superoxide dismutase (SOD1) is an endogenous antioxidant enzyme that defends against oxidative stress. We have reported that overexpression of SOD1 has a neuroprotective role against focal cerebral ischemia (Fujimura et al, 2000; Kinouchi et al, 1991). Transcription of SOD1, the gene coding for copper/zinc SOD1, is regulated in response to stimuli including oxidative stress and cytokines (Chang et al, 2002; Kong et al, 1993). However, the signaling cascades that govern SOD1 expression in the brain are not well described.
The Rel/nuclear factor-κB (NF-κB) family of transcription factors has been implicated in the regulation of genes involved in immunity and inflammation, and of processes such as cell survival, apoptosis, and cell growth. Nuclear factor-κB has a key role in the regulation of cellular responses to oxidative stress (Denk et al, 2000). Opposing roles for NF-κB in the nervous system have been proposed, namely, neuroprotection versus neurodegeneration. Activation of NF-κB during cerebral ischemia has been reported to promote proapoptotic as well as antiapoptotic mechanisms (Irving et al, 2000; Schneider et al, 1999). We showed that NF-κB activation increased in mouse brains after transient focal cerebral ischemia (tFCI) (Huang et al, 2001; Song et al, 2005, 2007). Differences in these findings may result from the nature of the ischemic injuries (permanent versus transient, duration and severity of ischemia, and reperfusion) or from the interaction between phosphatidylinositol 3-kinase (PI3K)/Akt and oxidative stress. We hypothesized that PI3K/Akt signaling and NF-κB activation are directly linked with mild ischemic oxidative stress, whereas during severe ischemic insult, NF-κB activity is associated with high levels of oxidative stress, which leads to neuronal death. The relationship of oxidative stress to Akt/NF-κB signaling and cell survival/death in cerebral ischemia is largely unknown.
Akt, a serine/threonine protein kinase, has a critical role in controlling the balance between apoptosis and cell survival in response to extra- and intracellular signaling. Three isoforms, Akt1, Akt2, and Akt3, are homologous, but differ slightly in the localization of their regulatory phosphorylation sites in mammals. Akt1 is the predominant isoform in most tissue and requires phosphorylation at Ser474 and Thr308 for activation. The principal role of Akt is to facilitate growth factor-mediated cell survival and to block apoptotic cell death, which is achieved by phosphorylating and deactivating pro-apoptotic factors such as BAD, caspase-9, and murine double minute-2 (MDM2) (Blume-Jensen et al, 1998; Cardone et al, 1998; del Peso et al, 1997; Mayo and Donner, 2001). Akt also phosphorylates and inactivates glycogen synthase kinase-3β (GSK-3β), the inactivation of which prompts upregulation of cyclin D and enhances cell cycling (Srivastava and Pandey, 1998). Akt is regulated by oxidative stress for cell survival (Wang et al, 2000), and phosphorylates IkB kinase-α/β. Activated IkB kinase-α/β, in turn, causes activation and nuclear translocation of NF-κB-dependent prosurvival genes (Kane et al, 1999).
We pharmacologically studied the role of oxidative stress in the interplay of Akt activation and NF-κB signaling using Akt inhibitor IV (N-((E)-2-(5-(benzo[d]thiazol-2yl)-3-ethyl-1-phenyl-2-(N-methyl-N-vinylbenzenamino)1H-benzo[d]imidazolium iodide). This inhibitor was developed for selective blocking of Akt phosphorylation/activation by targeting the adenosine triphosphate binding site (Kau et al, 2003). Although antioxidant effects of PI3K/Akt were reported in central and peripheral neurons (Brunet et al, 2001), a direct regulatory role in antioxidant defenses remains unclear. The goal of this study was to elucidate the mechanisms underlying the interplay among oxidative stress, Akt, and NF-κB activity in neuronal survival and death after 30 mins of mild cerebral ischemia in mice.
Materials and methods
Focal Cerebral Ischemia
Experiments were performed in accordance with National Institutes of Health guidelines and were approved by Stanford University's Administrative Panel on Laboratory Animal Care. CD1 mice were purchased from Charles River Laboratories (Wilmington, MA, USA). Male mice (35 to 40 g) were subjected to 30 mins of tFCI and reperfusion. An 11.0-mm 5 to 0 surgical monofilament nylon suture, blunted at the tip, was introduced into the left internal carotid artery through the external carotid artery stump (Yang et al, 1994). The mice were anesthetized with 2.0% isoflurane in 30% oxygen and 70% nitrous oxide using a face mask. Rectal temperature was controlled at 37°C with a homeothermic blanket. After 30 mins of middle cerebral artery (MCA) occlusion, blood flow was restored by withdrawal of the nylon suture. The left femoral artery was cannulated for measurement of blood pressure and arterial blood gases, with samples for analysis being taken immediately after cannulation. Blood gas was analyzed with a pH/blood gas analyzer (Chiron Diagnostics Ltd, Essex, UK). The left common carotid artery was exposed and the left external carotid artery and its branches were electrocoagulated. Sham-operated mice did not undergo surgery. Physiologic parameters were monitored through-out the studies and values were the same as previously reported (Fujimura et al, 1999).
Akt Inhibitor IV: a Benzimidazole Derivative
To examine the role of the Akt pathway after tFCI, we injected Akt inhibitor IV intracerebroventricularly (i.c.v.) 1 h before tFCI. This inhibitor, purchased from Calbiochem (La Jolla, CA, USA), was dissolved in 0.5% dimethyl sulfoxide. The scalp was incised on the midline and the skull was exposed. Akt inhibitor IV (100 μmol/L in 0.5% dimethyl sulfoxide) and the vehicle (0.5% dimethyl sulfoxide) were injected i.c.v. (2 μL, bregma; 1.0 mm lateral, 0.2 mm posterior, 3.1 mm deep). For measurement of ischemic infarct volume, Akt inhibitor IV (100 μmol/L, 2 μL, i.c.v.) was injected three times 1 h before the onset of 30 mins of tFCI and 1 and 3 h after tFCI.
Western Blotting
The animals were decapitated after reperfusion under deep anesthesia with isoflurane (n = 4). Samples were obtained from the MCA territory brain tissue on the ischemic side, including the striatum and cortex, and were quickly frozen in powdered dry ice and kept at −80°C until use. Cytosolic and nuclear fractions were prepared from the ischemic brains using ProteoExtract (Calbiochem). A lysate was run on a sodium dodecyl sulfate gel, subsequently transferred to a polyvinylidene difluoride membrane, and incubated with primary antibodies for 24 h at 4°C, and then with secondary antibodies. The primary antibodies used were polyclonal antibodies against p50 (1:200; Abcam, Cambridgeshire, UK), p65 (1:200; Cell Signaling Technology, Cambridge, MA, USA), GSK-3β (1:300; Cell Signaling Technology), phosphor-MDM2 (1:300; Cell Signaling Technology), phospho-BAD (1:300; Cell Signaling Technology), Akt (1:500; Cell Signaling Technology), phospho-Akt (1:500; Cell Signaling Technology), SOD1 (1:1000; Stressgen, Victoria, BC, Canada), cyclooxygenase-2 (COX-2) (1:500; Cayman, Ann Arbor, MI, USA), TFIID (1:400; Santa Cruz Biotechnology, Santa Cruz, CA, USA), or β-actin monoclonal antibody (1:5000; Sigma-Aldrich, St Louis, MO, USA). After washing, the membrane was incubated with horseradish peroxidase-conjugated anti-mouse immunoglobulin G (Amersham International, Buckinghamshire, UK) or horseradish peroxidase-conjugated anti-rabbit immunoglobulin G at a 1:5000 dilution for 60 mins. The signal was then detected with a chemiluminescent kit (Thermo Scientific, Rockford, IL, USA). Multi-Analyst 1.0.2 software (Bio-Rad Laboratories, Hercules, CA, USA) was used for data analysis.
Akt Kinase, Phospho-Specific (Ser473) Enzyme-Linked Immunosorbent Assay
To detect phosphorylated Akt1 on Ser473, samples prepared from the MCA territory were added to microtiter wells, and the plate was incubated for 2 h at room temperature. After washing the plate, anti-Akt detector antibody solution was added to each well and incubated for 1 h at room temperature. An anti-rabbit immunoglobulin G-horseradish peroxidase working solution was added to each well after washing the plate and incubated for 30 mins at room temperature. After the stabilized chromogen was added to each well in the dark, the liquid in the wells began to turn blue. After 30 mins, we stopped adding the solution and mixed the contents of the plate. The solution in the wells changed from blue to yellow. The absorbance was read at 450 nm, having blanked the plate reader against a chromogen blank. The unit (units per mL) was calculated from the standard curve.
Microwell Colorimetric Nuclear Factor-κB Assay
The ability of NF-κB to bind to DNA consensus sequences was measured using a commercially available kit (NF-κB p65 EZ-TFA transcription factor assay; Upstate, Cambridge, MA, USA). This method was used as an alternative to electrophoretic mobility shift assay and has been reported to be sensitive and specific (Shen et al, 2002). According to the manufacturer's protocol, nuclear extracts were tested for their ability to bind to a biotinylated double-stranded oligonucleotide probe containing the consensus binding sequence for NF-κB. Nuclear factor-κB activity was determined by the sample's ability to bind to consensus sequences (5‘-GGGACTTTCC-3’) in a 96-well plate. A primary antibody recognizes an epitope on p65 and is accessible only when NF-κB is activated and bound to its target. DNA was added to the wells, followed by a secondary horseradish peroxidase-conjugated antibody. Developing solution (tetramethylbenzidine) was added and the colorimetric reaction was stopped by adding stop solution (0.5 mol/L HCl). After stopping the reaction, absorbance was measured on a spectrophotometer at 450 nm with a reference wavelength of 650 nm. HeLa whole-cell extract was used as a positive control for NF-κB activation. An unlabeled oligonucleotide containing the identical NF-κB consensus sequence as the capture probe was used to monitor the specificity of the assay as a competitor control. A negative control probe was used to ensure that the signal obtained from the normal assay setup was specific to the NF-κB consensus sequence.
In Situ Detection of Superoxide Anion Production
The early production of superoxide anions in cerebral ischemia was investigated using hydroethidine (HEt) by a previously described method (Saito et al, 2003). Hydroethidine is diffusible into the central nervous system parenchyma after an intravenous injection and is selectively oxidized to ethidium by superoxide anions, but not by other reactive oxygen species such as hydrogen peroxide, hydroxyl radicals, or peroxynitrite. Hydroethidine solution (200 μL; 1 mg/mL in phosphate-buffered saline) was administered intravenously 15 mins before ischemia induction as described. In the brains of the animals intravenously injected with HEt, fluorescence was assessed microscopically at excitation = 355 nm and emission > 415 nm for HEt detection or at excitation = 510 to 550 nm and emission > 580nm for ethidium detection. Animals were killed 1 h after tFCI by transcardial perfusion as described (n = 3 each). After fixation with 3.7% paraformaldehyde for 12 h the brains were sectioned at 50 μm on a vibratome. Subsequently, the slides were covered with Vectashield mounting medium with 4,6-diamidino-2-phenylindole (Vector laboratories, Burlingame, CA, USA). These sections were observed with a microscope under fluorescent light.
2,3,5-Triphenyltetrazolium Chloride Staining
After 24 h of reperfusion, the brains were removed and sliced coronally into five 1-mm sections in a mouse brain matrix. The slices were then incubated in 2% 2,3,5-triphenyltetrazolium chloride in saline for 10 mins at 37°C, followed by 3.7% formalin. Stained sections were scanned with a GS-700 imaging scanner (Bio-Rad).
Cell Death Assay
For quantification of apoptotic-related DNA fragmentation, we used a commercial enzyme immunoassay to determine cytoplasmic histone-associated DNA fragments (Roche Molecular Biochemicals, Mannheim, Germany), which detect apoptotic but not necrotic cell death. Samples were obtained from the entire MCA territory of the ischemic brain and from sham-operated brains (n = 4 each). Fresh brain tissue was cut into pieces after 4 and 24 h of reperfusion and homogenized. Cytosolic supernatants collected at 10,000g for 20 mins were used for the enzyme-linked immunosorbent (ELISA) assay following the manufacturer's protocol.
Quantification and Statistical Analysis
The data are expressed as mean ± s.d. Comparisons among multiple groups were performed by one-way analysis of variance with appropriate Bonferroni or Dunnet tests (GraphPad Prism; Oberlin, San Diego, CA, USA). P-values less than 0.05 were considered statistically significant.
Results
Mild Cerebral Ischemia Causes Increased Akt Phosphorylation
Mice were subjected to 30 mins of transient MCA occlusion followed by 1 and 6 h and 1 and 7 days of reperfusion (n = 6 each). Phospho-Akt (Ser473) and Akt were constitutively expressed in the cortex and striatum of the sham-operated brains. Phosphorylation of Akt was significantly increased and peaked at 1 h in the cortex and striatum after 30 mins of tFCI (Figure 1A). The increased levels of phospho-Akt returned to basal levels 24 h after tFCI. Akt showed no significant increase or decrease after reperfusion in either the striatum or cortex. Detection of phosphorylated Akt1 on Ser473 using a protein kinase B, phospho-specific (Ser473) ELISA kit also showed 3.1- and 4.7-fold increases, respectively, at 1 h in the cortex and striatum (Figure 1B; **P < 0.01). The ELISA did not detect increases in Akt phosphorylation at 6 h because of detection sensitivity. However, at 1 h, the increasing pattern of phosphorylated Akt shown by ELISA was the same as in the Western blot analysis of phosphorylated Akt in the ischemic brain. These data show that phosphorylation of Akt is temporarily accelerated in the cortex and striatum as early as 1 h after mild tFCI.
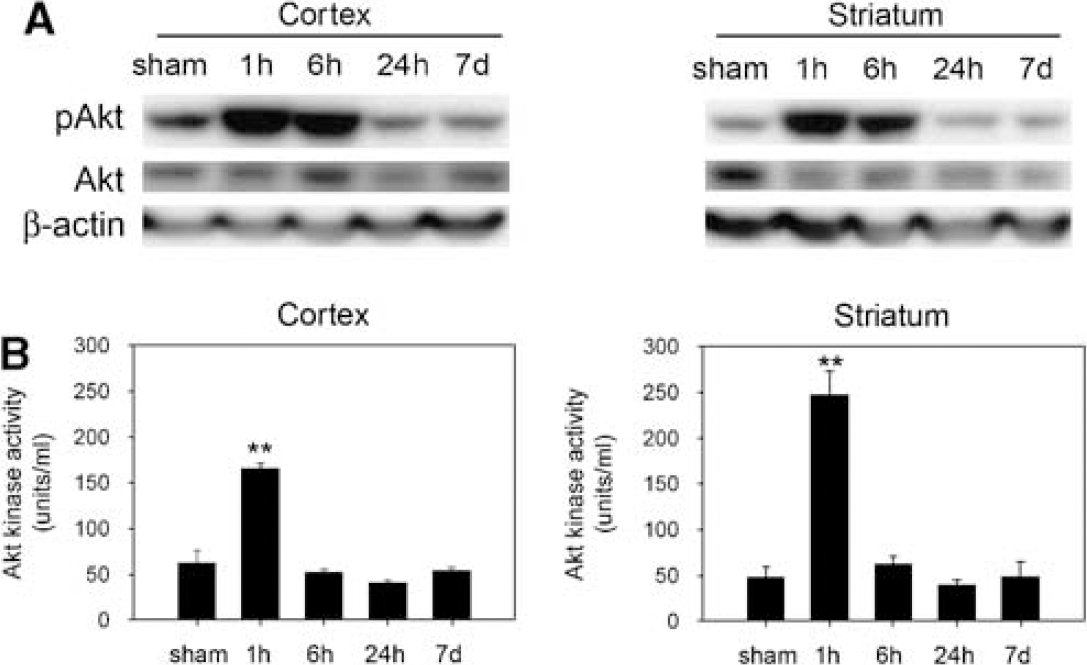
Mild cerebral ischemia induced phosphorylation of Akt at Ser473 in the cortex and striatum after 30 mins of tFCI. (
Akt Inhibitor IV Decreases Levels of Akt Phosphorylation and NF-κB Signaling
To investigate the role of Akt signaling in mild cerebral ischemia, we administered the specific Akt inhibitor IV to the mice. The drug was injected i.c.v. (100 μmol/L, 2 μL) 1h before the onset of 30 mins of MCA occlusion. In our preliminary studies, we used several different dosages and found that 100 μmol/L, 2 μL were effective in reducing phosphorylated Akt expression in the ischemic brain. As phosphorylation of Akt increased at an early time after 30 mins of tFCI, we investigated changes in Akt and its down-stream signaling 1 and 4 h after tFCI in vehicle- and Akt inhibitor-treated mice after 30 mins of tFCI. In :he vehicle-injected mice, 30 mins of tFCI induced increased levels of Akt phosphorylation at 1 and 4 h (Figure 2A). Akt inhibitor IV significantly blocked early Akt phosphorylation in the ischemic brain at 1h (Figure 2B; **P < 0.01), whereas, there was no change in its expression at 4 h, which may indicate that a single injection 1 h before tFCI is too little to maintain an effective drug concentration for blocking Akt phosphorylation up to 4 h. There was no significant increase or decrease in Akt with vehicle treatment or Akt inhibitor IV treatment after reperfusion.
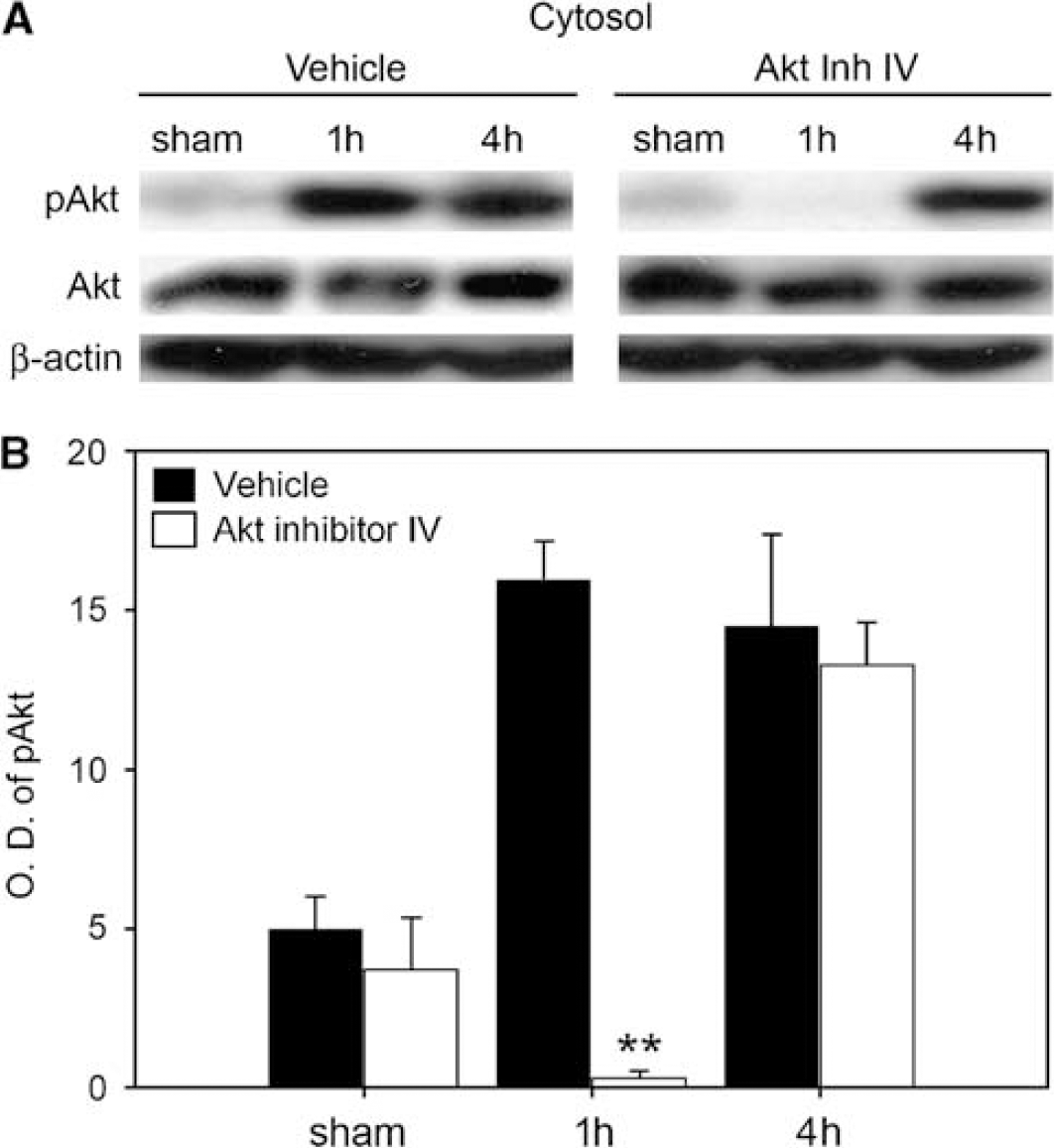
The specific Akt inhibitor IV induced decreased levels of Akt phosphorylation. The vehicle and Akt inhibitor (100 μmol/L, 2 μL, i.c.v.) were injected 1 h before onset of 30 mins of tFCI. Brain tissue extracts were subjected to 4% to 20% sodium dodecyl sulfate—polyacrylamide gel electrophoresis and blotted with antibodies against each protein. (
We examined activation of NF-κB signaling, one of the Akt downstream signaling pathways, after 30 mins of tFCI. In the vehicle-treated ischemic brains, phosphorylation and degradation of IκBα increased in the cytosol. In the Akt inhibitor-treated mice, blocking Akt phosphorylation induced decreases in phosphorylation of IκBα, nuclear translocation of p65 and p50, and NF-κB binding activity at the same time (1 h) as the decrease in Akt phosphorylation (Figure 3). This result suggests that Akt inhibition reduces NF-κB activation through its association with the NF-κB pathway in mild cerebral ischemia.
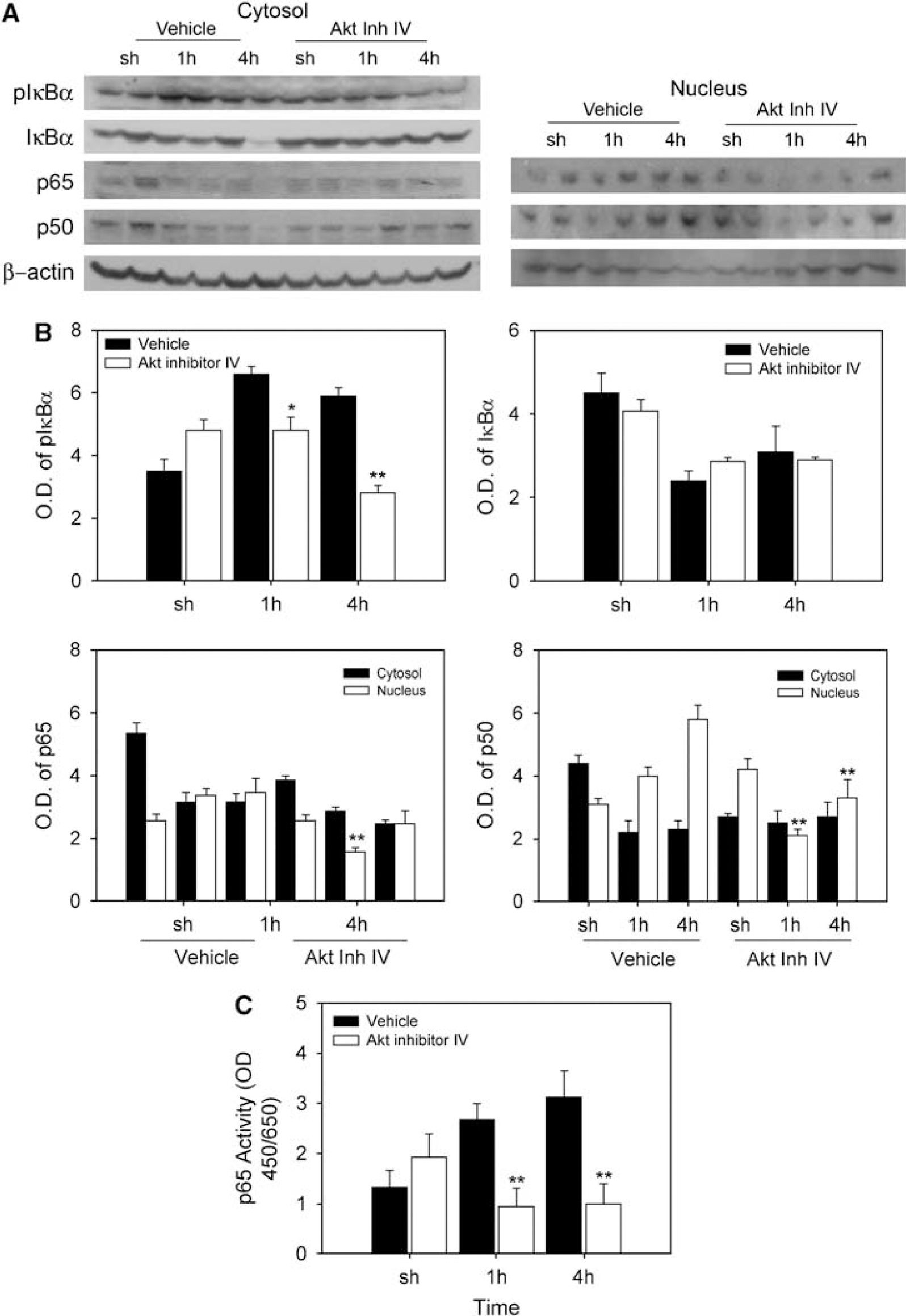
Akt inhibitor IV induced decreases in sequential NF-κB signaling, including phosphorylation of IκBα (plκBα), nuclear translocation of p65 and p50, and NF-κB activation. (
Inhibition of Akt Phosphorylation Reduces Antiapoptotic Signals, but Increases Cyclooxygenase-2
We also examined the antiapoptotic effects downstream of the Akt pathway. After reperfusion, phosphorylation of GSK-3β and MDM2 was increased in the cytosol and nucleus after 30 mins of tFCI in the vehicle-treated mice (Figure 4). In the Akt inhibitor-treated mice, phosphorylation of GSK-3β and MDM2 was decreased in the cytosol and nucleus after tFCI. Similarly, the increases in phospho-BAD in the vehicle-treated mice were also decreased in the Akt inhibitor-treated mice at 1 and 4 h, but the level of COX-2 (an inflammatory protein) in the Akt inhibitor-treated mice was higher than in the vehicle-treated mice after tFCI.
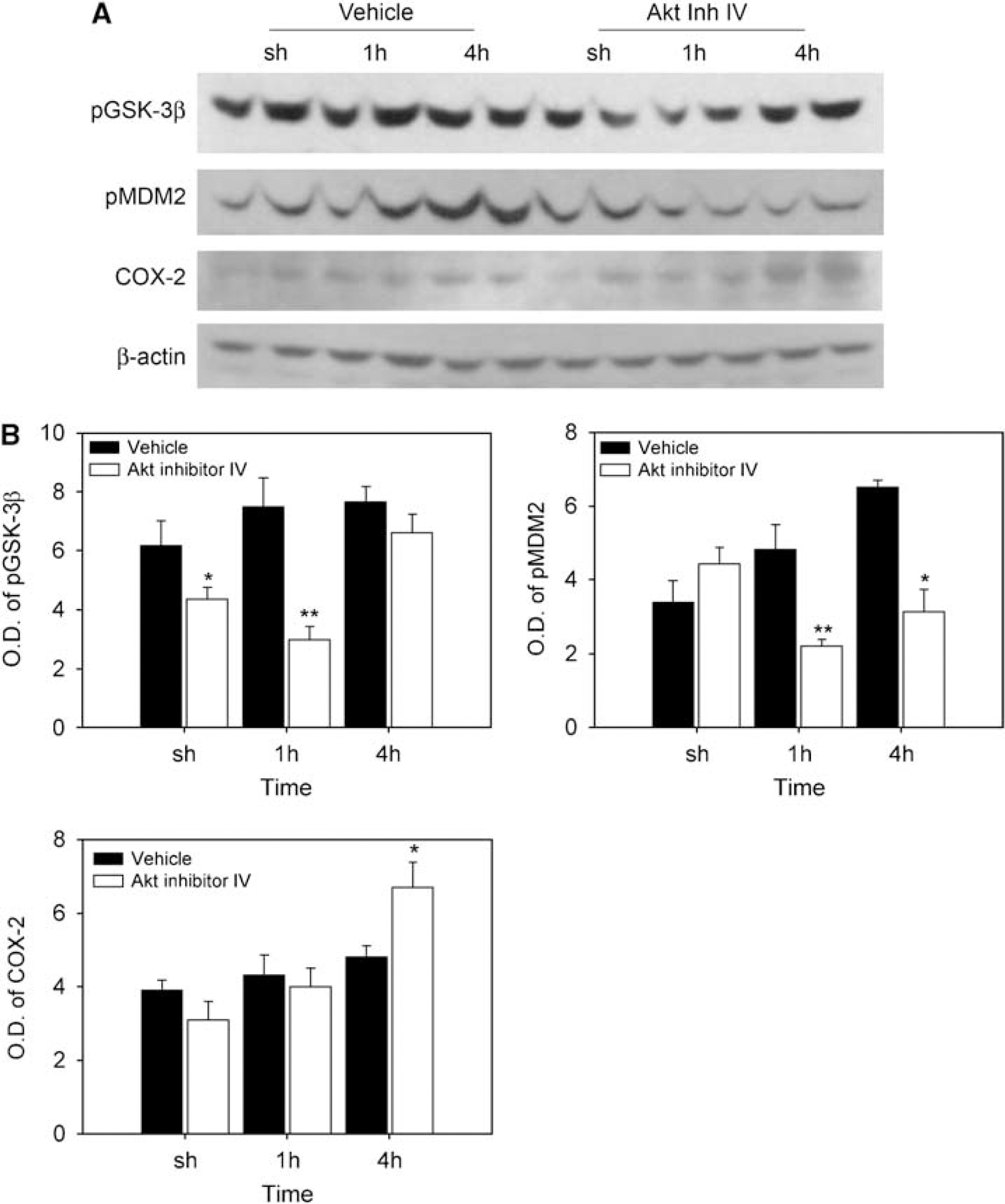
Changes in phosphorylation of GSK-3β, MDM2, and COX-2 in brain extracts after 30 mins of tFCI. (
Ischemic Infarction and Apoptotic-Related DNA Fragmentation Increase in Akt Inhibitor-Treated Mice
Akt has been implicated in the activation of NF-κB, whose role in the ischemic brain is not clearly understood. To examine the influence of Akt signaling in mild cerebral ischemia, we treated mice with the specific Akt inhibitor IV, and measured brain infarction volume and apoptotic cell death after 30 mins of tFCI. An ischemic lesion in the MCA territory was larger in the Akt inhibitor-treated mice than in the vehicle-treated mice, by 2,3,5-triphenyltetrazolium chloride staining, 24 h after reperfusion (Figure 5A). A cell death assay revealed that apoptotic-related DNA fragmentation in the Akt inhibitor-treated group increased by 52% and 21% in the striatum and cortex, respectively, compared with the vehicle-treated mice 24 h after reperfusion. A statistical study showed there was a significant difference (Figure 5B; *P < 0.05). Ischemic infarction and cell death were exacerbated more by treatment with Akt inhibitor IV than with the vehicle.
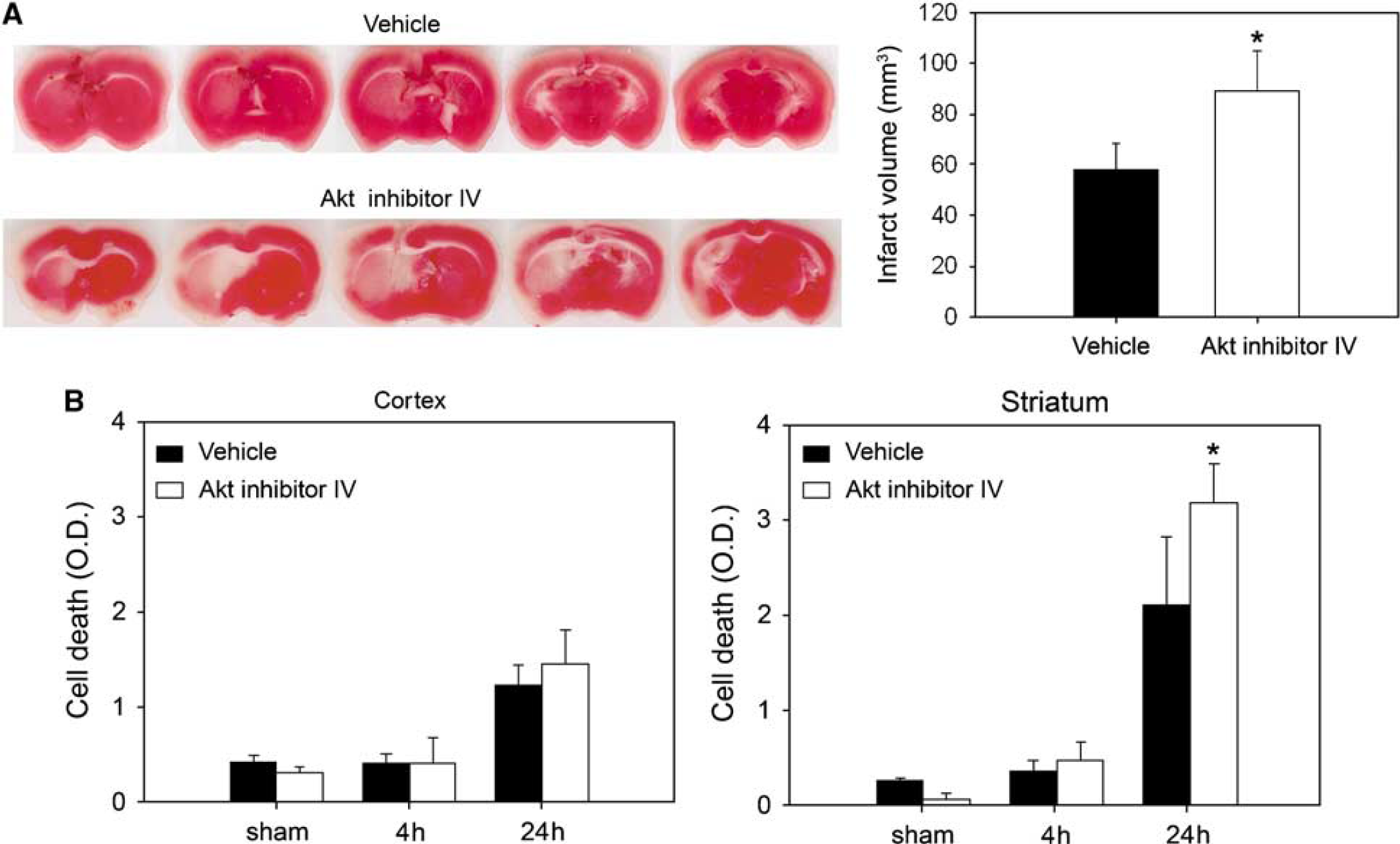
Akt inhibitor IV caused increases in infarct volume and apoptotic-related DNA fragmentation. (
Akt Inhibition Induces Superoxide Accumulation and Reduction in SOD1 Expression
The Akt signaling cascades involved in cell survival are complex and modulation by reactive oxygen species is not well understood. We examined production of superoxide and changes in expression of SOD1, the most abundant and ubiquitous antioxidant enzyme, after 30 mins of tFCI. The vehicle and Akt inhibitor IV were injected 1 h before onset of 30 mins of tFCI. We determined superoxide anion production using HEt in both the vehicle- and Akt inhibitor-treated mice. Oxidized HEt signals were strongly observed in the ischemic caudate 1 h after reperfusion in the vehicle-treated mice and were increased in the Akt inhibitor-treated mice compared with the vehicle-treated mice (Figure 6A). Superoxide dismutase expressed constituently in the sham-operated mice and the SOD1 band was observed at 19 kDa. Western blotting showed that the level of SOD1 expression in the vehicle-injected mice was decreased by injection of Akt inhibitor IV 1 h after 30 mins of tFCI. The graph in Figure 6B shows 33% inhibition of SOD1 expression in the Akt inhibitor-injected mice compared with the vehicle-treated mice 1 h after tFCI. These results suggest that Akt signaling has a neuroprotective role by reducing the level of superoxide against oxidative stress.
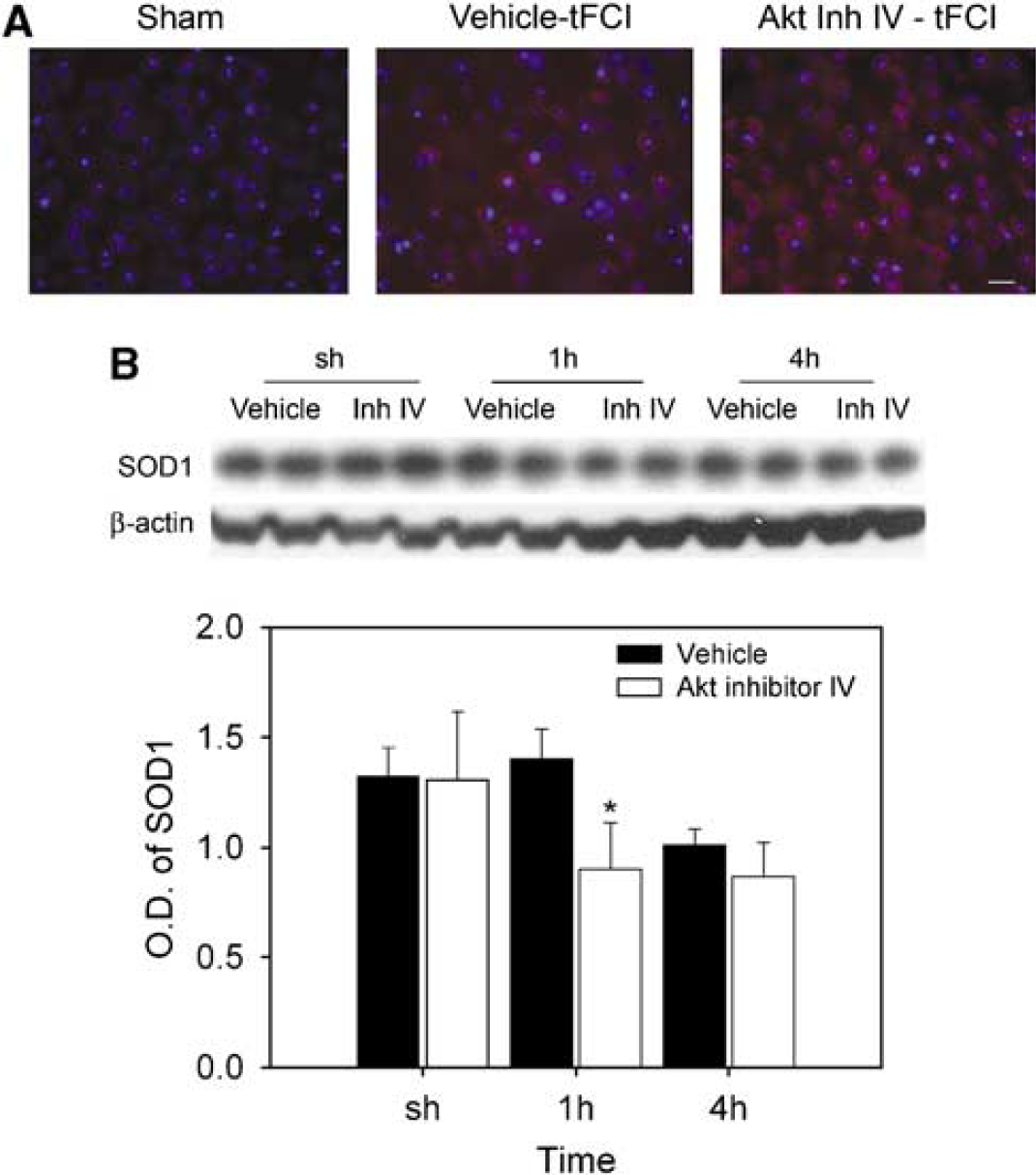
Effect of Akt inhibitor IV on the production of superoxide and SOD1 protein expression. (
Discussion
The intricate balance between the cell survival and death pathways has an important role in determining the fate of a cell. The Akt pathway and its downstream effectors are key players in preventing apoptosis and are necessary for cell survival (Kosmidou et al, 2001; Pham et al, 2000). It has been suggested that oxidative stress and Akt act in concert to modulate NF-κB activity, which determines the survival or death outcome. We have shown that PI3K, Akt, and NF-κB are involved in neuronal survival or death after cerebral ischemia and reperfusion (Chan, 2004; Noshita et al, 2003; Song et al, 2007). Regulation of Akt activation is significantly mediated by the level of reactive oxygen species. In the present study, 30 mins of tFCI induced increases in Akt phosphorylation at 1 and 4 h, which was different from decreases in Akt phosphorylation by 60 mins of tFCI (Noshita et al, 2001). Mild cerebral ischemic oxidative stress has been linked to Akt activation and transiently induced phosphorylation of Akt at an early time point. To elucidate the link between Akt signaling and NF-κB in mild cerebral ischemia, we investigated Akt and its downstream NF-κB signal in Akt inhibitor IV-treated mice after tFCI. Despite the greater understanding of the role of PI3K/Akt with the use of inhibitors such as wortmannin or LY294002, the study of Akt signaling has been limited by the lack of pharmacological specificity of the inhibitors. Unlike other phosphatidylinositol analog-based Akt inhibitors, Akt inhibitor IV does not affect PI3K and selectively blocks phosphorylation of Akt (Kau et al, 2003). Akt functions as a major downstream target of PI3K, and after phosphorylation, it phosphorylates IkB kinase-α/β. After cerebral ischemia and reperfusion, pharmacological inhibition of Akt phosphorylation induced decreases in sequential NF-κB signaling, including phosphorylation of IκBα, nuclear translocation of p65 and p50, and NF-κB binding activity, and increased brain infarct and neuronal apoptosis after cerebral ischemia. These results suggest that the linking of oxidative stress to Akt/NF-κB signaling is involved in a neuroprotective effect.
Recently, we showed that reduced oxidative stress increased NF-κB-related rapid defenses, such as immune response and antiapoptosis after tFCI (Song et al, 2007). A dual role for NF-κB has been suggested in neurodegenerative diseases; activation of NF-κB in neurons promotes their survival, whereas activation in glial cells mediates pathological inflammatory processes (Camandola and Mattson, 2007). The reasons for such a difference are the different stress, time points, brain regions, and cell types being investigated in the nervous system. Emerging research requires dissecting the pathways that lead to activation of the different NF-κB proteins and the gene targets of NF-κB in established experimental conditions. In PC12 cells, the PI3K/Akt pathway targeted the NF-κB site in the SOD1 promoter, resulting in upregulation of SOD1 gene expression, which decreased reactive oxygen species levels and protected cells against oxidative stress (Rojo et al, 2004).
We previously reported that the level of Akt phosphorylation was significantly increased in surviving neurons in mice that overexpressed SOD1 after tFCI (Noshita et al, 2003). In the present study, we showed that inhibition of Akt phosphorylation reduced production of superoxide and the level of SOD1 expression after 30 mins of tFCI. Although our study did not show a causal relationship between regulation of Akt and induction of SOD1 by mild cerebral ischemia, it suggests that one of the major mechanisms is that mild oxidative stress upregulates SOD1 by the activation of Akt and NF-κB. Overexpression of the SOD1 protein would reduce neuronal susceptibility to oxidative stress during reperfusion. In addition to NF-κB activation by phosphorylation of IkB kinase-α/β, Akt also phosphorylated GSK-3β, BAD, and MDM2, and decreased apoptosis (Blume-Jensen et al, 1998; Cardone et al, 1998; Cross et al, 1995). After administration of Akt inhibitor IV, the downstream survival proteins, including phosphorylated GSK-3β, and MDM2, were also decreased in the mildly ischemic brain, whereas COX-2 was increased. Therefore, the downregulation of Akt activation correlated with increases in brain infarct and apoptosis after 30 mins of tFCI. The present study reports on a novel molecular event, the decrease in NF-κB activation by inhibition of Akt activation after tFCI, and shows that inhibition of NF-κB activity triggers oxidative stress in cerebral ischemia. We propose that NF-κB exerts neuroprotection after oxidative tFCI in mice, in part, by Akt signaling mechanisms that involve downstream survival signals in the Akt pathway.
Disclosure/conflict of interest
None of the authors has a conflict of interest.
Footnotes
Acknowledgements
We thank Liza Reola and Bernard Calagui for technical assistance, Cheryl Christensen for editorial assistance, and Elizabeth Hoyte for assistance with figures.